


Abstract
The eye is a complex organ with a series of anatomic barriers that provide protection from physical and chemical injury while maintaining homeostasis and function. The physiology of the eye is multifaceted, with dynamic flows and clearance mechanisms. This review highlights that in vitro ocular transport and metabolism models are confined by the availability of clinically relevant absorption, distribution, metabolism, and excretion (ADME) data. In vitro ocular transport models used for pharmacology and toxicity poorly predict ocular exposure. Although ocular cell lines cannot replicate in vivo conditions, these models can help rank-order new chemical entities in discovery. Historic ocular metabolism of small molecules was assumed to be inconsequential or assessed using authentic standards. While various in vitro models have been cited, no single system is perfect, and many must be used in combination. Several studies document the use of laboratory animals for the prediction of ocular pharmacokinetics in humans. This review focuses on the use of human-relevant and human-derived models which can be utilized in discovery and development to understand ocular disposition of new chemical entities. The benefits and caveats of each model are discussed. Furthermore, ADME case studies are summarized retrospectively and capture the ADME data collected for health authorities in the absence of definitive guidelines. Finally, we discuss the novel technologies and a hypothesis-driven ocular drug classification system to provide a holistic perspective on the ADME properties of drugs administered by the ocular route.
Introduction
According to the Centers for Disease Control and Prevention, as of 2015, approximately 3.2 million people in the United States have been reported to have vision impairment as defined by the best-corrected visual acuity in the better-seeing eye, 8.2 million people had vision impairment from an uncorrected refractive error, and approximately 1 million people were blind (https://www.cdc.gov/visionhealth/risk/burden.htm Date accessed: March 30, 2018, Page last updated: October 30, 2017, content source: Division of Diabetes Translation, National Center for Chronic Disease Prevention and Health Promotion, maintained by: Centers for Disease Control and Prevention). By the year 2050, these numbers are estimated to double. Currently, at least 3.4 million are blind (visual acuity of 20/200 or less or a visual field on 20 degrees or less) or visually impaired (visual acuity of 20/40 or less), although other estimates indicate that this number may be as high as 21 million. Additionally, nearly 80 million people have diseases such as cataracts, glaucoma, diabetic retinopathy, age-related macular degeneration, and others, that may potentially lead to blindness (https://www.cdc.gov/visionhealth/basic_information/vision_loss.htm Date accessed: March 30, 2018, Page last updated: September 29, 2015, content source: Division of Diabetes Translation, National Center for Chronic Disease Prevention and Health Promotion, maintained by the Centers for Disease Control and Prevention). Blindness and severe vision impairment are devastating for patients, negatively impact their quality of life, and place a great burden on their caregivers owing to dependence for many activities, including seeing the ophthalmologist for treatment of their ocular disorders. Even a lower degree of vision impairment negatively impacts a patient’s quality of life. A large number of drugs, which include small molecules, biologics (antibodies and other proteins), gene/cell or other therapies (digital medicines) are currently being explored for their potential to treat ocular diseases. A variety of routes of administration, including topical ocular (eye drops), intravitreal, sub-retinal, or oral are being evaluated for administering such drugs. Additionally, a number of approved medicines are currently administered via topical ocular or intravitreal administration. Especially in cases of topical or local delivery, the eye is the site of “first pass” and consideration of drug disposition within the eye is of great importance to evaluate a drug’s ability to treat ocular disorders.
Most absorption, distribution, metabolism, and excretion (ADME) research has focused on the hepatic metabolism and transport of drugs, whereas the extrahepatic metabolism and transport of drugs have been relatively less understood. Within the field of extrahepatic metabolism, the eye is studied to a lesser extent than other organs, such as the kidney, intestines, or brain. Although relatively few drugs are administered directly to the eye, it is worth noting that many orally administered drugs can distribute from the bloodstream into the eye, as evidenced by ocular toxicities after oral or intravenous administration of certain drugs and the use of the vitreous as a matrix in forensics. Reasons for lack of attention to ocular metabolism/transport studies may include a lack of readily available ocular samples, the small size of the eye, the complexity of the tissue, the challenges in describing the pharmacokinetics (PK) of a drug in the eye, and a lack of awareness of the metabolic capability of the eye.
The objective of this review is to highlight ocular disposition and the underlying human-relevant in vitro models, case examples, in silico approaches, and current technologies. Strengths and limitations of each section are discussed in the context of research, development, and regulatory guidances. This article focuses predominantly on low-molecular-weight (LMW) drugs and new chemical entities (NCEs). Biologics, cell, and gene therapies are out of the scope of this review since the factors that drive their distribution and clearance differ markedly from the chemical modalities. Similarly, the role of ocular drug disposition in relation to a decision tree in pharmaceutical research and development or with respect to the regulatory guidance is a subject worthy of discrete and focused attention.
Challenges Associated with Investigating Ocular Drug Delivery and Disposition
In the field of ophthalmology, regardless of drugs’ modality and route of administration, significant challenges exist in describing the ocular PK of drugs. Not surprisingly, it is very difficult (if not impossible) to describe human ocular drug disposition. For laboratory animals, it is not possible to serially sample ocular tissues, with the possible exception of the aqueous humor. Consequently, for in vivo studies, one animal is used per point in time, which results in the use of a large number of animals. To improve the inherent sampling challenges, one technology includes in vivo imaging approaches to describe the ocular PK of drugs over the course of a few hours. These methods are described later in this article. The precise collection of in vivo parameters, such as drug exposure, distribution, and metabolism, constitutes the benchmarks researchers use to develop in vitro models to describe ocular drug delivery and disposition. The inherent challenges associated with ocular ADME studies are highlighted herein before a detailed discussion of in vitro transport/metabolism models, the lack of in vitro toxicity models, approaches to human relevant ADME studies, enabling technologies, and in silico analyses.
Anatomic Complexity.
The eye is an organ that has a complex anatomy and physiology, with numerous different cell types and tissues, each of which has its own metabolic capability and barriers to distribution of drugs. The anatomy and physiology of the eye (Fig. 1) are intricate, with multiple anatomic barriers and clearance mechanisms that are described elsewhere (Duvvuri et al., 2004; Ghate and Edelhauser, 2006). The complexities of the different ocular barriers as they relate to ocular drug transport are described later in this review.

Macroscopic and microscopic anatomy of the human eye (not to scale).
Multifaceted Drug Passage Routes.
The most common delivery systems to get a drug to a target in the eye are topical (least invasive), intravitreal (most invasive), and oral administration (assumed highest level of patient compliance). Although it may be reasonable to expect that intravitreal administration would accurately administer a dose, this route of administration is relatively inaccurate (+/-30%) because of the small volume administered (50 µl from a 1 ml syringe) and reflux of the dose solution out of the eye. The topical route of administration is preferred for many classes of drugs when treating diseases associated with the anterior segment because of ease of administration and patient convenience. Penetration across the cornea is proposed to be the primary pathway for the distribution of a drug from the surface of the eye to the aqueous humor and anterior segment, followed by the posterior segment. Reaching the posterior segment tissues by topical administration is challenging as the drug needs to penetrate through the anterior structural barriers (e.g., cornea, conjunctiva, sclera) and the vitreous humor.
Misrepresentations in the Ocular Drug Metabolism and Pharmacokinetics Literature.
For most types of ocular administrations, a key piece of information used to determine ocular PK and calculate safety margins is the volume of the vitreous in animal species and in humans. The article which has been most frequently cited reports the vitreal volumes of the rabbit and cynomolgus monkey as 1.5 and 1.5–3.2 ml, respectively (Short, 2008). This review article cites other publications for the vitreal volume in rabbits (Leeds et al., 1997) and in cynomolgus monkeys (Pearson et al., 1996; Leeds et al., 1997). Unfortunately, on closer inspection of the cited articles, it seems the original articles may have been misinterpreted. The original article assumed an average vitreous volume of 1.5 ml for rabbits as opposed to an experimental determination (Leeds et al., 1997). The higher vitreous volume value for cynomologous monkey (3.2 ml) is the distribution volume of cyclosporine administered intravitreally (Pearson et al., 1996). This apparent volume of distribution differs from the actual volume of the distribution since there is no evidence that cyclosporine is restricted to the eye. Recently, this error was acknowledged (Emami et al., 2018) and a direct measurement of vitreal volume in the monkey and other species has been made. The most reliable estimates of the vitreal volumes are now considered those reported by Covance (Covance: Comparison of ocular tissues weights (volumes) and tissue collection techniques in commonly used preclinical animal species. Date accessed: May 23, 2018. https://www.covance.com/content/dam/covance/assetLibrary/posters/StrubleEVER14.pdf); where rabbit and cynomolgus monkey vitreal volumes are 1.4 and 2.0 ml, respectively. The estimate of the human vitreal volume (∼4 ml) is similarly doubted as textbooks and research articles do not indicate how the volume was determined. Another cautionary note in the ocular literature includes investigational research articles that have been retracted or received an editorial note of concern. These articles continue to be cited even though they have been retracted (falsification of data) or their veracity questioned. The titles of these articles are listed in Supplemental Table 1 so that the ocular drug metabolism and pharmacokinetics (DMPK) community is aware of the compromised nature of these articles.
Different Metabolic Capabilities.
Understanding the metabolism of a drug in such a complex organ can be challenging because of the many different tissues with different metabolic capabilities. For example, the simplest approach is to homogenize the whole eye and use the homogenate to describe the metabolism of a drug. However, the utilization of whole homogenates may result in the dilution of the metabolism of a particular NCE in a particular tissue that represents a small portion of the eye. Additionally, by homogenizing the whole eye, it may be possible to create metabolic pathways that do not exist in the individual tissues, a disadvantage that exists for most hepatic and extrahepatic drug metabolism models involving homogenization of the entire organ. High levels of nonspecific binding of the drug and its metabolites, low signal-to-noise ratio, and small sample volumes may pose added analytical challenges.
Requirement for a Large Number of Animals or Subjects.
Ideally, the eye is dissected, and each individual tissue is isolated. This approach can be a tedious task and is challenging for small animals (e.g., mice and rats). Dissection of eyes obtained from larger animals (e.g., rabbits, dogs, monkeys) and humans is more straightforward. Although it may be feasible to source a large number of eyes from large animals, it is extremely difficult to obtain human eyes. The lack of availability of human eyes greatly limits the in vitro experiments that can be performed. This means that the data generated is usually from samples obtained from only one or two individuals, which may limit its utility. There are many tissues that can be isolated from the eye and used for in vitro experiments; however, it is not advisable to perform in vitro experiments on all tissues. For a drug that is administered by oral, IV, or SC administration it is almost certain that it will distribute to the liver and hence performing in vitro metabolism experiments using hepatocytes or hepatic sub-cellular fractions are appropriate. However, not all drugs will distribute to all tissues within the eye. Distribution to different tissues of the eye will depend on the drug’s intrinsic properties and route(s) of administration. In such cases, in vitro metabolism should be performed in the relevant tissues to which the drug distributes. Unlike the study of hepatic metabolism, in which a large amount of high- protein homogenates or microsomes can be made from one liver, the amount of homogenates or microsomes that can be generated from one eye is relatively small. Consequently, it may be necessary to sacrifice a large number of animals to generate sufficient material to perform in vitro studies.
Melanin Binding.
A further confounding factor is the presence of melanin in certain ocular tissues, such as the iris and retinal pigment epithelium (RPE). The wide variation in the constitution of melanin is a result of varying proportions of eumelanin and pheomelanin, melanin’s two chemical forms. Because of this property, precise translation of in vitro melanin binding data to and across laboratory animals is extremely difficult. Small lipophilic or cationic drugs may bind to melanin, thereby reducing their distribution to other tissues (Salminen et al., 1985; Zane et al., 1990). Binding to melanin may act as a sink for the drug. In vivo, binding of the drug to melanin can give the impression that the drug has been distributed to a particular tissue at high concentrations, although the free concentrations of drug may be low. Similarly, in in vitro homogenized pigmented ocular tissues, the binding of the drug to melanin may reduce the amount of drug available to be metabolized. Thus, the measurement of drug tissue levels can overestimate the amount of available drug for interaction with its pharmacologic target or intraocular metabolism since most of it might be bound to melanin. In addition, melanin binding can lead to slow release or formation of depot, leading to prolonged drug exposure in vitreous humor over time, which may also have an impact on fraction available for metabolism. Consequently, binding to melanin must be considered when describing the ocular PK of drugs in the eye and the in vitro metabolism/transport of drugs in ocular tissues.
In Vitro Models of Ocular Transport
A drug targeting the eye encounters several barriers, which are shown in Fig. 1 and described herrein in relation to ocular transport. To study drug transport across these barriers, a few primary and immortalized cell lines of animal and human origin are mentioned. Whereas most of the cell lines have been used to determine the toxicity of ocular drugs and the scope of formulations or as pharmacology models, some have also been used to determine the permeability of drugs intended for ophthalmic administration. A list of human cell lines derived from various ocular barriers is compiled in Table 1.
Summary of human ocular tissue derived cell lines listed with their respective transepithelial electrical resistance (TEER) values and commonly reported applications
Tear Film and Cornea.
The tear film is the first barrier that a topically administered drug encounters. Dilution by the tear film and the subsequent drainage (lacrimal and eyelid movement) reduce the bioavailability of all topically administered drugs. Although there is no in vitro model to study the effect of tear dilution, in vivo studies are conducted to understand the effect of formulations that can improve retention on the eye and thus improve ocular bioavailability. After topical ocular administration, the drug encounters the cornea and conjunctiva. The cornea is a multilayered structure posing both a physical and biochemical barrier to the movement of molecules across it. There are approximately six to seven layers in the human corneal epithelium, but only the superficial two to three layers express tight junctions. These tight junctions, coupled with transporters and enzymes of the cornea, act as barriers. Below the epithelium are the collagenous stroma and the endothelium. Although the epithelial layers are easily permeated by lipophilic molecules, the hydrophilic collagenous stroma presents a barrier to such molecules. Additionally, because of the tight junctions in the epithelial layers, only small molecules and small fragments of biotherapeutic agents, such as single-chain variable fragments, can easily penetrate the intact cornea.
Although presenting a large surface area for absorption of drugs, the cornea is an important barrier to the entry of molecules owing to its multilayered and diverse structure (lipophilic epithelium and hydrophilic stroma) after topical ocular administration. Several animal and human corneal cell culture models are available to study drug permeability, ranging from primary cells to immortalized cell lines (Kahn et al., 1993; Mohan et al., 2003; Ranta et al., 2003; Sunkara and Kompella, 2003; Toropainen et al., 2003). Models to study toxicity are also available from SkinEthic Laboratories (Nice, France) and MatTek Corporation (Granby, QC). Instead of monolayers, multilayered cultures are typically grown on collagen-coated plates to mimic the in vivo scenario. Of late, corneal structures with the epithelium, stroma, and endothelial cells are being built as three-dimensional models to mimic what happens in vivo (Kruszewski et al., 1997). Whereas primary animal and human corneal epithelial cells have been cultured to express tight junctions, the transepithelial electrical resistance (TEER) values do not always reflect the high-resistance scenario encountered in vivo. Therefore, these models have been used more to study toxicity rather than permeability and to reconstruct the cornea in ocular surface disorders (Ward et al., 1997). Immortalized corneal cell lines from rabbits, rats, cows, and humans have been grown as multilayered cultures with tight junctions and high TEER values for studying permeability. The permeability across these monolayers demonstrated dependence on lipophilicity, molecular size and weight, and transporter dependence. Most theoretical corneal models of corneal permeability, such as those based on physicochemical properties of drugs (Fu and Liang, 2002; Zhang et al., 2004), can be applied to rank-order corneal permeability and enhance ocular penetration of virtual compounds. Chemically synthesized drug candidates can be further tested in vitro in the immortalized cell models while keeping in mind the inherent differences in drug-metabolizing enzymes and transporters between each of the models and human cornea. Finally, little information is available on the translatability of in vitro models for corneal irritability and toxicity.
Conjunctiva.
The conjunctiva, which covers the anterior portion of sclera (bulbar conjunctiva) and part of the eyelids (palpebral conjunctiva), is highly vascularized and more porous than the cornea. Like the cornea, it has two to three epithelial layers with tight junctions; however, the intercellular pores are larger, thus allowing transcellular transport of larger and more hydrophilic molecules. Beneath the epithelial layers is the vascular tissue through which drugs are absorbed into the systemic circulation. This systemic absorption of topically applied molecules reduces ocular bioavailability and represents another clearance mechanism from the eye. The underlying sclera consists mainly of collagen, through which hydrophilic molecules can easily permeate. LMW drugs gain access to the retina, choroid, and vitreous humor through this pathway. Although cell lines from animal species are available, those from humans are fewer, and most are used as pharmacology models (Diebold et al., 2003; Gipson et al., 2003; Garcia-Posadas et al., 2017), and little work has been done to study permeability using these cell lines.
Blood-Aqueous Barrier.
The blood-aqueous barrier, located in the anterior segment of the eye, is formed by the endothelial cells in the blood vessels of the iris and ciliary body (ICB). This barrier’s characteristic tight junctions restrict the flow of molecules from the systemic circulation into the aqueous humor. Few in vitro models are available to study the blood aqueous barrier. Part of the difficulty comes from isolating ICBs with intact endothelial cells, which can express tight junctions and other proteins when cultured. Rabbit (Cilluffo et al., 1997) and human (Noske et al., 1995) models of this barrier have been described in the literature, but both suffer from poor tight junction expression, resulting in leaky monolayers with low TEER. Therefore, these monolayers suffice for studying safety endpoints, but their usefulness in studying permeability is limited.
Blood-Retinal Barrier.
The blood-retinal barrier is in the posterior segment of the eye and is formed by the endothelial cells of the retina blood vessels and the RPE. The endothelial cells of the retinal blood vessels control the movement of molecules between the blood and the retina. This barrier separates the neural retina from the vascular choroid and is responsible for maintaining homeostasis in the neural retina. It is often known as the inner blood-retina barrier and sometimes compared with the blood-brain barrier. The RPE contains tight junctions and expresses several transporters to ensure the supply of nutrients to the retina while preventing injury. Thus, the blood-retinal barrier restricts entry of molecules into the retina, including the movement of plasma proteins, ions, and drugs (barring a few exceptions, such as mitogen-activated protein kinase inhibitors). Small and lipophilic molecules can permeate this barrier better than large, hydrophilic molecules. Molecules administered into the vitreous humor are cleared through this route into the systemic circulation, in addition to the clearance from the anterior chamber.
Because of the complex nature of the blood-retinal barrier and the RPE, appropriate cell lines or cultures are challenging to develop. Culture medium composition, culture conditions, cell source, time of culture, and such factors affect the nature of barrier properties, making it harder to replicate in vivo–like properties in vitro. Both primary and immortalized human cell lines of the RPE and blood-retinal barrier express tight junctions, enzymes, and transporters and are being used for studying transport and cytotoxicity, as well as for in vitro pharmacology studies (Lu et al., 1995; Dunn et al., 1996; Holtkamp et al., 1998; Urtti et al., 2000). A list of the transporters expressed in the human eye is presented in Table 2. They act in concert to ensure proper functioning of the eye, supplying nutrients to various parts of the eye while removing waste and keeping out toxins. Therefore, studying drug transport across these barriers helps inform the drug distribution to various parts of the eye. The blood-retinal barrier has been recently reviewed in detail (Kubo et al., 2018).
A comprehensive table of transporters identified in human
 indicates presence detected as either protein, mRNA, or functional activity by any relevant analytical methodology.
indicates presence detected as either protein, mRNA, or functional activity by any relevant analytical methodology.  indicates lack of evidence.
indicates lack of evidence.  indicates conflicting literature reports, i.e., not detected by one or more laboratories in contrast to presence detected by others.
indicates conflicting literature reports, i.e., not detected by one or more laboratories in contrast to presence detected by others.  indicates absence, i.e., not detected as either protein, mRNA or functional activity by any relevant analytical methodology. Transporters have not been identified in aqueous humor, vitreous humor, RPE, optic nerve, and sclera. These tissues are therefore not included in the table.
indicates absence, i.e., not detected as either protein, mRNA or functional activity by any relevant analytical methodology. Transporters have not been identified in aqueous humor, vitreous humor, RPE, optic nerve, and sclera. These tissues are therefore not included in the table.
Advantages and Limitations of Ocular Transport Models.
Although data from animal cell lines do not translate to human, they can still be used to rank-order the permeability of compounds. Most human ocular cell lines are used as pharmacology models and to investigate toxicity, but their use in understanding and predicting ocular drug bioavailability has been limited. The biggest drawback in the DMPK ophthalmology is the lack of ocular tissue exposure data from the clinic resulting from the invasive and destructive nature of the current bioanalytical techniques. Without clinical exposure, it is not possible to determine in vitro–in vivo correlation; thus, the utility of these cell lines for predicting human ocular tissue exposures or PKPD is limited. In addition, many human cell lines suffer from low TEER, an inability to grow as uniform monolayers, and difficulty in replicating in vivo conditions. These caveats the utility of the data generated. Certain features, like tear film, rapid movement of eyelids, and lack of expression or functionality of transporters and enzymes, cannot be reproduced in vitro or ex vivo. Although the role of transporters can be studied in preclinical species and in human cell lines, the translatability of such data to humans and their role in human ocular drug transport is less studied. Additionally, there is no guidance in place about organ-related drug-drug interaction as it applies to the eye. Compared with the limited availability of human eyes, the continuous supply of tissues from animals makes them more dependable for isolating different cells; however, cells from human eyes are preferred because of species differences. Finally, modeling can be used to overcome some drawbacks but not all.
In Vitro Models of Ocular Metabolism
In vitro models for studying ocular metabolism, an important disposition mechanism, have generally been overlooked compared with the academic and industry gold standards for drug metabolism, including hepatic subcellular fractions, hepatocytes (plated or suspension), and other cell line models, including, hepatopac, Kupffer cells, and ADMET in vitro hepatocytes. Many topical ocular drugs have low hepatic turnover. This trend may be prominent as many ophthalmic treatments were developed by repurposing older medicines for ocular indications; therefore, ocular metabolism was assumed to be similar to that observed for oral treatment once the compound reached systemic circulation (Zimmerman, 1993). Oral cardiovascular β-blockers were repurposed as a topical ocular treatment to lower intraocular pressure, to reduce systemic exposure to the drug and its metabolites, and consequently improve the systemic safety profile of the drug (e.g., reduce adverse cardiovascular side effects) (Frishman et al., 2001). Additionally, ocular research has been focused on hormone/endobiotic metabolism or drug-metabolizing enzyme superfamilies by using general fluorescent probe substrates. These aspects have been well summarized elsewhere (Nakano et al., 2014; Argikar et al., 2017a). As a result, ocular metabolism literature and research have focused on basic enzymology research and used readily available in vitro models or ex vivo tissues.
More recently, the description of xenobiotic metabolism has included radiolabeled in vivo studies to demonstrate low systemic exposure to parent drug and metabolites rather than assume low systemic exposure and metabolic turnover. Minimal in vivo metabolism of lifitegrast was observed in rabbits and dogs (Chung et al., 2018). For ocular sustained release therapies, reported metabolism studies were limited by analytical challenges caused by the need to use different analytical methods to measure the conjugated drug, released drug, and its metabolites (Lv et al., 2017). Ocular tissues sections can be obtained from animals after topical ocular or intravitreal administration. Using such sections for metabolite identification and detection is feasible when standard materials are available (i.e., the metabolism is already known). Successful examples of in vivo ocular metabolism examples include, but are not limited to, tafluprost (Fukano and Kawazu, 2009), nepafenac (Chastain et al., 2016), and carbonic anhydrase prodrugs (Huang et al., 2015). Although the in vivo quantitation of metabolites can be helpful for physiologically based PK modeling of the data (if needed), its utility is limited due to the low doses and the need for high specific activity of radiolabeled compounds.
Analytical challenges and sample availability have led to questions regarding how, why, and when it is appropriate to study ocular metabolism in vivo and in vitro (Argikar et al., 2017b). The review notes a resurgence in xenobiotic ocular metabolism, yet a comprehensive in vitro model to study the eye for metabolism and subsequent transport is not available as compared to the way one would traditionally characterize hepatic metabolism. Relevant human ocular drug-metabolizing or housekeeping (implicated in cell/organ survival and function) enzymes from reported mRNA, protein, or functional activity have been collated in Table 3. This information was compiled between 2013 and 2018, from PubMed searches of the literature (1950s–present) using key words that included ocular metabolism, ocular tissue(s), enzymes (from superfamilies to isoforms), and enzymes modulated in ocular diseases.
Comprehensive table of ocular drug-metabolizing enzymes and housekeeping enzymes (i.e., enzymes implicated in cell/organ survival and function) identified in humans
 The presence detected as protein, mRNA, or functional activity by any relevant analytical method.
The presence detected as protein, mRNA, or functional activity by any relevant analytical method.
 Lack of evidence.
Lack of evidence.  Conflicting literature reports (i.e., not detected by one or more laboratories, in contrast to presence detected by others.
Conflicting literature reports (i.e., not detected by one or more laboratories, in contrast to presence detected by others.  The absence (i.e., not detected as protein, mRNA, or functional activity by any relevant analytical method. Drug-metabolizing enzymes have not been identified in aqueous humor and vitreous humor. These tissues are therefore not included in the table.
The absence (i.e., not detected as protein, mRNA, or functional activity by any relevant analytical method. Drug-metabolizing enzymes have not been identified in aqueous humor and vitreous humor. These tissues are therefore not included in the table.
Cell Lines.
Ocular cell lines are sometimes used to describe the transport and permeability of topical therapies to assess NCE. These studies determine whether a candidate or drug could be dosed topically and delivered to the back of the eye based on permeability assessments (De Saint Jean et al., 2000; Majumdar et al., 2009). A few notable studies have recognized the knowledge gap between ocular transporters and drug-metabolizing enzymes; transporters have been more routinely characterized than their DME counterparts. These studies present primarily an mRNA perspective. mRNA levels of various oxidative and conjugative enzymes from human corneal epithelial, human keractocyte, human corneal endothelial cell lines, and human corneal tissue sections (n = 16, subjects aged 18–102 years) were measured and compared with liver tissue and Caco-2 cell line (Kolln and Reichl, 2012). This research noted that mRNA levels from the human cornea cell line were equivalent to the mRNA measured in individual human cornea tissues and lower than those measured in human liver or Caco-2 in vitro model. In addition, in a human ciliary epithelial cell line, mRNA levels of CYP1A1, CYP1B1, and aryl hydrocarbon receptor and low levels of CYP2D6 have been reported (Volotinen et al., 2009). This research also noted induction of CYP1B1. CYP1A2 and cytochrome P450 (P450) superfamilies CYP2 (except CYP2D6) and CYP3 were not detected in the ciliary cell line. Although the evaluation did not assess the corresponding functional activity, metabolism of known CYP1B1 substrates, steroids, and retinoic acid have been independently measured (Doyle et al., 1995).
Xiang and coworkers measured mRNA levels of P450s (CYP3A4 and 5) and UGT1A1 in an immortalized corneal cell line (human corneal epithelial (cHCE) cells) by Cambrex BioScience (East Rutherford, NJ) that has been used to study ocular permeability. Functional activity was measured through the hydrolysis of latanoprost and reduction of levobunolol, but the responsible drug-metabolizing enzyme(s) are not characterized beyond their esterase and reductive functionality. In cHCE, overall drug turnover was low compared with corneal tissue (Xiang et al., 2009). cHCE cell line could be used as a surrogate to rank-order esterase substrates instead of using ex vivo animal corneal tissues. Despite the overall low enzymatic activity in cHCE cells, it would be interesting to examine the overall ocular disposition of topical therapies with a cell line that is well characterized for transporters and that has a strong prediction of in vivo ocular permeability. Research has only begun to scratch the surface of a holistic in vitro ocular metabolism and disposition model.
Ocular Tissue Homogenates and Tissue Sections.
The use of isolated ocular tissues for metabolism studies has been a relatively common practice in both industry and academia. Ocular P450 activity was quantified via mRNA for 10 P450s in the human cornea, ICB, and retina choroid tissues compared with the liver and extrahepatic tissues, small intestine, and kidney (Zhang et al., 2008). Like observations in cell lines, ocular tissues have markedly less mRNA compared with the liver, and many of the common P450s responsible for xenobiotic drug metabolism were, in general, absent. Early metabolism research in ocular tissues investigated specific drug-metabolizing enzyme activity via homogenates from various animals—including rats, rabbits, and cows. The presence of N-acetyltransferase activity in rats and the impact of light cycles or circadian rhythm on the enzyme’s activity were measured in retinal homogenates (Miller et al., 1980). Later, because of the ease of obtaining eyes from a slaughterhouse, N-acetyltransferases were further explored in pooled bovine RPE homogenates (Gaudet et al., 1993). The relevance and translatability of bovine ocular metabolism to humans are unknown, but research in bovine eyes or ocular tissue sections may serve as an easy surrogate in vitro test system compared with human eyes. Overall, these in vitro models provide information about specific and narrow metabolism questions but do not help improve characterization of potential NCE as ophthalmic targets as often done for orally dosed compounds.
More recently, ocular tissues were often used to measure the direct conversion of prodrugs to active drugs as they were directly related to the in vivo physiology and disposition. Fresh rabbit corneas in an Ussing chamber were tested to measure hydrolysis with a select general substrate lantanoprost (Xiang et al., 2009). Enzyme activity was monitored as a function of hydrolytic turnover over 4 hours, supplemented with O2:CO2 (95%:5%) gas to mimic biologic conditions. Alternatively, tissues have also been extracted and weighed from animals before transferring the tissues sections to in vitro tubes. The tissue sections are incubated (without added cofactors) with NCE for up to 6 hours before extraction protocols and analysis (Ke et al., 2000). For studying human in vitro metabolism, cadaver eyes are carefully dissected before in vitro incubation with compound (Ke et al., 2000). Similar to the disadvantages of using liver slices to study metabolism, these studies are limited to single donors, require extensive skills in tissue dissection, a large number of animal resources, or access to human tissue shortly after donation. Limited in vitro hydrolysis of nepafenac to amfenac in rabbits compared with humans was noted (Ke et al., 2000). Rabbits have been well characterized for their extensive esterase activity in the liver. The results may be limited by the surface area or exposure of compound to the ocular tissue slices. For reference, the Ussing chamber (Warner Instruments Corporation, Hamden, CT) surface area is 0.2 cm2/well (Xiang et al., 2009). Alternatively, nepafenac may be a poor esterase substrate in rabbits compared with humans, or ocular esterases may differ from those in the liver (e.g., isoforms, total protein expression, functional activity). Further characterization of relevant laboratory animal models and human in vitro models are needed to identify an appropriate in vitro model to study ocular metabolism.
Measuring both parent and metabolites in ocular tissues comes with various analytical challenges. Once the lens is removed, homogenization can be difficult. Coupling high- background noise and low topical ocular doses often results in insufficient lower limits of quantitation to measure metabolites with standards, especially if the turnover in the ocular tissues is low. Unless the parent drug is radiolabeled, measuring metabolism via a bioanalytical approach captures only a portion of ocular metabolism and subsequent metabolism is otherwise unknown. Measurements in ocular tissue sections are valuable for physiologically based PK predictions. Yet, these predictions can be verified only after conducting clinical trials. Depending on the ocular tissue type, however, it is difficult, if not impossible, to collect the samples and confirm these predictions. It is possible to collect aqueous humor from patients, although the collection is restricted to patients receiving intravitreal administration of drugs such as ranibizumab. In rare circumstances, vitreous humor can be collected when a patient undergoes a planned vitrectomy and is willing to be dosed with an investigational agent. Obtaining retinal tissue or samples of cornea, conjunctiva, etc. is not feasible. Human ocular drug and metabolite concentrations have also been measured in rare instances before enucleation surgery (Hollo et al., 2006). New technological advancements such as the smart contact lens and “Focal View” smart phone application, provide a fascinating new angle of digital technologies to improve ophthalmology clinical research (https://www.reuters.com/article/us-novartis-digital/novartis-digital-drive-continues-with-eye-disease-app-idUSKBN1HW0LI. Date accessed: May 04, 2018, Technology News/April 25, 2018 Maintained by Reuters).
Ocular Subcellular Fractions.
Over the past 50 years, subcellular fractions have been used in a scattered fashion throughout the literature compared to the use of traditional hepatic subcellular fractions. Ocular microsomes or cytosol were relatively commonly used to investigate functional activity across ocular tissue sections. From the 1960–1990s, studies focused on fractions from animals where it was easy to obtain ocular tissues. These models focused on ranking the activity of endobiotic metabolism in animals, including arachidonic acid metabolism in pigs (Asakura et al., 1994) and prostaglandin synthesis in rabbits (Bhattacherjee and Eakins, 1974; Kass and Holmberg, 1979) across ocular tissues. Detailed studies of an enzyme superfamily or multiple families were reported in bovine ocular tissues because of the ease of obtaining eyes from local butchers. Examples include characterization of gluthatione-S-transferases in bovine microsomes by affinity chromatography (Saneto et al., 1982), whereas P450 (Kishida et al., 1986; Schwartzman et al., 1987) and aldehyde oxidase (Shimada et al., 1988) functional activity were measured in the ICB. Although these drug-metabolizing enzymes were noted for functional activity, most research focused on a single species that often lacks translatability to the pharmaceutical industry.
To overcome the inability to scale to humans and the lack of general comparisons across preclinical species, industry methods have used ocular subcellular fractions to establish a flexible “whole-eye” S9 model. The lens is removed, and the eye is homogenized using a technique similar to that used for other tissues, such as the liver, lung, kidney, intestine, and others. The ocular subcellular fractions were pooled from nonsmoking individual male and female donors who were free of ophthalmic disease. The model is flexible and can be customized with the cofactors used to study ocular metabolism. The in vitro ocular S9 model has been used to explore new metabolism of historical topical drugs, such as levobunolol, that demonstrated subsequent metabolism of parent and active metabolite, dihydrolevobunolol (Argikar et al., 2016). Such a model could represent an economical and speedier in vitro assessment before extensive in vivo studies. Similar to high-throughput assays or identification of metabolic soft spots during lead optimization, the in vitro S9 model has the potential to be applied to early discovery metabolic screening to triage compounds. The model can also be used in lead optimization to drive structure-metabolism relationships by asking the right question at the right time. Examples at both ends of the drug discovery process may expand the way industry studies new ophthalmic treatments (Argikar et al., 2017b; Khojasteh et al., 2017).
To date, a few examples have been published from an early drug-discovery mind set. The S9 fractions were used to characterize the ocular disposition of betaxolol, a commonly used topical therapy (Bushee et al., 2015). These S9 fractions have also been used to evaluate in vitro ocular metabolites of ketoconazole at clinically relevant concentrations (Cirello et al., 2017). These studies have also noted cross-species comparison of laboratory animals compared to human. For example, S9 fractions were used to detect the activity of the rodent CYP2D family, which had not been previously reported by mRNA or protein activity (Dumouchel et al., 2017). The results indicate the role of rodent CYP2D2, 4, and/or 18 in the oxidation of timolol to the major oxidative metabolite observed in vivo. Similar to hepatic subcellular fractions, the ocular S9 model has been shown to be reproducible, with the use of levobunolol as a positive ocular metabolism control (Argikar et al., 2016; Dumouchel et al., 2017). Ocular S9 fractions are comparable across in vitro species; however, the timolol investigation in rats is the only direct comparison with in vivo metabolite identification studies using the same liquid chromatography-tandem mass spectrometry (LC-MS/MS) analytical technology.
Although S9 fractions provide new insights to older therapies and can be customized to study specific ocular metabolism questions, there are disadvantages to their use. The ocular S9 fractions require time and resource investment, experience in ocular dissection techniques, and procurement of enough human eyes, which may take months to years to yield small pooled human lots. In contrast, commercially available human hepatic subcellular fractions are derived from large donor pools to represent an average human (>100 donors, 20 mg/ml protein). Extrahepatic subcellular fractions are limited small donor lots (<20 pooled donors, 5 mg/ml protein). The published examples in ocular S9 support functional activity by a marker substrate; however, extensive characterization of ocular drug-metabolizing enzymes has not been assessed by an orthogonal approach (i.e., mRNA and/or protein quantification). Also, the eyes obtained from laboratory animals are nonpigmented compared with human eyes used to prepare the S9 fractions. The role of melanin binding and differences in metabolism of nonpigmented versus pigmented eyes may be worth exploring further. Furthermore, the S9 fractions noted do not include mitochondrial drug-metabolizing enzymes and as such cannot be used to study mitochondrial β-oxidation, which has been reported for current ophthalmic therapies (i.e., prostaglandins). Comparable to well established scaling factors used for the liver, the ocular S9 fractions require scaling factors to assess more completely the impact of ocular metabolism across species and the translatability of the in vitro model to in vivo data. Overall, the S9 fractions are a static model and do not account for transport of ocular therapies from the front to back of the eye or vice versa. These shortcomings are common for all in vitro models to date, and further research is needed to find an ideal in vitro ocular metabolism model that can predict a clinical outcome.
Although a complete ocular model for studying drug metabolism is not available, renewed emphasis on the eye could provide future model improvements. A question not answered by many models is the importance of the lens in xenobiotic metabolism in addition to its protective role, where glutathione S-transferase and N-acetyltransferase activities are important (Argikar et al., 2017a). Transitioning from healthy donors and nonpigmented animal models may or may not be an ideal model for studying ophthalmic disease state in humans. Recent reports have shown overexpression of drug-metabolizing enzymes in various ophthalmic disease-state models or from clinical proteomics assessments. High expression levels of CYP1B1 have been studied in glaucoma patients (Volotinen et al., 2009). Also, altered expression and activity of CYP4V2, which mediates fatty acid metabolism, is linked to Biett’s retinal dystrophy (Nakano et al., 2012; Astuti et al., 2015). The role of CYP4V2 in xenobiotic metabolism of NCE has not been explored in depth. Additionally, soluble epoxide hydrolases and CYP2C8 were overexpressed in murine choroidal neovascularization models and consequently identified as research areas of interest (Hasegawa et al., 2017; Sulaiman et al., 2018).
Bioactivation.
Although the eye has been studied for its overall metabolic capacity, it has not been considered for its bioactivation potential. To complicate searching the literature, the term “bioactivation” has been previously misused for “metabolism” (e.g., the hydrolysis of nepafenac) (Ke et al., 2000). In the context of drug metabolism, bioactivation refers to metabolic activation via formation of reactive intermediates. To date, only few publications are available on ocular metabolic bioactivation of the drug via reactive intermediates. It has been demonstrated that human ocular subcellular fractions metabolized ketoconazole similarly to subcellular fractions from human liver, including bioactivation of ketoconazole via an iminium ion at therapeutically achieved concentrations (Cirello et al., 2017). In contrast, a reactive aldehyde intermediate was observed only for timolol in hepatic but not ocular S9 fractions, despite similar biotransformation pathways. As a counterbalance to bioactivation, ocular enzymes in rabbit ICB and cornea were shown to metabolize an administered timolol-ketoxime to an inactive ketone via hydrolysis and reduction (Bodor et al., 1997). Especially from an ocular toxicity perspective, ocular bioactivation may represent an underexplored area.
Ocular Toxicology
At present, no regulatory guidances specifically address ophthalmologic topics, other than the guidance for reformulated products. Ocular toxicity is not a common occurrence during the conduct of general systemic nonclinical toxicology studies, but when encountered, it can pose a major hurdle to further development of the drug candidate (Brock et al., 2013). In vitro ocular metabolism and transport models may be used as a method to understand adverse events observed in vivo; however, the link between ocular bioactivation and toxicity is circumstantial at best. There are ample examples in the literature of systemically administered compounds, particularly anticancer drugs that have caused a wide range of ocular toxicities, including corneal thinning or opacity, glaucoma, cataracts, retinal degeneration, optic neuritis, conjunctivitis, uveitis, periorbital edema, etc. (Renouf et al., 2012; Onodera et al., 2015). As discussed earlier, ocular toxicities are not limited to topical dosing. Practolol, an orally administered β-adrenergic blocker, was withdrawn from the market in 1975 for severe ocular toxicity affecting many tissues of the eye (Garner and Rahi, 1976; Rahi et al., 1976). Toxicity onset ranged from a few months to a few years and was reversible in only some cases. The importance of the eye as a sensory sense organ makes it imperative that ocular endpoints are included in general systemic nonclinical toxicology studies, yet little guidance is available to direct the toxicologist on how best to design and interpret these studies. Ophthalmologic examinations should be conducted in all animals, including vehicle controls, at least once before dosing, during the dosing phase, and during the recovery if findings are observed during the dosing phase. In-life ophthalmologic examinations should include pupillary reflex, direct or indirect ophthalmoscopy, biomicroscopy (slit lamp), tonometry and histology of the eye, and adnexa at study termination. Electroretinograms for retinal function and optical coherence tomography for detailed imaging of the retina can be used on a nonroutine basis based on a knowledge of drug class, target, and/or previously observed findings. Nonclinical study designs with a detailed explanation of ophthalmologic endpoints (including fixation and processing for microscopy) and toxicology species differences can be found in previously published reviews (Short, 2008; Brock et al., 2013; Onodera et al., 2015; Novack and Moyer, 2016). A significant advantage of ophthalmologic examinations is that inflammatory processes during a study can be readily evident and monitored for reversibility via routine and advanced ophthalmologic endpoints. It is important for the toxicologist, ophthalmologist, and pathologist to integrate proactively all data to understand the relevance of an observed finding and translatability to the clinic based on the type and severity of the finding, reversibility, species differences, safety margins, and risk-benefit (Onodera et al., 2015). Even with detailed study designs, the ability of nonclinical safety studies to predict clinical ocular toxicity remains variable. In a review of 20 anticancer drugs approved by the Federal Drug Administration between 2012 and 2016, ocular toxicity was poorly predicted by nonclinical safety studies (Ahuja et al., 2017). In contrast, a review of 142 approved drugs in Japan from 2001 to 2010, which excluded anticancer drugs, indicated that 72% of ocular adverse drug reactions in humans were predictable based on nonclinical safety assessment (Tamaki et al., 2013).
Nonclinical safety studies for ophthalmic products can vary considerably in design. The International Conference on Harmonization (ICH) guidance for ophthalmologic drugs gives only minimal guidance, and study design thus depends on many factors, such as clinical route (topical or injected), frequency and duration of dosing, target homology, relevance of toxicology species, and whether the drug is an NCE or is being repurposed or reformulated as a marketed drug (Short, 2008; Novack and Moyer, 2016). In general, drugs need to be evaluated by the ocular and systemic routes of administration. For systemic studies, one species may be appropriate if the drug or drug class has well understood pharmacology and toxicology (ICHS6(R1), Parent Guideline dated July 16, 1997, Addendum dated June 12, 2011, http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S6_R1/Step4/S6_R1_Guideline.pdf; Short, 2008; Novack and Moyer, 2016). Separate systemic nonclinical toxicology studies have not been required for some intravitreally administered biotherapeutics (Bantseev et al., 2018). Selection of species can depend on the type of drug (small molecule or biotherapeutic), route (topical or injected), metabolism, and sequence homology (pharmacologically active). For topical ocular drugs, rabbits and dogs are used most frequently. The nonpigmented eyes of the New Zealand white rabbit is most commonly used, but in cases where the drug binds to melanin, the pigmented Dutch-belted rabbits may be used. Please refer to the Melanin Binding section for details. As dogs have a nictitating membrane that can affect the topical absorption of a drug, nonhuman primates at times are used instead of dogs (Novack and Moyer, 2016). Two nonrodent species can be used for topical ocular studies (rabbits and dogs or nonhuman primates) for LMW compounds. The duration of nonclinical safety studies needs to be as long as the intended clinical dosing: up to 6 months in the rabbit and 9 months in the dog or nonhuman primate (6 months for biotherapeutics). For injected drugs, the small size of the rat eye limits dosing volume, making the rat of limited value. Dosing in nonhuman primates is usually maximum feasible dose in a volume up to 100 µl (two separate 50 µl injections). In instances where there is cross-reactivity in rabbits, the onset of antidrug antibody responses can limit the use of rabbits for repeated-dose studies (de Zafra et al., 2017). Ophthalmologic assessments are more detailed than those used in general systemic nonclinical safety studies and can include direct and indirect ophthalmoscopy, biomicroscopy, tonometry, fluorescein angiography, corneal pachymetry, and electroretinograms if the drug is expected to reach the back of the eye.
Approaches to Human Relevant Ocular ADME Studies
At present, ADME studies in humans for ocular drugs are usually not required by regulatory agencies unless the drugs are administered systemically or orally. Most preclinical conducted ADME studies are limited to absorption and distribution, and only a limited number of ocular ADME studies are published in the literature. In general, it is challenging to identify the ocular contribution to metabolism in vivo as it is quite difficult to determine in vivo whether a metabolite was formed locally in the eye and released in the circulation or formed systemically afterward. Sometimes, unilateral dosing to one eye and monitoring of the other eye compared with systemic dosing or the use of in vitro ocular metabolism models can indicate whether a metabolite might be formed locally in the eye. The first three case studies illustrate the need for accurately designing in vivo ADME studies with the intention of addressing appropriate endpoints while highlighting the difficulties and caveats. The fourth case study documents the application of low-microtracer [14C]-labeled dose to humans topically, to accurately estimate single and multiday PK properties, and exemplifies that routine human ADME studies are not possible because of limitations on the allowed exposure to the eye and the inaccurate loss of drug (contamination on face, clothing, ingestion, etc.). Although these cases highlight the performed in vivo metabolism studies, it is unknown whether they were requested by a health authority or conducted by the sponsor out of an abundance of caution and what impact they had on the health authority’s review of the marketing application.
Case Example 1.
The absorption and distribution of brimonidine into anterior and posterior ocular tissues of monkeys and rabbits after topical dosing and intraperitoneal administration to rats were investigated (Acheampong et al., 2002). Results from unilateral dosing have shown high drug levels in the treated eye versus the nontreated eye, which indicated that brimonidine penetrates the posterior tissues by a local route, not by systemic absorption. As described earlier, on the way to posterior segment, metabolism is possible; however, the influence of metabolism in comparison with transporters and physiologic clearance mechanisms was assumed to be small. In general, only limited information is available about metabolism within the vitreous, retina, choroid, and sclera, and the overall impact of drug metabolism is generally unknown. In this study, the authors assumed that the posterior tissues were exposed mainly to brimonidine.
Case Example 2.
Chung et al. (2018) reported the distribution and pharmacokinetics of [14C]-labeled lifitegrast, an approved drug for the treatment of dry-eye disease, in rabbits and mass balance excretion in dogs recently. After repeated topical dosing of radiolabeled lifitegrast to rabbits, radioactivity was greatest in anterior segments, such as conjunctiva and cornea, whereas only low concentrations were observed in the posterior segments. This distribution correlated well with the site of action. In dogs, after a topical dose, most of the radioactivity was lost down the snout spillage through the nasal passage, highlighting the challenging nature of ocular ADME studies. After single intravenous administration to dogs, fecal excretion was the primary route of elimination, and urinary excretion was minor. The excreted radioactivity consisted mainly of unchanged lifitegrast, which indicated only minor in vivo metabolism. The results of this study were in line with observations from previous studies, which reported similar tissue distribution of lifitegrast (Murphy et al., 2011). A different distribution profile was reported in the eyes of rats, which could be explained by ocular anatomy differences in rats and dogs (Rao et al., 2010).
Case Example 3.
The disposition and metabolism of [3H]-tafluprost, an antiglaucoma prodrug, were investigated in rats after ocular administration by instillations to the cornea of both eyes (Fukano and Kawazu, 2009). After repeated ocular dosing, the radioactivity remained highest in cornea, followed by the ICB and aqueous humor. In female rats, after a single ocular dose, radioactivity was excreted mainly in urine and feces. Tafluprost was extensively metabolized in rats. No intact prodrug was detected in tissues and excreta, and the resulting acid metabolite was the most commonly detected component in the cornea, aqueous humor, ICB, and plasma, along with uncharacterized minor metabolites. In excreta, the major detected components were the respective glucuronide or sulfate conjugates of the acid. The authors did not investigate the main enzymes responsible for the hydrolysis in additional detail, but they mentioned that this reaction could also be mediated by P450.
Case Example 4.
A successful case example of the application of [14C]-labeled microtracer dose to humans has been documented (Iyer et al., 2012). The primary objective was to characterize the PK of AL-8309B, an extensively metabolized drug, after single- and multiple-day topical dosing in healthy human subjects. ADME was described after repeated dose administration. Each subject received a total radioactive dose of 13 µCi (500 nCi per dose/eye, twice daily) over 6.5 days, which is approximately 10-fold lower than the conventional therapeutic radioactive single dose (125 µCi). The low radioactive dose, coupled with the sensitive analytical method, such as accelerator mass spectrometry, provided well characterized PK of [14C] AL-8309B in healthy male subjects. The low radioactive dose minimized the amount of radioactivity exposure to study subjects and at the same time enabled measurable levels of [14C] radioactivity. The light-labeled human ADME studies are not applicable for all topical ocular instilled products and should be considered with a clear understanding of the PK (including ADME) behavior by administering the test article after different routes of administration. In addition, cost-effectiveness should be considered. Over the last decade, the use of microtracer approaches combined with accelerated MS has gained remarkable attention for ADME studies (Lappin and Garner, 2004), especially for drugs administered intravenously or orally.
Advanced Technologies for Studying Ocular PK and Drug Distribution
Traditionally, the most common methods for evaluating ocular PK and drug distribution involve the administration of radiolabeled or nonlabeled compound locally to the eye, followed by collection of selected ocular tissues for processing and analysis. In the case of radiolabeled studies, analytical methods include sample digestion or combustion [14C], followed by liquid scintillation counting ([14C], [3H] labeled small molecules) or γ- counting ([125I] labeled proteins). One alternative to so-called “cut-and-burn” studies or γ-counting, is ocular autoradiography (ARG), involving flash-freezing and sectioning of the eye, followed by exposure to photographic film or phosphor-imager plates to generate an image of ocular distribution of radioactivity. Applying more controlled freezing techniques reduces the formation of ice crystals, yielding higher-resolution images. The clear advantage of ARG is the preservation of spatial orientation of ocular structures since the eye is sliced, not dissected; however, dissection and ARG studies both use a radiolabeled compound, which requires a dedicated synthesis effort. In addition, there is the potential loss of label in the case of [3H] (by tritium exchange) or [125I] (by deiodination), and total drug-related radioactivity (i.e., the sum of parent and all metabolites) is typically measured.
With the advent of highly sensitive mass spectrometry (MS) instrumentation, it has become more common to collect and process tissues for analysis using LC-MS/MS. In the case of protein analysis, ligand binding methods, such as enzyme-linked immunosorbent assay, can be particularly useful. The chief disadvantage of this approach is the need to develop bioanalytical methods capable of measuring low concentrations (typically with sensitivity of sub-ng/ml) in the smaller tissues of the eye. In addition, slight variations in the assay may be required for each tissue type owing to matrix effects. Since the eye is dissected, even small structures must be collected either in whole or as macroregions; otherwise, significant spatial resolution is lost. There is also the risk of cross contamination between tissues during dissection. More recent advances in imaging MS have allowed for substantial improvement in visualization and relative quantification of a compound or drug distribution in the eye while maintaining spatial resolution of the intact anatomic structures. These newer techniques are further discussed to follow.
Matrix-Assisted Laser Desorption/Ionization Imaging Mass Spectrometry.
Although not new, imaging mass spectrometry (IMS) and matrix-assisted laser desorption/ionization (MALDI) were introduced roughly five and two decades ago, respectively; their marriage and application to analysis of particularly endogenous biologic compounds greatly expanded the use of MALDI IMS (Norris and Caprioli, 2013; Cornett and Scholle, 2017). With the advent of increasingly more sensitive MS and specialized tissue-processing methods, as well as advanced image analysis software, application has expanded to monitoring exogenous and small and large molecules intended as therapeutics. MALDI IMS has become a valuable tool in pharmaceutical and biotechnology research, with application to pharmacology, toxicology, PK studies, and drug metabolism, including ocular PK/distribution studies. MALDI IMS has the advantage of minimal sample preparation; with increasing sample throughput, it is continuing to expand application to discovery research. Unlike radiolabeled methods, MALDI IMS can image parent compound and multiple metabolites, as well as endogenous compounds, simultaneously. Most importantly, MALDI IMS preserves the spatial and regional integrity of the ocular anatomy for visualization of drug distribution. As a multiplex method, it allows for monitoring of arrays with hundreds of MS spectra. Distribution in the eyes of endogenous and exogenous compounds and pharmacodynamic markers and localization of tissue structures based on protein or lipid markers are possible.
MALDI IMS has been reviewed in detail elsewhere (Norris and Caprioli, 2013). Briefly, the method involves first the collection of the whole eye or selected ocular segments, which are flash-frozen and cryosectioned into thin slices (e.g., 20 µm). Once placed on slides, matrix is applied to the sections. This matrix facilitates the laser-induced desorption and ionization of the analytes, which are then analyzed using MS. The soft ionization process enables analysis of a wide range of molecular weights, typically 0.1 kDa to >100 kDa (i.e., small molecules to large biologics; e.g., antibodies or other therapeutic proteins). As mentioned, endogenous compounds can be monitored to assess pharmacologic or toxicologic markers. MS analysis creates an array of spectra that can provide specific fragmentation information or, with imaging mode, visualization of the analyte’s tissue distribution. Resolution depends on the speed and discrimination of the laser and the sensitivity of the MS instrument and typically ranges from 20 to 250 µm. With research-grade instruments, resolution can be as low as 1 µm; however, sensitivity is generally lower than what can be achieved with traditional LC-MS/MS on dissected tissue and can limit utility, particularly in the eye, where drug concentration can be low and have a wide range of concentrations from gradient-driven distribution. That said, sensitivity is continuing to improve, and this technology may someday replace dissection methods entirely. The results also tend to be semiquantitative because these are principally based on differential mass spectrometric ionization properties of the parent drug and its metabolites (Hatsis et al., 2017). An analogous technique that has been applied to tissue sections of the brain and appears promising for the eye is surface sampling microliquid chromatography tandem MS (Chen et al., 2016). The spatial resolution for this technique is lower than that of MALDI, but the distinct advantages are ease of sample preparation and the ease of possible absolute quantification of metabolites given the availability of analytical reference standards. As an example of MALDI IMS applied to the eye, Grove et al. (2017) assessed the ocular distribution of topical ocular brimonidine in rabbits. MALDI IMS was particularly well suited to investigating spatial and temporal distribution locally in the eye from the anterior segment to posterior segment. With a resolution of 80 µm, the study was able to demonstrate absorption or distribution in the cornea, aqueous humor, and iris, with some drug detected in the retina. Researchers have also used MALDI IMS to image specific protein and lipid markers in the rodent optic nerve and the neural retina (Anderson et al., 2017). A spatial resolution of 10 µm allowed by the high signal intensities was obtained.
Imaging Mass Cytometry.
Another imaging MS method that has the potential for application in ocular PK/distribution studies of proteins is mass cytometry, also referred to as imaging cytometry time-of-flight mass spectrometry (CyTOF), indicating the use of time-of-flight MS for analysis. This is also a multiplex assay, which could preserve the spatial arrangement of ocular structures since, like MALDI IMS, it involves the flash-freezing of the eye followed by cryosectioning of tissue. Unlike MALDI IMS, CyTOF requires labeling of the molecule of interest, which uses heavy metals with an atomic weight sufficient to differentiate from the numerous lower atomic weight endogenous metals present in the tissues. The advantage is little loss of label and a highly sensitive analysis of the metal label, down to one part per trillion, using inductively coupled plasma (ICP) MS. Although CyTOF does not allow specific analysis of compound, much like radioanalysis, this is outweighed greatly by the sensitivities achieved, a clear advantage in the assay of low concentrations in small ocular structures.
Imaging CyTOF has been used (Giesen et al., 2014) to image tumor tissues with subcellular resolution, allowing for the discrimination of cellular populations and cell-cell interactions. Before the advent of imaging CyTOF, mass cytometry had only been used to sort and analyze cell suspensions. The new imaging method maintains the spatial morphology of the tissue in a manner similar to that using MALDI IMS. For example, CyTOF was applied to evaluate the tissue distribution of cisplatin in tumor and normal tissues in cisplatin-treated mice with pancreatic cancer patient-derived xenografts (Chang et al., 2016). Distribution was determined using ICP analysis of platinum in tissue. Application to ocular studies has yet to be fully realized, but ICP has been used for evaluating the distribution of compounds in the eye. An example includes determining the ocular distribution of Gd-labeled albumin (Molokhia et al., 2009). Magnetic resonance imaging was used to image the distribution of Gd, but direct analysis of tissues was by ICP-optical emission spectroscopy. Imaging CyTOF, although still in its infancy, may prove to be a valuable technique for studying the biodistribution of metal-labeled proteins in the eye. The sensitivity of this method, without the use of radioactivity, and the relatively little method development requirement, make CyTOF particularly attractive as a research tool.
Ocular Classification System
Complex ocular anatomy implies that a single relevant model that classifies ocular drugs according to their physicochemical or disposition properties will be difficult to create. Historically, endogenous and exogenous ocular compounds have been classified by either in silico properties or by ex vivo permeability models. We have applied a compound categorization method, known as the extended clearance concept classification system (EC3S), originally developed for prediction of systemic elimination pathways and potential transporter effects, for its use in ocular drug disposition anticipation. EC3S classifies drug compounds based on their drug transport and metabolic turnover potential (Camenisch, 2016). Since transporter expression dramatically varies throughout the different layers of the eye, and to reflect the barrier role of a biologic membrane without the complication of overexpressed Pgp, we used permeability data evaluated in Madin-Darby canine kidney low-efflux cells as a surrogate parameter for drug transport (Perm,pas). Metabolic turnover (CLmet) refers to the highest value determined in human microsomes, hepatocytes, or S9 fractions. Scaling has been performed according to commonly accepted approaches, detailed in previously reported papers (Camenisch and Umehara, 2012; Umehara and Camenisch, 2012).
A subset of 22 chemically and pharmacologically diverse topical ocular dugs that are clinically used worldwide, in a variety of dosage forms, was used to illustrate observations on ocular drugs (Supplemental Table 2). The resulting two-dimensional scatterplot with CLmet on the x-axis and Perm,pas shown on y-axis is given in Fig. 2. To differentiate between “low” versus “high” permeability and “low” versus “high” turnover compounds, in alignment with EC3S, hypothetical thresholds were introduced at a Perm,pas value of about 5 × 10^−6 cm/s and a CLmet value of about 50 ml/min/kg. In Fig. 2 these thresholds are demarcated by solid lines. All drugs in the current data set were classified as “known as transporter substrates” (squares, Fig. 2) and “not known as transporter substrates” (triangles, Fig. 2; Supplemental Table 2). Using these definitions as a starting point, keeping in mind that a hepatic drug classification system may not necessarily be directly applicable to ocular disposition, the following conclusions and inferences can be drawn:
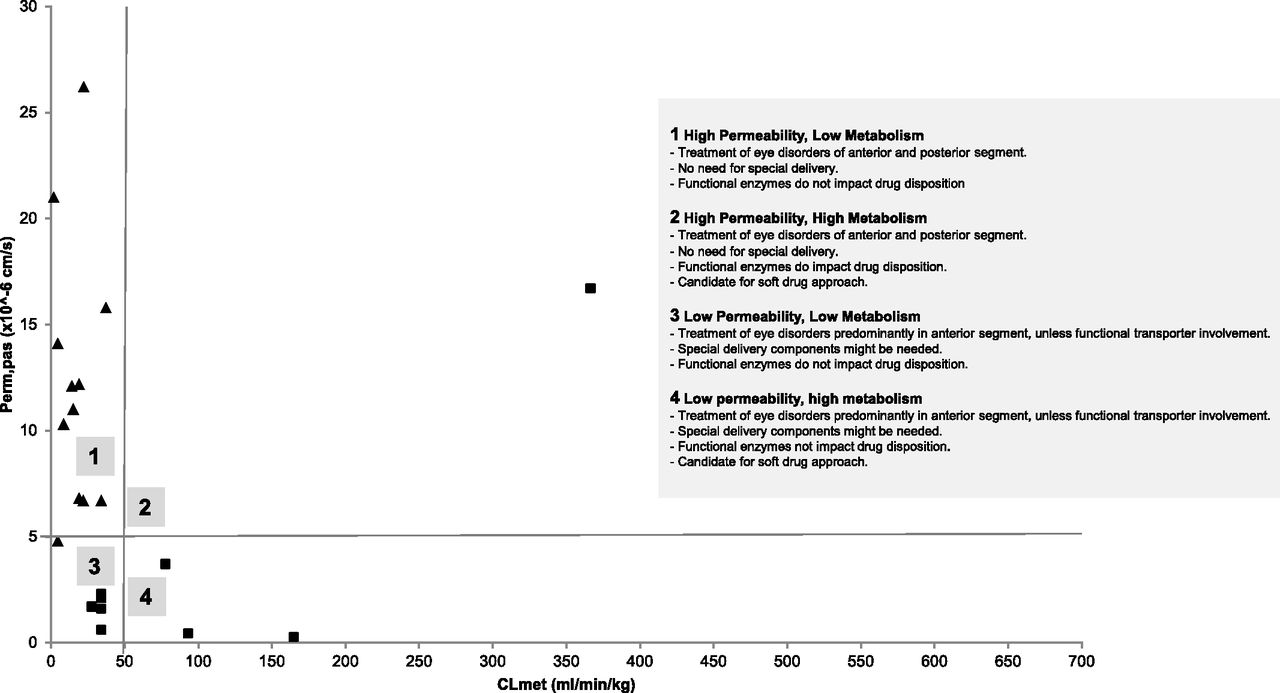
Two-dimensional scatterplot of topical ocular drugs. Similar to hepatic drug classification system plots, metabolic turnover (CLmet) is shown on the x-axis and permeability (Perm,pas) is shown on the y-axis. Clear separations can be drawn between “high” and “low” permeability (around Perm,pas of 5 × 10^−6 cm/s) and “high” and “low” turnover (CLmet = 50 ml/min/kg) drugs. Squares mark all “known transporter substrates,” and triangles represent compounds for which transporter affinity has not been demonstrated.
Ocular compounds largely fit into the “high permeability, low metabolism” category (EC3S class 2). These drugs are widely used for the treatment of anterior and posterior ocular disorders, without the use of specialized drug-delivery systems. Transporters or drug-metabolizing enzyme(s) have little to no impact on the ocular disposition of such drugs.
Several drugs from the current subset also belong into “low permeability, low metabolism” category (EC3S class 4). Despite low passive permeability, most of these compounds are well known solute-carrier substrates (predominantly OATs and OCTs). As such, it can be observed that this type of compound is used mainly for the treatment of the disorders of the anterior segment of the eye unless functional transporter involvement allows further passage. Specialized delivery systems that increase permeability or lead to sustained drug release might be needed, but drug-metabolizing enzymes are not expected to impact ocular disposition of such drugs.
High-turnover drugs (i.e., drugs for which enzymes are expected to impact ocular drug disposition) do not seem to be commonly used for the treatment of eye disorders. Referring to our data set, a handful of compounds have a metabolic turnover value greater than 50 ml/min per kilogram. Miconazole, erythromycin, and cyclosporine were identified assigned to the “low permeability, high metabolism” category (EC3S class 3). For such drugs, special delivery systems might be needed because the dosing interval is generally small (i.e., the frequency of dosage form of administration is generally high, typically every few hours and up to six times a day). Please refer to Supplemental Table 1 for details. Drug-metabolizing enzymes do impact ocular disposition and in theory EC3S class 3 drugs are candidates for soft-drug approach.
Finally, diclofenac was identified as the only member of the “high permeability, high metabolism” category (EC3S class 1). In this category, special delivery systems are likely not needed, and drug- metabolizing enzymes remarkably impact ocular disposition. Such drugs are also candidates for a soft-drug approach and are frequently dosed. In the case of diclofenac, despite the compounded topical formulations in a hospital pharmacy setting, the preferred dosing route for the treatment of ocular inflammation and uveitis is oral.
The EC3S-based compound categorization might be able to categorize ocular drug disposition and hence streamline, tailor, and speed up drug discovery and development efforts while investigating the potential of an NCE. It is important to note that metabolism in this section of the review refers to turnover in human liver-based cellular or subcellular fractions. Differences in ocular metabolism between laboratory animals and human species and differences in metabolic rates and profile between the eye and the liver are widely noted (Bushee et al., 2015; Argikar et al., 2016; Cirello et al., 2017) and may preclude direct preclinical translation of this data set. Lastly, considering the limited size of the data set used (n = 22), this initial approach requires further follow-up and refinement.
Summary
For topically dosed drugs, the notion of understanding the mechanisms of ocular drug disposition has moved away from “discretionary” to develop novel, target-specific, and locally acting improved therapeutic agents. Many of the models discussed here are immensely helpful for investigating the concepts, including ocular metabolism and transport, concentration-effect relationships, formation of active metabolites, etc. of drugs and drug-like compounds. Although these models are helpful, further research (i.e. accurate scaling factors, a single model that enables the study of transport and metabolism, understanding the differences in health and disease) is needed to extrapolate in vitro ocular drug-disposition parameters to in vivo. The complex and dynamic nature of the eye, in addition to interspecies differences in ocular disposition in laboratory animals and humans, currently present a remarkable challenge. The role of melanin in ocular drug distribution is possibly one of the most controversial topics in ocular pharmacology and drug disposition. A detailed deliberation of melanin binding of drugs and the subsequent impact has been written elsewhere (Argikar et al., 2017b). Therefore, extrapolation of ocular drug exposure, disposition, and potency/efficacy data from in vitro models with or without melanin to in vivo animal models must be conducted with caution. If needed, risk assessment of melanin binding can be conducted early in an appropriate in vitro model that relates to pharmacology to avoid unnecessary issues at a later stage.
The in vitro models discussed herein provide information about specific and narrow metabolism questions, but do not help improve on the characterization of potential NCEs as ophthalmic targets as is often done for orally dosed compounds. EC3S-based ocular drug classification described earlier may anticipate ocular drug disposition once such an approach is expanded to include a larger number and diversity of drugs. Another major gap in the entire in vitro metabolism models discussed is the lack of an ocular distribution component. The understanding surrounding the blood-retinal barrier, including kinetics, metabolism, and transport of topical ocular drugs or metabolites out of the eye and transport of a systemic drug into the eye, is limited. In the scope of drug discovery and lead optimzation, scientists must rely heavily on the use of in vitro cell lines and laboratory animals for in vivo data for prediction to human. As conducting ocular in vivo experiments means sacrificing an animal for each time point, developing appropriate in vitro models will help reduce the number of in vivo studies conducted. These in vitro models may also be used in a high-throughput screening capacity to optimize lead candidate ADME properties and formulations. Currently, such models are readily available for investigating ocular drug absorption, efficacy, and safety. Further advancement of a lead candidate from preclinical through clinical development to a drug approved to treat ophthalmic disease(s), presents many challenges that do not exist for drugs administered by other routes of administration. There are no regulatory guidances which are dedicated to the development of new ophthalmic drugs; however, the US Food and Drug Administration has issued a guidance that describes the development of an approved drug by an alternative route of administration (https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm079245.pdf. Date accessed: March 30, 2018, Page last updated: October 30, 2017, Content source: U.S. Department of Health and Human Services Food and Drug Administration, authored By: Center for Drug Evaluation and Research). The strategic role of ocular drug disposition and the placement of in vitro models in a decision tree in pharmaceutical research and development environment is a subject outside the scope of the present review and will be tackled separately in the future. Subsequent ocular disposition research will build on the established in vitro models and add to the industry and academic approaches to designing topical ocular therapies.
Acknowledgments
We thank past and present ophthalmology project teams at Novartis and Alcon for the collaborative discussions. We appreciate the support from Olivier Kretz.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Dumouchel, Chemuturi, Milton, Camenisch, Chastain, Walles, Sasseville, Gunduz, Iyer, Argikar.
Footnotes
- Received June 11, 2018.
- Accepted August 10, 2018.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- ADME
- absorption, distribution, metabolism, and excretion
- ARG
- autoradiography
- cHCE
- Cambrex BioScience immortalized human corneal epithelium
- CyTOF MS
- cytometer time-of-flight mass spectrometer
- DMPK
- drug metabolism and pharmacokinetics
- EC3S
- extended clearance concept classification system
- HCE
- human corneal epithelial cells
- HCJE
- human conjunctival epithelial cells
- HREC
- human retinal microvascular endothelial cell line
- ICP
- inductively coupled plasma
- IMS
- matrix-assisted laser desorption/ionization imaging mass spectrometry
- LC-MS/MS
- liquid chromatography-tandem mass spectrometry
- LMW
- low molecular weight
- MALDI
- matrix-assisted laser desorption/ionization
- MS
- mass spectrometry
- NCE
- new chemical entities
- P450
- cytochrome P450
- PK
- pharmacokinetics
- RPE
- retinal pigment epithelium
- TEER
- transepithelial electrical resistance
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics
References
In this issue





{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Introduction
- Challenges Associated with Investigating Ocular Drug Delivery and Disposition
- In Vitro Models of Ocular Transport
- In Vitro Models of Ocular Metabolism
- Ocular Toxicology
- Approaches to Human Relevant Ocular ADME Studies
- Advanced Technologies for Studying Ocular PK and Drug Distribution
- Ocular Classification System
- Summary
- Acknowledgments
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters
- PDF + SI