


Abstract
The minipig has become an animal of considerable interest in preclinical drug development. It has been used in toxicology research and in examining/establishing regulatory guidelines as a nonrodent animal model. We have reviewed some basic issues that one would want to consider in the development and testing of any animal model for humans. The pig is a reasonable alternative to the dog, but there are some clear limitations and unexplained disparities in the literature, which require further study; primary among these is the need for standardization in choice of breed and sex and routine protocols. The minipig offers numerous advantages over other established animal models, and it has similarities to the human with regard to anatomy, physiology, and biochemistry. The gastrointestinal tract is structurally and functionally similar to humans. This appears to be true for enzymes and transporters in the gut as well, but more study is needed. One major concern is assessment of oral drug absorption, especially with regard to potential food effects due to gastric emptying differences, yet this does not appear to be a consistent observation. Hepatic metabolism seems to reflect enzymatic patterns in humans, with some differences. Kidney function seems similar to humans but requires further study. We have analyzed literature data that suggest the pig would offer a reasonable model for human oral bioavailability and for allometric predictions of clearance. The minipig appears to be the model for dermal absorption in humans, and we discuss this in terms of literature data and our own in-house experience.
Introduction and Historical Perspective
Modern breeds of swine (hog, pig) are derived from wild boar, Sus scrofa, which were domesticated about 10,000 years ago in the Middle East, about 7000 years ago in East Asia, and later in Europe (Ganderup, 2016). There are early references to the use of pigs in medical research, especially in physiologic and anatomic investigations by such historical luminaries as Galen (130–200 AD), Harvey (∼1628), and Bernard (∼1865) (Bollen et al., 2010). Although the pig has become an invaluable animal model for a wide variety of biomedical research investigations, the farm pig is generally not suitable for such purposes because of its size, rapid growth, and lack of control of a safe and healthy environment from the time of birth. Many authors cite the similarities between pigs and humans with regard to anatomic, physiologic, and biochemical characteristics (Doyle et al., 1968; Swindle and Smith, 1998). Among these similarities are included the following: cardiovascular system, gastrointestinal (GI) tract (GIT), liver, adrenals, skin, and kidney. In recognition of the remarkable and numerous similarities between the pig and humans, there was a concerted effort, beginning in the late 1940s, to develop small strains of the species to accommodate the needs of experimental medical and biologic research. The Hormel minipig was first bred in 1949 (Hormel Institute, Austin, MN), and from it were derived several popular breeds, including the Göttingen (∼1961) and Sinclair minipigs (∼1965), which belong to the smaller weight minipigs, along with the Yucatan micropig (i.e., 35–70 kg mature weight). The Hanford and Yucatan (∼1960) minipigs have larger adult weights (∼70–90 kg) (Bollen et al., 2010). Although there are numerous other minipig breeds (e.g., Vietnamese potbelly), the most popular breeds have been the Göttingen, Yucatan, Hanford, and Sinclair. In contrasting numerous biologic metrics, Bollen et al. (2010) list miniature swine having two categories, 35–70 and 70–90 kg, as noted above. The term “micro-pig” seems to be of recent vintage and appears to refer to animals less than about 35 kg; however, we have not found a working definition.
Doyle et al. (1968) summarized various aspects of and comparisons among domesticated farm animals for use in medical research and notes the greater interest in selecting an animal model on the basis of its similarity to humans or because of a specific feature related to the human medical or disease condition (i.e., “pathological appropriateness”). The pig is similar to humans with regard to, at least, the nervous and cardiovascular systems and the GIT (Earl et al., 1964), and others have noted that the pig is the accepted model for human dermal studies (e.g., “The minipig is the species of choice to investigate dermal absorption in preclinical drug development”; Preube and Skaanild, 2012). Whereas the GIT blood circulation in the chimpanzee is quite similar to that of humans, Noer (1943) has noted that the circulation in the small intestine of the dog is quite different from humans. Importantly, there has been an extensive accumulation of biologic data in farm animals over many years, which provides useful basic information relevant to the choice of animal models.
Driven in part by regulatory agencies requiring the use of a rodent and nonrodent model for a wide range of testing prior to submission of new chemical entities (NCE) to inform the design of safe clinical studies as well as support the safety evaluation included in the regulatory registration of new drugs and chemicals, there has been interest in nonrodent species other than the dog, which has more or less become the default animal species of choice, somewhat surprising as it is a popular companion animal. Other species having a closer genetic relationship to humans, especially primates, such as the monkey and chimpanzee, while possibly offering the greatest potential similarity to humans, have significant disadvantages, including ethical and cost issues. The pig, especially the minipig and micropig, offers significant advantages as a nonrodent model for all phases of drug testing, from early preclinical studies to toxicology investigations (e.g., Svendsen, 1998) and food additives and medical device testing; indeed, the minipig appears to have become the standard animal model for toxicology studies in Western Europe. Among these advantages are as follows: similarities to humans in numerous body systems with regard to anatomy, physiology, and biochemistry; small size and docile characteristics; ease of breeding; long-term study (crossover designs) and sampling; all dosing routes used in humans are accessible in pigs; and not generally considered a companion animal. The pig is generally regarded as the animal of choice for xenotransplantation, and it is rapidly becoming so for human risk assessment and, we predict, for preclinical drug studies (Walters et al., 2011).
An attempt will be made to review, in a limited way, our current state of knowledge with regard to the role of minipigs in biomedical research, especially with respect to drug development and testing issues and use as a species for predicting human drug behavior and disposition. To the extent possible, given space limitations, we will attempt to summarize and reinterpret/reanalyze the literature (where necessary), highlight the current status and utility, and offer commentary on limitations of our understanding and future directions. As noted by Box (1976, 1979), with regard to statistical models, “All models are wrong, but some are useful”; we strongly believe that the same admonition applies to animal models used for the purposes of describing and predicting drug disposition and behavior in humans.
Some relevant general reference texts to which the interested reader might want to refer (including some cited above), include the following: Porter, 1993; Bustad et al., 1966; Swindle, 1983, 1992, 1998; Tumbleson, 1986; Tumbleson and Schook, 1996; Pond and Mersmann, 2001; Bollen et al., 2010; McAnulty et al., 2012; and Swindle and Smith, 2016. There are also several reviews comparing minipigs and humans in terms of drug testing and pharmacokinetics (PK) (e.g., Witkamp and Monshouwer, 1998; van der Laan et al., 2010; and Lignet et al., 2016). Furthermore, reflecting the interest in the pig as a useful animal model, the National Institutes of Health maintains a National Swine Resource and Research Center, established in 2003 (www.nsrrc.missouri.edu). The stated purpose of the Center is “…to develop the infrastructure to ensure that biomedical investigators across a variety of disciplines have access to critically needed swine models of human health and disease.” More recently, a Pig PeptideAtlas has been made available as a resource for proteome research (Hesselager et al., 2016).
Physiologic and Anatomic Considerations
Swindle and Smith (1998) have provided a relatively short review of the comparative anatomy and physiology of the pig. More in-depth reviews of specific body systems in the pig can be found in McAnulty et al. (2012). As with any other animal model, the pig is not a duplicate of the human (and, hence, a model), but it appears to share substantial similarities with humans with regard to certain, but not all, body systems. Differences are expected to exist; the degree of those differences and their expected impact on the function of interest are what determine the model’s potential limitation. The cardiovascular system is sufficiently similar to that of the pig that it is considered a standard model for purposes of drug and device testing and for studies of the diseased heart (e.g., Swindle and Smith, 1998; Gootman, 2001; and Myers et al., 2016). Differences, however, do exist and need to be considered (Bode et al., 2010). The GIT, an essential system to consider for oral drug absorption and bioavailability (BA) studies as well as for preclinical experiments involving the oral route, is physiologically similar to the human GIT, but there are some important anatomic differences. Kararli (1995) has provided a useful compilation comparing aspects of the GIT among commonly used laboratory animals, including the pig. The buccal epithelium of the pig is similar to that of humans, and absorption from the buccal cavity has been examined in numerous studies (e.g., Meng-Lund et al., 2016). Pigs are true omnivores, unlike rodents, rabbits, and carnivores, and it is that behavior that may result in shared similar physiologic functions. Although able to vomit, the pig is less prone to emesis than the dog, an advantage following oral drug dosing (Bode et al., 2010). The GIT in the pig differs in several ways from that in the human. The presence of a pharyngeal diverticulum needs to be considered when oral gavage tubes are placed to avoid problems upon intubation. Whereas the stomach has the same basic shape and regions (and function) as in humans, there is a muscular pouch (torus pyloricus) near the pyloric sphincter, which facilitates the closure of the pylorus. The adult Yucatan (∼50 kg) stomach volume is about 1300 ml. The primary secretory cells include the following: mucous, parietal (HCl secretion), and chief (protease secretion; primarily pepsin) cells. Gastric fluid pH ranges from 1.6 to 4.3; mean liquid retention time, 0.8 to 0.9 hours; mean solid retention time, 1.0 to 1.3 hours; half-emptying time (semisolids), 0.75 to 0.96 hours (Van Ginneken, 2012). Others have suggested an emptying time of 2–8 hours, depending on several variables, or 2–4 hours if fasting before measurement (Casteel et al., 1998). Because of its potential importance in drug absorption, the literature concerning gastric emptying is discussed more completely in a later section.
The long (as in humans) small intestine is located primarily on the right side of the abdomen. The duodenum, jejunum, and ileum occupy about 5%, 90%, and 5%, of the small intestine, respectively, with a diameter of about 20 mm in a 50 kg Yucatan pig (McCrackin and Swindle, 2016). The human colon exists in an inverted U-shape to accommodate the ascending, transverse, and descending colon. In contrast, the pig large intestine is tightly coiled (spiral colon) and is located in the left upper quadrant of the abdomen. The large intestine is about 20% the length of the small intestine with a diameter of about 25 mm. The total estimated surface area of the small intestine in the pig (47 kg) ranges from 168 to 210 m2 (vs. 252 m2 in a 70 kg human) (DeSesso and Williams, 2008). Total blood flow to the GIT is estimated to be 22% of cardiac output in the Yucatan pig (McKirnan et al., 1991), comparable to humans. Descriptions of the small intestine and colon indicate essentially the same physiologic and biochemical functions to those of humans. These organs are designed to secrete biochemicals, process food, and provide efficient absorption of digested materials, just as in humans and other mammals. The small intestinal fluid pH ranges from 5.7 to 7.2; mean liquid retention time, 3.9–4.4 hours; mean solid retention time, 3.7–4.3 hours. The colon has a fluid pH of about 7.1; mean liquid retention time, 24.9–41.3 hours; mean solid retention time, 35.6–44.4 hours (Van Ginneken, 2012). Most of these numerical values are similar to those measured and reported in humans, although the colonic retention times in the pig seem quite long. The pH ranges noted above are those most often cited for human gastric and intestinal fluids. Intestinal transit times in humans are generally considered to be about 4 hours, regardless of the presence or absence of food. The presence of metabolizing enzyme systems and transporters in the gut wall is discussed later.
Swindle (2016) notes the similarity of the pig and human liver from a surgical perspective; however, the pig’s liver is made up of six lobes and a gall bladder, compared with the four lobes in a human liver. The liver, from a functional, physiologic, and biochemical standpoint, is comparable between the two species. Indeed, the pig is the most often cited source of liver cells when considering an engineered animal-derived liver for transplantation into humans or an extracorporeal device (e.g., Schuurman et al., 2012). Similar considerations apply to numerous other engineered pig-derived organs (Waltz, 2017). Composition of pig bile is similar to that of most mammals, with the exception of having less cholic acid and a bile salt:phospholipid concentration of 9:1 (Swindle, 2016). Total hepatic blood flow in the chronically catheterized conscious farm pigs (8–16 weeks; 20–70 kg) was determined to be about 1100 ml/min (∼20%–25% of cardiac output), a value essentially the same as in adult humans (Drougas et al., 1996). An important consideration is that the porcine liver is more similar to the human liver than most other species with regard to enzymatic metabolic potential (Swindle, 1996). An extensive listing of cytochrome (CYP) P450 liver enzymes can be found in Preube and Skaanild (2012). Total liver microsome CYP P450 in Göttingen minipigs (∼0.81 nmol/mg protein) is higher than estimates in Caucasians (∼0.43 nmol/mg protein). Whereas the pig liver has essentially the entire complement of CYP P450 isozymes, with some exceptions [e.g., low CYP2C (Puccinelli et al., 2011) and high CYP2D (Thorn et al., 2011)], one needs to be aware of differences among farm pigs and breeds of minipigs and between males and females. Bode et al. (2010) conclude, “All main metabolic activities typical for human CYP enzymes are found in porcine liver microsomes” (Skaanild and Friis, 1997; Anzenbacher et al., 1998; Monshouwer et al., 1998). Preube and Skaanild (2012) have presented a useful and extensive comparative table of CYP450s found in the liver of humans and pigs. The readers are referred to that source for specific comparisons. Phase II metabolism has not been thoroughly studied to date. One report indicates that liver microsomes of the Yucatan minipig are able to glucuronidate nicotine and cotinine, whereas mice, rats, rabbits, and dogs showed no such activity (Ghosheh and Hawes, 2002). More information about enzymes and transporters is provided in a later section.
The pig kidney, as with most mammals, is similar to that of the human in terms of function, physiology, and anatomy. The kidney has been well characterized as a result of its use in transplantation surgery (Conn, 2008). Importantly, access to the male urinary tract is not possible as a result of the sigmoid flexure of the penis. Urine pHs, which can influence the excretion of ionizable drugs, encompass the same range in humans and pigs (∼7–9) depending on nutrition. Glomerular filtration rates in adult humans and pigs are essentially the same (∼126–175 ml/min in pigs vs. 130 ml/min in humans per 70 kg) as is renal blood flow, 3.0–4.4 ml/(min × g) in pigs versus 4.0 ml/(min × g) in humans (Sachs, 1994).
Gastric Emptying and Intestinal Transit
Gastric emptying (GE) is an important physiologic variable that may impact drug absorption from the human GIT, as is well known in the field (e.g., Mayersohn, 2009). It is not surprising that the same has been found in the pig, although there remains considerable lack of consensus about numerical values because of substantially different findings reported in the literature, and, therefore, questions remain about the utility of the pig for oral absorption studies in the absence and presence of food. (Some caution here to the reader: values for GE in the literature are presented in different metrics: GE rate or rate constant, GE time, GE half-time.) During fasting, as in humans, GE is controlled by the migrating myoelectric complex (MMC). Numerous factors will affect emptying rate in the fed condition, including type and volume of meal, viscosity, osmolarity, etc., as is the case in humans. It is important to note that the feeding pattern will have an important impact on GE. Small frequent meals do not appear to interfere with the MMC during the day, whereas two large meals interrupt the MMC for about 6 hours and a fed pattern is seen (Argenzio and Monteiro-Riviere, 2001). Differences in feeding patterns may explain some of the disparity in the literature with regard to food effects on GE and drug absorption. GE of food follows a bimodal pattern; 30%–40% empties within 15 minutes of eating with subsequent emptying during the next hour. However, emptying may be incomplete, with food remaining in the stomach for the entire day (Witkamp and Monshouwer, 1998). Davis et al. (2001) were among the first investigators to study GE and intestinal transit of pharmaceutical dosage forms in the pig (large white/Landrace/Duroc cross; 90–100 kg; fed twice a day) using external γ scintigraphy. Animals were fasted for 18 hours prior to dosing and 6 hours following dosing. The mean emptying half-time for a test liquid and solid pellets were 1.4 and 2.2 hours, respectively. In three pigs tested, the time to complete emptying of a tablet was between 5 and 6 hours in two animals and 1.5–2 hours in another. Estimates of small intestinal transit times were 3 to 4 hours and considerably longer time for total transit (∼24–72 hours, with wide variability). These values appear to be consistent with those reported by other investigators (see Table 4 in Davis et al., 2001), suggesting that the pig would be a good representative model of humans with regard to GE. In sharp contrast with these findings, however, are reports by other investigators who determined long and sometimes extraordinarily long GE times (Hossain et al., 1990; Aoyagi et al., 1992; Kabanda et al., 1994), all of whom conclude that the pig is not a good model to assess drug absorption and BA in humans. Prolonged gastric residence (>5 days) was found for enteric-coated nondisintegrating magnesium hydroxide caplets (Hossain et al., 1990). Aoyagi et al. (1992) found that the dog is a better animal model for BA studies under fasting conditions than the pig by examining GE among human, dog, pig, and rabbit with nondigestible tablets and granules. Kabanda et al. (1994) also observed a long stomach residence time of erodible matrix tablets in pigs.
It is difficult to reconcile these disparities reported in GE; however, one notes experimental differences (e.g., fed/fasted, frequency of feeding, type of food ingested, breed, sex and age/weight of animals, etc.) that could account for at least some of the differences measured. What is wanting in this study, clearly, as should be made obvious from this review in general, is standardization of procedures and, to the extent possible, choice of pig breed. More recent attempts to investigate this issue using, for example, acetaminophen as a marker compound for GE have rather convincingly demonstrated, under the reported experimental conditions, that pigs have a prolonged GE, food remains in the stomach (even after fasting for many hours; confirmed by postmortem inspection), and protocols to resolve this concern (e.g., administration of GIT stimulants, such as metoclopramide) may not be appropriate or always work (Suenderhauf et al., 2014; Christiansen et al., 2015; Henze et al., 2018). Lack of a food effect, when expected or noted in humans for specific drugs and not found in pigs, using the Food and Drug Administration–approved high-fat content meal (often used in the dog), would suggest that the pig is not, under current protocols, a reliable animal model.
Is the pig still a viable model for examining drug absorption, dosage form, and route of administration effects? We believe, as do others, that the answer to this question is yes, with the qualification of the need for additional studies under well-controlled conditions to establish standard protocols. Aspects of this issue will be discussed in several of the following sections.
Hepatic and GIT Enzymatic Metabolism and Transporters
Substantial effort has been expended in attempts to characterize metabolizing enzymes in various body systems of the pig, especially the liver and GIT, with special interest in comparison with humans. Achour et al. (2011) conducted a thorough study of the CYP P450 liver enzyme system obtained from two pig livers (adult Suffolk White). (As an aside, this publication illustrates the extent to which the pig can be instrumented by conducting surgical procedures that are able to address complex questions of absorption and metabolism.) It is likely that the results can be generalized to minipig breeds, but further study may be required. The investigators identified the P450 subfamilies to be (% total P450): CYP1A (2%), CYP2A (34%), CYP2C (16%), CYP2D (26%), CYP2E (8%), and CYP3A (14%). CYP3A is present in a larger percentage in human liver and is the major drug-metabolizing enzyme and responsible for the majority of drug–drug interactions. The investigators also found sequence homology between the pig and human P450 isoforms ranged from 72% to 95% with a similarity of about 98%. These findings are in agreement with numerous previous investigations using Göttingen minipig livers as the source of microsomes (e.g., Anzenbacher et al., 1998), which leads to the general conclusion that the pig offers an excellent representation of the human liver complement of CYP450s (e.g., see reviews by Skaanild, 2006; Puccinelli et al., 2011). Higher levels of Flavin-Containing Monooxygenase 1 (FMO1) have been observed in the minipig liver microsomes compared with humans (Yamazaki et al., 2014).
This general conclusion, however, should not imply the same identity, activity, or substrate specificity to all human CYP isoforms. For example, Thorn et al. (2011) noted qualitative differences in the metabolite pattern of testosterone (3A4); whereas diclofenac (CYP2C) metabolism was low in the pig, dextromethorphan metabolism by CYP2D6 was extensive and rapid compared with the human isozyme. Verapamil, a 3A4 substrate, exhibited similar metabolism between pigs and humans (Petri et al., 2006). Similar observations were made by others; human CYP2C substrate, S-mephenytoin, was not metabolized by the pig; diclofenac and tolbutamide, also CYP2C substrates, were metabolized to a much lower extent in pigs (Anzenbacher et al., 1998; Puccinelli et al., 2011). Such differences help to explain deviations of predictions of metabolic clearance (CL) or estimates of oral BA in humans based on the pig (see later section). Metabolic differences are further complicated by both breed of pig and sex. The Landrace pig has shown substantial CYP2D6 and CYP2C9 activity, whereas the Göttingen breed exhibits minimal activity of those isoforms. The Göttingen minipig illustrates more sex-dependent enzyme activity than other breeds; CYP1A2 and CYP2E1 activity was about four times greater in the female versus the male Göttingen, and there was a 70-fold higher CYP2A activity in female compared with male minipigs, whereas similar activity was observed in Landrace pigs (Skaanild and Friis, 1997; Bogaards et al., 2000). Similar differences are not seen in humans, which raises significant issues with regard to harmonization in developing the pig model to reflect the human condition. Any differences between the species in intrinsic hepatic CL will affect hepatic extraction ratio and, therefore, the extent of the hepatic first-pass effect and systemic BA. BA and prediction of species-based PK parameters are discussed in later sections.
There have been limited studies examining phase II metabolic processes. It seems that glucuronidation is greater, whereas sulfation is lower in the pig compared with humans (Nielsen and Dalgaard, 1978). Hepatic UDP-glucuronosyltransferase has been found in minipigs and shown to glucuronidate numerous drugs similar to what is seen in humans (Higashi et al., 2014). Acetylation (NAT1 and NAT2) has been observed in the pig, which is absent in the dog (Trepanier et al., 1997; Loureiro et al., 2013). Clearly, more in-depth study of phase II metabolism is called for, given the importance of conjugating enzymes in drug metabolism.
Aldehyde oxidase (AOX) is involved in metabolic processes of many drugs, including those containing heterocyclic nitrogen. Although dogs lack hepatic AOX, pigs have been shown to have an active form (Diaz and Squires, 2000; Garattini and Terao, 2013). In vitro–in vivo extrapolation (IVIVE) of the CL of substrates involved in the AOX metabolic pathway was studied in minipigs using AOX substrates. The results showed that in vivo CL was underpredicted by IVIVE, generally similar to that observed for compounds with elimination mechanisms via other metabolic pathways. In vivo–based allometry, with an exponent fixed at 0.75 (between minipigs and humans), seemed to predict well the human in vivo CL, supporting the use of minipigs for predicting human PK for AOX substrates (Wilkinson et al., 2017). The in vitro intrinsic CL (CLint) of five AOX substrates in multiple species, including humans and minipigs, were evaluated. An allometric model of in vitro CLint was examined using single (exponent fixed at 0.75) or multiple species. The single-species method seemed to predict the human in vitro CLint reasonably well. It should be noted that the prediction of the in vivo human CL still needs determination of whether the IVIVE within species is predictive. Nevertheless, the allometric model for the in vitro CLint demonstrated a good correlation of CLint for AOX-based metabolism (Crouch et al., 2018).
Enzyme-mediated hydrolysis reactions represent an important metabolic route for, as an example, prodrugs (often esters) that require hydrolysis prior to or after reaching the systemic circulation following oral, transmucosal, or transdermal administration. Relatively little information is available about these reactions in the pig, although they have been widely studied in humans. Important among these enzymes is the carboxylesterase family. 1,3-Dichloro-9,9-dimethyl-9H-acridin-2(7)-one (DDAO) benzoate, which is a specific probe for human carboxylesterase 2, was shown to have higher activity in pig liver microsomes compared with humans and dogs (Ma et al., 2017). Minipig skin appears to be similar to human skin with regard to ester hydrolysis of dermally applied drugs (Prusakiewicz et al., 2006; Jewell et al., 2007); observations further supporting the pig as a model for dermal drug absorption are discussed later. In vivo studies, corresponding to these in vitro studies, need to be conducted and the resulting correlation established to fully understand and quantitatively use the pig as a model for compounds undergoing enzymatic hydrolysis. Jones et al. (2016) present a strategy to identify drugs that may not be suited for study in the minipig because of excessive amide hydrolysis.
An additional aspect of presystemic metabolism involves GIT first-pass effects. The GIT is the most important extrahepatic site of drug metabolism (Helke and Swindle, 2013). A further complication is that of influx and efflux transporters in the small intestine, which work in tandem with GIT enzymes. Quantitative information about metabolizing enzymes in the GIT of the pig appears sparse, although CYP3A has been shown in the small intestine of the developing Göttingen breed along with the presence of P-gp (Van Peer, 2014). Shulman et al. (1988) have suggested that the young minipig (Hanford) reflects the GIT membrane content of selected enzymes (lactase, sucrase, maltase, glucoamylase, and acid β-galactosidase) in the human infant and, therefore, would be a useful model in pediatric GIT research. Tang et al. (2004) examined the distribution pattern of P-gp protein levels along the entire GIT of the Yucatan micropig. The amount of P-gp protein increased distally from the duodenum to the ileum over an approximate 10-fold range with much lower amounts in the colon. This expression pattern is similar to that reported in humans (Mouly and Paine, 2003). It is noted that the intestinal permeability using the pig model was studied by Westerhout et al. (2014), who showed that the pig intestinal tissue (mounted in the InTESTine system) Papp values are more comparable to human Papp values compared with Caco-2 Papp values.
Oral Absorption and BA
Much of this previous review, along with literature to be cited, allows these writers to reach the conclusion that the minipig would be a good candidate animal model to explore drug absorption and disposition with the reasonable expectation that the data will reflect those processes in humans. Others have expressed the same opinion, which is based on measurable similarities between the two species in most aspects of physiology and biochemistry of many, if not all, body systems. The caveat, of course, is that our current state of knowledge is far from being complete, and, as with any model, refinements and adjustments need constantly to be made to achieve better (but not perfect) predictability.
Animal models are typically used to assess the absorption, BA, and PK behavior of orally dosed NCEs during drug discovery and early preclinical phases of drug development. Toxicity and safety assessment are determined in parallel studies. Oral dosing experiments are designed to provide an early signal about potential limited absorption and exposure. This, of course, is done in conjunction with an understanding of the physical chemical properties of the drug (e.g., pKa, ClogP, aqueous solubility, etc.). Although the pig has gained great popularity in recent years, current animal models continue to include the mouse, rat, rabbit, dog, and monkey (to some extent). The rat and dog remain the most extensively used, if for no other reasons than the existence of large data bases (and appropriate experimental facilities and experience) and their use in toxicity studies. The dog allows oral dosing of conventional dosage forms used in humans, unlike the rat. We conducted a review of the literature some years ago to obtain an estimate of BA in the dog, expressed as the fraction of the dose absorbed systemically (F), and we calculated an r2 value of about 0.24 in a plot of Fhuman versus Fdog (Zhang, Tang, and Mayersohn, unpublished data). More recently, Musther et al. (2014) reported on extensive analyses correlating oral BA in humans and mice, rats, dogs, and nonhuman primates. The results were disappointing, with r2 values of 0.253, 0.287, and 0.374 for the mouse, rat, and dog, respectively. An r2 of 0.694 was obtained for the nonhuman primates, perhaps as good a correlation as one might expect to reasonably achieve. Correlation did not improve when considering ionic class (acid, base, neutral, Zwitterion). One would expect that these correlations would improve, perhaps dramatically, if: standard experimental procedures were used in all cases, the same strain of animal was examined, the same laboratory (and technician) conducted the work, and identical sensitive and selective analytical methods were applied.
In preparing to write this minireview of minipigs, we did an analysis of the literature to determine the correlation of oral BA in pigs (Fpig) with Fhuman, as done for other species by, for example, Musther et al. (2014). We collected and evaluated all comparable F data in humans and pigs, recognizing that such a data set would be quite limited (with the hope of it expanding with time as minipig use increases). We were able to obtain 31 pairs of data (Supplemental Material Table A1). The details of this analysis can be found in the Supplemental Material (further analysis will be submitted for publication). These data are shown in Fig. 1 (left), which illustrates the line of identity with lines that define the 2-fold and 1-/2-fold ranges (i.e., a total 4-fold range). Overall, it appears that a low, but not unreasonable correlation exists (r2 = 0.408); in contrast, the highest r2 found by Musther et al. (2014) was for the nonhuman primate, 0.694. The data from that publication, analyzed in the same manner, are illustrated in Fig. 1 (right), offering a side-by-side comparison of the two species. Both animal species appear to underestimate Fhuman. For the pig, 6 of 31 drugs are outside the 4-fold range (20%), whereas 15 of 41 drugs (37%) are outside that range for the nonhuman primate. As a tentative general rule, if the BA in the pig is over 10%, the corresponding BA in humans will be over about 30%. A similar conclusion for the nonhuman primates suggests that BA values greater than 10% correspond to a BA in humans greater than about 20%. Clearly, this is not a rigorous rule, but it deserves testing until additional data are generated. What is missing in this analysis are insights into the BA differences between the two species: what is the causative mechanism(s), especially for those drugs having more than about a 2-fold difference between the two species? It is this mechanistic understanding that will ultimately provide the insight needed to design the appropriate studies and assist in resolving practical issues of poor BA in humans.

(Left) Comparison of the fraction of the oral dose absorbed systemically (F) in humans and pigs; (right) comparison of the fraction of the oral dose absorbed systemically (F) in humans and nonhuman primates (NHP). The lines shown are the lines of identity and the 2-fold and 0.5-fold ranges. The NHP data were taken directly from the tabular data of Musther et al. (2014). Reproduced with permission of the copyright owner (Saguaro Technical Press, Tucson, Arizona).
We consider in this work, briefly, those drugs from the above analysis having a greater than 2-fold difference between human and pig. Diclofenac is an exceptional example, as it is the only drug in which Fpig was much greater than Fhuman (viz., 100% vs. 42%). This is likely to be explained by a greater hepatic first-pass effect in humans due to the higher CYP2C activity in humans (Willis et al., 1979; Oberle et al., 1994). Midazolam BA in pigs (∼5%–14%), although similar to that in the rat and dog, is much lower than that in humans (∼34%). This appears to be due to the greater systemic (primarily hepatic) CL that approaches liver blood flow in pigs (Lignet et al., 2016). Cimetidine has a higher metabolic CL in minipigs, resulting in a higher hepatic and/or intestinal first-pass effect, which may be the cause for lower oral BA in the pig (∼33 vs. 78%; Lignet et al., 2016). In pigs, finasteride has an oral BA of 40% compared with 80% in humans, and, again, this may be explained by greater hepatic/intestinal extraction in the pig (Lundahl et al., 2011). Antipyrine has a low systemic CL in both pigs and humans, and, furthermore, a sex-related difference was noted in both species. Whereas the in vitro hepatocyte CL was low and similar in the two species, the oral BA of about 30% in both male and female pigs was much lower than that in humans (∼100%). This discrepancy can be explained by either lower absorption in pigs and/or higher extraction in the GIT. Because antipyrine is a Biopharmaceutical Classification System (BCS) class I compound, differences in absorption between the species are unexpected. It is possible, as with midazolam, which undergoes GIT extraction (Thummel et al., 1996), that antipyrine is also extracted by the GIT preferentially in the pig. This possibility needs to be explored. The tetracycline derivatives (tetracycline, oxytetracycline, and chlortetracycline) have significantly lower BA in both fed and fasted pigs compared with humans. Because tetracycline and oxytetracycline are eliminated primarily by renal excretion and have good solubility at the pH of the GIT fluids, first-pass effects cannot explain the disparity. These compounds are well known to form insoluble complexes with heavy metals present in the gut that decrease absorption, which, if the cation composition of the gut fluids of the pig is different from humans, could explain the lower BA, but this is speculation.
Although the above review and analysis provide a reasonable basis to view the minipig as a promising animal model to examine human drug absorption and BA, there is need for harmonization and standardization to the extent possible. Ideally, a single breed of minipig or micropig could be used, possibly the Göttingen strain, because of its past and current widespread utilization (especially in toxicity studies). Their small size is a real advantage to housing and handling as well as requiring less drug, an important consideration in early discovery programs. Protocols should be standardized as to feeding (throughout the day or twice per day), type of food, and the choice of male (castrated or noncastrated) or female animals, unless, of course, the study intention is to examine sex-related effects. The age of the animal is a consideration, most likely selected at sexual maturity, unless younger or aged animals can be shown to reflect maturation or aging effects in humans. Because pigs have the advantage of placement of vascular access ports (e.g., Bailie et al., 1986; Myers et al., 2016) that can remain patent for long periods, the experimenter has the advantage of considering multiple crossover studies, each requiring frequent blood sampling of long duration. It is important that there be robust data analysis and clarity in reporting the study design and results to maximize the information provided by the study and to minimize the bias or loss of information. Much of the literature is devoid of these important details. Specifically, there should be examination of drugs with low BA in humans because the current data analysis and interpretation were limited to compounds with BA values greater than 10% in humans. We encourage research in comparing the oral BA for drugs with lower BA in humans (<10%). We also suspect that there is a considerable amount of in-house data that might address this issue, and we encourage the industry to publish such data (especially those that appear to be failures in correlating with human results).
In Vitro and In Vivo Porcine Prediction of In Vivo Human CL
There have been generally two schools of thought when attempting to predict human CL: in vitro-based (usually referred to as IVIVE) and in vivo–based empirical methods (Obach, 1999; Tang and Mayersohn, 2005a; Houston, 2013; Rostami-Hodjegan, 2018). Although physiologically-based PK (PBPK) models are generally recognized as a more mechanistically-based methodology in describing and predicting human PK, these models depend upon IVIVE values for estimates of CL. Those estimates are critical to the success of the model, as the CL parameter depends upon organ extraction ratios and organ blood flows, the driving force behind all PK processes that drugs undergo in the body. In reality, both methodologies have been widely used in practice because neither method is completely satisfactory (Hosea et al., 2009; Ring et al., 2011). In addition, the real situation with regard to predicting human PK parameter values is usually more complicated than what is typically portrayed in most publications. For instance, the totality and quality of the data vary with the project stage (e.g., discovery screening data are usually much more limited) and vary also with the nature of the compounds (e.g., current NCEs are more complex and lipophilic, which results in variable in vitro and in vivo PK behaviors). High plasma protein-binding estimates in vitro are often associated with quality of measurement issues. In vitro metabolic incubation, especially with hepatocytes, is often associated with high variability from batch to batch and technician to technician working on the project and numerous other uncontrolled factors. Many of these complexities, both in vitro and in vivo, are not reflected in many scientific publications. Therefore, based on the authors’ experience with several large pharmaceutical companies and personal communications among active scientists in the field, both in vitro– and in vivo–based methods are always considered for incorporation into appropriate models for predicting human PK.
IVIVE.
Lignet et al. (2016) conducted a comprehensive IVIVE study in minipigs (adult male and female Göttingen) with seven reference compounds (antipyrine, atenolol, cimetidine, diazepam, hydrochlorothiazide, midazolam, and theophylline) following i.v. and oral dosing. CL values ranged from low to high (near liver blood flow). In vitro data were generated on hepatocellularity, metabolic CL in hepatocytes, and blood and plasma protein binding. The estimated in vitro CLint, CLint, and CLint from in vivo experiments illustrated an overall good correlation between minipigs and humans, with only one significant outlier (cimetidine, 11-fold difference). When incorporating plasma protein binding, in vitro binding in incubations, and blood/plasma ratio into the IVIVE model, the predictions seemed to get much worse. This has been observed by other investigators, in which consideration of plasma protein binding made the predictions worse (e.g., Poulin, 2013). Researchers have been making modifications to existing models, and progress continues to be made in the IVIVE arena, as with the introduction of the unbound liver fraction idea (Poulin, 2013; Poulin and Haddad, 2018). However, the IVIVE approach remains associated with uncertainties due to the complexity of the true mechanistic models, which may differ, in some aspects, among compounds and the measurement of uncertainties in the in vitro assays. Overall, the prediction performance of IVIVE in minipigs is generally similar to what has been observed in humans.
In Vivo–Based Methods.
Yoshimatsu et al. (2016) conducted PK studies with 14 compounds in three species: mouse, rat, and male NIBS (Nippon Institute for Biological Science) minipigs (3 months of age). Single-species allometry of CL with or without correcting for fraction unbound in plasma (fup) was used to predict human CL by fixing the allometric exponent at 0.75. The more conventional allometry using three species was also examined with or without correction for brain weight or maximal life span potential (Mahmood and Balian, 1996). Overall, the predictions based on either the minipig single-species method or conventional allometry, which included all three species, performed better than other in vivo methods. The single-species method performed similarly or better than the three-species method. This is not a surprising observation by virtue of the mathematical nature of the analysis; the conventional multiple-species allometry method depends on the predictability of the species having the largest weight, in this case the minipig (Tang and Mayersohn, 2005b). Adding more species to the allometric analysis, especially a species having less predictability or error among the lower body weight species, may amplify the error of prediction. Correcting for brain weight or maximal life span potential, the rule of exponents (Mahmood and Balian, 1996) is equivalent to applying some constants to the predictions, which are not based on any mechanistic foundation (Tang and Mayersohn, 2005c). It should also be noted that Yoshimatsu et al. (2016) found an excellent, predictive relationship for unbound apparent volume of distribution in humans from the minipig data (all data within a 4-fold range of values). With few exceptions, the unbound fractions of drug in the plasma were essentially the same when comparing the minipig and human. The i.v. data reported in this study will be used in the following section to evaluate an in vivo–based method for predicting human CL.
It should also be noted that there exists a large body weight gap between a typical dog (such as the Beagle, ∼10 kg) and a human adult (∼70 kg) in a conventional allometric model. The pig will represent a larger animal species, closer in body weight to humans, which mathematically exerts the largest impact in predicting human parameters in the allometric model (Tang and Mayersohn, 2005b). As discussed in previous sections, pigs resemble humans in many important PK aspects (physiologic/metabolic/transport, etc.). Thus, pigs as a predictive species with a closer body weight to humans are expected to improve the predictability of human parameters with the allometric model.
Data Collection and Reanalysis.
This section presents the development of a single-species allometric analysis for predicting human CL based on CL measured in minipigs. Literature data for i.v. dosing were collected and evaluated (n = 40 drugs; see Supplemental Material Table A2), including the data from Yoshimatsu et al. (2016) (n = 14), Lignet et al. (2016) (n = 7), and Mogi et al. (2012) (n = 5). A simple allometric relationship was employed, where the coefficient is optimized by minimizing the absolute average fold error (AAFE),
where the coefficient is optimized by minimizing the absolute average fold error (AAFE), the resulting equation, after optimizing the coefficient, was determined to be
the resulting equation, after optimizing the coefficient, was determined to be Overall, the prediction performance based on the in vivo minipig data appears to be acceptable, better than the single-species methods based on rat and dog, and similar to the single-species method based on the monkey, which were developed with the same methodology (n = 102; same model and optimization). The overall AAFEs from our analysis of the minipig data were 1.96 versus 2.35, 2.52, and 1.90 for the rat, dog, and monkey, respectively. The single-species data showed the same AAFE as found in the monkey data, but improved values over the rat or dog (Table 1). The coefficient from the pig analysis, 0.314, was lower than that from the dog (similar body weight), 0.410, and the monkey (generally lower body weight), 0.467, indicating that the metabolic activities were generally higher in pigs than those in the dog or monkey. The latter conclusion is consistent with our observations in the previous section that oral BA was generally lower in pigs, most likely due to the relatively greater value of CL in pigs. Results of the allometric analysis are illustrated in Fig. 2, which shows the line of identity along with the 3-fold and 1-/3-fold ranges.
Overall, the prediction performance based on the in vivo minipig data appears to be acceptable, better than the single-species methods based on rat and dog, and similar to the single-species method based on the monkey, which were developed with the same methodology (n = 102; same model and optimization). The overall AAFEs from our analysis of the minipig data were 1.96 versus 2.35, 2.52, and 1.90 for the rat, dog, and monkey, respectively. The single-species data showed the same AAFE as found in the monkey data, but improved values over the rat or dog (Table 1). The coefficient from the pig analysis, 0.314, was lower than that from the dog (similar body weight), 0.410, and the monkey (generally lower body weight), 0.467, indicating that the metabolic activities were generally higher in pigs than those in the dog or monkey. The latter conclusion is consistent with our observations in the previous section that oral BA was generally lower in pigs, most likely due to the relatively greater value of CL in pigs. Results of the allometric analysis are illustrated in Fig. 2, which shows the line of identity along with the 3-fold and 1-/3-fold ranges.
Comparison of the prediction performance of the single-species methods among rat, dog, monkey, and pig
The rat, dog, and monkey data were taken from Tang et al. (2007). Percentage fold error is predicted/observed.

The comparison of predicted vs. observed systemic CL in humans based on the optimized model in pigs. Lines shown are the line of identity and the ranges from 3-fold to 0.33-fold. Data compiled in Table A1 (Supplemental Material). Reproduced with permission of the copyright owner (Saguaro Technical Press).
It is worth noting that the single-species models above are the other mathematical forms of the allometric models with fixed exponents (Tang et al., 2007). The single-species methods have been extended to the scaling of unbound total CL (CL/fup) by some investigators as well as in practice in the industry. The thinking behind such corrections by fup is based on the hypothesis that plasma protein binding, which does not scale by body weight, may vary across species; thus, excluding such a confounding factor may better reflect the allometric relationship of body weight and organ elimination capacity (a hybrid of organ weight and enzymatic/excretion activity). There have not been robust data and analyses done to evaluate this approach, however. It should also be noted that, for high plasma protein-binding drugs (approximately >95%), the measurement errors associated with such binding are high; thus, correcting for such a parameter introduces another substantial random error.
Although the results of the above analyses are promising with regard to the use of minipigs for predicting human CL, the database currently available is limited and needs to be extended and, as noted above, standardized experimental procedures need to be applied. Of greatest interest and challenge is a mechanistic understanding of the reasons behind those drugs that are outside (often substantially outside) the 9-fold range of prediction.
PBPK Models
PBPK models, since they were first conceptually elaborated and applied by Bischoff and Dedrick (1968), have, in recent years, become increasingly important tools for drug development by the pharmaceutical industry in understanding and predicting PK behavior of NCEs (e.g., Rostami-Hodjegan and Tucker, 2007; Rowland et al., 2011; and Zhao et al., 2011). PBPK models integrate experimental PK-relevant data with current knowledge of physiology, assuming certain mechanistic models, and attempt to quantitatively describe and predict the PK behavior of drugs. Such a model is essentially an expression of all of the information and methodologies discussed in the previous sections of this minireview (i.e., from basic physiology, in vitro metabolic and transporter processes, in vivo PK data, etc.). This section will focus on a review of the current application and limitations of these PBPK models in pigs and propose future work. All of the effort invested in developing a PBPK model is directed toward the prediction of PK behavior of drugs, especially NCEs, in humans.
Publications dealing with PBPK models in pigs have been limited, and they are directed to the following: 1) data compilation and model development; 2) evaluation of formulations (typically oral); and 3) predicting human PK.
Data Compilation and Model Development.
Although there have been summaries of various physiologic values in pigs (e.g., Hannon et al., 1990; Upton, 2008), Suenderhauf and Parrott (2013) took the initiative in reviewing and compiling basic physiologic, biochemical, and PK data in Göttingen minipigs and, from that, developed a PBPK model used to evaluate two drugs, moxifloxacin and griseofulvin (both were dosed i.v. and orally). This is an important reference, which offers the basis for development of any PBPK model in Göttingen minipigs. Suenderhauf et al. (2014) further tested a PBPK model with a frequently used reference compound, acetaminophen (paracetamol), to evaluate gastric emptying in Göttingen minipigs. The drug was given orally as solution, capsule, and tablet forms, and the in vivo GE rates were estimated using a deconvolution method. Consistent with previous discussion, the GE was slower than found in humans, possibly due to the presence of food in the stomach. In a fed state, the GE was prolonged and more variable, as has been noted in humans. Sjögren et al. (2012) used another test drug, repaglinide, to investigate the interplay of metabolism and transport and its effect on the drug’s PK based on in vitro and in vivo data obtained from pigs (Yorkshire and Swedish Landrace). The drug is eliminated primarily by CYP3A4 and 2C8 metabolism and glucuronidation. It is also a substrate for organic anion-transporting polypeptide 1B1. Drug–drug interactions have been reported involving both uptake transport and metabolic inhibition in humans, in which cyclosporine or gemfibrozil could markedly increase the plasma concentrations of repaglinide. Based on the in vitro metabolic data from microsomes and hepatocytes and transport data, the investigators developed a PBPK model, which was able to quantitatively predict the in vivo PK.
There are several considerations: first, it is important to note that the compilation of the data by Suenderhauf and Parrott (2013) establishes the need for a unified pig model because differences are likely to exist, which will impact the results, such as the following: breed, food and feeding pattern, age and sexual maturation, sex, etc. Body weight, tissue composition, growth curve, and metabolic enzyme activities are also likely to vary among breeds. Several of these differences have been noted in previous sections. Importantly, to the extent possible, these variables need to be controlled for and standards established for experimental designs.
A second important note is with respect to the unique features of gastric emptying in the pig compared with humans. This process may control rate and perhaps extent of oral absorption for some classes of drugs (e.g., class I of the BCS and poorly soluble weak basic drugs). We have discussed this issue previously.
The third note of caution is with regard to all PBPK models being developed for any animal species, and that is to clearly understand the assumptions that are going into the model. Many physiologic and biochemical parameters were assumed in model development for minipigs; some were made between species, and some were made between the breeds of pigs. A sensitivity analysis around these assumptions should be conducted to evaluate the impact of the assumptions on the outcomes of the endpoints of greatest interest. This is generally true of all PBPK models; however, it may be even more important and mandatory for PBPK models developed in pigs because data and experience are much more limited than, for example, rats and dogs. It is useful to keep in mind that PBPK model development and its applications require a considerable amount of rich experimental data (in vitro and in vivo), derived from extensive labor-intensive efforts, the need for rigorous quantitative methods of analysis, an understanding of multiple scientific disciplines, and clarity and effectiveness in communicating PBPK information (especially assumptions made) for decision making. Such requirements limit the wide application across all drug discovery and development stages. With regard to the development of PBPK models in the pig, consideration should be given to the relative value(s) obtained from the required scientific/time/cost effort. In that regard, the following sections examine the potential value that investigators may want to offer by applying the PBPK approach in pigs: formulation assessment and human PK predictions.
Formulation Assessment.
Dogs are typically used for preclinical evaluation of formulations, especially for the prototype formulations intended for humans. However, for many reasons, some of which have been discussed above, the dog is not the most appropriate nonrodent species for many oral dosing scenarios, mostly based on substantial differences in the GIT compared with humans. For example, the dog has a much shorter small intestinal transit time compared with humans (which may be inappropriate for evaluating extended-release products), and they have a higher and more variable intestinal pH than in humans (Dressman, 1986). Exploration of the pig model as an alternative may be necessary. Kesisoglou et al. (2014) used the Yucatan minipig to develop a level C in vitro–in vivo correlation (IVIVC) for a BCS class III extended-release matrix tablet. The food–effect study, however, was shown to be challenging. Kesisoglou et al. (2016) developed a PBPK model for the dog to describe absorption in the large intestine for a BCS class I drug dosed via ileal and colonic ports to evaluate region-dependent absorption. A minipig absorption PBPK model was also developed by using the default minipig ACAT model in “Gastro-Plus” and assuming regional absorption differences to be the same as in the dog. (The grossly different GIT anatomy between the two species makes this a questionable assumption.) Two extended-release formulations were tested in minipigs, and the PK results were reasonably well predicted by the PBPK model along with the in vitro dissolution data. This work generally showed the utility of the PBPK approach, especially that the PBPK-predicted extended-release PK profile predicted the observed data, reflecting a correlation between the in vitro profile and the regional absorption characteristics.
A few observations are in order. A full description of the proposed PBPK model is essential. Although it might be clear to those investigators using specific software packages for example (in this case Gastro-Plus), it is difficult for other investigators to follow and understand the work, nor is the methodology transferable if other tools are used to analyze the data. For example, the study used castrated male minipigs of about 50 kg. Questions that may arise are as folows: Are there specific GIT parameters for the 50 kg Yucatan minipig? What are the values for gastric emptying, because GE may be different from other species, as shown in many publications? What are the values for GI fluids? PBPK modeling is associated with many assumptions, and the evaluation of those assumptions is important. Missing or incomplete information about those assumptions will inevitably create problems of reproduction. The authors encourage that all modeling work, especially PBPK models, present the fullest and clearest set of assumptions used in creating the model (perhaps in some supplementary format).
Research allowing comparison with human data is encouraged. The only way to further develop and confirm the usefulness of a model is to continue to test it. Although this may provide an initial burden for the industry, which will benefit the most, establishing a useful animal model would go a long way in developing predictive models for humans. The animal model must be able to distinguish, for example, among dosage forms of the same drug. Those in vivo data in conjunction with in vitro data (especially for extended-release forms) would be expected to offer a useful a priori insight into the human profile. Having such confidence will only evolve from establishing human and pig comparability over a wide spectrum of drugs/dosage forms.
Human PK Predictions.
Shida and Yamazaki (2016) constructed a series of simplified PBPK models in monkey, dog, and pig. The purpose of their modeling work was to predict human PK. The approach was to first construct PBPK models in animals. They then used a scale-up strategy, which predicts human PK parameters (such as absorption rate constant, central volume of distribution, CLint, etc.) based on animal values estimated from the model and in vitro metabolic data. The test compounds were as follows: caffeine, S-warfarin, omeprazole, metoprolol, and midazolam, whose corresponding metabolic CYP isoforms are 1A2, 2C9, 2C19, 2D6, and 3A4, respectively. The investigators concluded that the PBPK-scaled models predicted the observed PK in humans. It should be noted that this approach is similar to simplified conventional compartmental (i.e., lumped) PK models. The most important human parameters (hepatic CL, volume of distribution, and absorption) were scaled or estimated with common approaches. For example, fixed-exponent allometry was used to scale CL in rodents to humans; IVIVE was used to estimate in vivo CL. The development of such simplified models may lend certain credibility to using a similar structured model for predicting human PK; however, the extent of any added value from such models may be limited because the prediction of key PK parameters (CL, volumes, and absorption) is still the fundamental challenge. Then the key questions that need to be asked are still the same in predicting human PK values: allometry, IVIVE, or scale-up strategy (a hybrid of allometry and IVIVE in animals). Whether pigs could serve as an alternative or as a more predictive model of human PK will require a great deal more data with systematic and robust analyses, as noted previously.
To conclude this section, there are some considerations for the future. Pig breeds need to be unified and standardized for PBPK development, as for any other purpose. This goal may require some coordinated effort from academia, industry, and government regulatory agencies. Gastric emptying seems to be a major unique feature associated with pigs that may offer an obstacle in establishing absorption/BA models and in examination of fed versus fasted states. Again, the best approach would seem to select one breed (e.g., Göttingen) for further examination or, in the alternative, determination of that breed which is least susceptible to GE limitations or food effects. Studies of different drugs/dosage forms are necessary with comparison with human data. A major value of a porcine model may well be as a screen for oral dosage forms and its relationship to human absorption, limitations discussed notwithstanding. Predicting human PK parameter values from a porcine-based PBPK model appears to have potential; however, the prediction of key driving parameters (for example, systemic CL) remains the major challenge.
Evaluation of Formulations
Development and assessment of drug dosage forms are essential functions of any research and development division and for which having a predictive animal model would be a desiderata. We have noted in several sections above numerous issues associated with the pig that may compromise or make challenging the use of a porcine model for assessment of (especially) oral dosage forms; advantages are also noted. Disadvantages also exist for the dog in this regard, which is the most often relied upon model. We have also cited several publications that have examined the drug absorption process in the minipig (e.g., Suenderhauf et al., 2014; Henze et al., 2018). In this section, we discuss several other specific examples in which drug dosage forms have been assessed in the pig and for which there are relevant human data. Many other studies have been published, but for which there are no comparative human data, making it difficult or impossible to assess the suitability of the animal model.
Christiansen et al. (2015) examined the effect of food on the oral absorption of pravastatin and atazanavir, two low aqueous solubility drugs in Göttingen minipigs. They used a protocol similar to one used in dogs (Lentz et al., 2007). GE was followed with coadministration of oral acetaminophen (as a marker of GE). As noted by others, GE times were prolonged in the pigs (2.3–8.4 hours); however, there was no food effect seen. Pravastatin and atazanavir have shown positive and negative food effects, respectively, in humans. The results of this study challenge the idea of using the pig as an animal model for studying food effects in humans. The investigators conducted a study to examine the impact of a pharmacological agent that promotes GE, metoclopramide, to normalize GE. That agent was found to have no effect on GE in a fed or fasted study, again questioning the pig as an appropriate animal model for examining food effects.
Drug absorption from modified (i.e., controlled or sustained) release dosage forms has been examined in numerous studies. Kulkarni et al. (2012) compared the absorption of immediate and modified release in immature farm pigs (∼3.5–4 months; 21–27 kg) and dogs (apparently under fasting conditions). The results suggested that the pig may be more suitable an animal model on the basis of smaller variance in area under the plasma concentration–time curve (AUC) and a higher BA (∼80%, similar to humans). The lower BA value in the dog may reflect the shorter GI transit times, not allowing sufficient time for absorption. Based on the similar plasma concentration–time profiles for the immediate and modified release forms in the pig, there was no suggestion of a delay in GE. Genissel et al. (2004) examined the absorption of trimetazidine from an oral solution, immediate release, and sustained release dosage forms in pigs (24.2–31.8 kg) and dogs. The i.v. data indicated greater CL in the pig, with a corresponding greater first-pass effect associated with the oral forms. The plasma concentration–time data in pigs suggested no delay in absorption, with mean absorption times being similar in the two species. Either species appeared to be an adequate model for the evaluation of the modified-release form. Ikegami et al. (2006) studied controlled-release theophylline absorption in dogs, monkeys, and pigs (Göttingen; 10–12 kg). Even after 20 hours of fasting, delay in absorption in pigs was seen. The relative BA were 33% and 47% in the dog and pig, respectively, compared with 80% in the monkey. There was no relationship between in vitro and in vivo release in dogs and pigs, and the authors concluded that the monkey is the better animal model. One reason for the lower BA in the pig may be incomplete estimation of AUC, whose value may be affected by GE. The low BA in the dog may be due to the short GI transit time, as noted before. In this study, as elsewhere, a more complete IVIVC and comparison with human data would be ideal.
Ramsay-Olocco et al. (2004) used Yucatan micropigs (25.3–51 kg) to examine the effect of vitamin E d-α-Tocopheryl polyethylene glycol 1000 succinate (TPGS) on intestinal permeability for P-gp efflux of a substrate with high solubility and low permeability. Although there was large variability with TPGS, there was a suggestion of improved BA with TPGS (25%–44%). In humans, no improvement was observed with TPGS. It should be noted, however, that the human dose was 5 mg, whereas that in pigs was 50 mg. Considering the approximately 3-fold difference in body weight, the actual dose/kg difference may be up to 30-fold. Thus, the actual amount of TPGS in humans was substantially lower per intestinal volume or area, which could lead to minimal impact of TPGS in P-gp inhibition. This point emphasizes the importance of quantitative analysis and design of studies (e.g., effective inhibitory concentrations in in vivo luminal contents).
The above brief presentation makes clear that the pig may not be an ideal (if such a thing exists) animal model for examination of dosage form performance, at least with regard to food–effect studies; it may prove to be at least as good as other animal models. The pig is likely to be useful as a preclinical screen for quantifying dosage form differences and to serve to reflect the human condition. At the current time there is too little rigorous information, especially in comparison with human data, to reach an unequivocal conclusion; time will tell.
Dermal Absorption
Dermally applied dosage forms for the transdermal delivery of drugs have become an increasingly important route of administration, having significant advantages in terms of convenience to the patient, slow and prolonged absorption profile, and avoidance of GIT first-pass effects (Paudel et al., 2010). Predicting human exposure, therefore, becomes an important task, as it is for other routes of administration. Excised human cadaver skin is considered the gold standard for predictive purposes (e.g., Franz et al., 2009). However, there are significant limitations to the in vitro use of human cadaver skin, including the high cost and availability (e.g., there is no human skin bank in China), experimental variations due to the limited supply of samples, and the potential differences between the in vitro skin-testing system and the in vivo performance of skin. In vitro models are only quantitatively useful for predictive purposes when a correlation exists between the in vitro and in vivo systems, reflecting the need for an IVIVC. Potential causes for a lack of correlation include blood flow (vs. sink conditions), local pH and temperature, and metabolism (due to cryopreservation, etc.) (Pershing et al., 1989; Auclair et al., 1991; Clarys et al., 1998; Manevski et al., 2015). The method of skin preparation can be critical to the experimental results with regard to lag times and permeability measures. For example, the lack of a blood flow causes the lower portions of the dermis to act as a rate-limiting step for transport, a barrier that would not exist in vivo (Takeuchi et al., 2012). There are numerous studies showing that human skin contains most of the enzymes found in other major elimination organs (e.g., CYPs, phase II enzymes such as glutathione S-transferase, UDP-glucuronosyltransferase, NAT, etc., and others, including Flavin-Containing Monooxygenase (FMO), cyclooxygenases, Alcohol dehydrogenase (ADH) and aldehyde dehydrogenase (ALDH), esterases/amidases, etc.) (e.g., Oesch et al., 2007). Information about skin metabolism in pigs is limited; however, substantial metabolism may exist for certain drugs, but this will depend upon the processing and preservation conditions of the skin. Yucatan micropigs resemble humans with regard to esterases, and albumin seemed to affect the preservation of its activity (Rangarajan and Zatz, 2001).
Although rats are readily available for such studies, significant differences have been found between rat and human skin: rodent stratum corneum is thinner, which may lead to the observed higher permeability noted in rodents. Pig skin is much more similar to human skin with regard to thickness of the stratum corneum, lipid components, etc. (Príborský and Mühlbachová, 1990; Stricker-Krongrad et al., 2017). As we noted above, “The minipig is the species of choice to investigate dermal absorption in preclinical drug development” (Preube and Skaanild, 2012). The intent of this brief review of dermal absorption is to focus on several publications that have evaluated transdermal drug absorption comparing in vitro and in vivo data in the pig (Yamamoto et al., 2017; Yoshimatsu et al., 2017). We also provide in-house data generated at Guangzhou Dazhou Biomedicines (unpublished data) to illustrate the development of an IVIVC to demonstrate the utility of this approach.
Transdermal IVIVC in Pigs.
Yamamoto et al. (2017) studied several drugs (nicotine, rivastigmine diclofenac, and lidocaine) in vivo in four minipigs (5 months; 10.0–14.4 kg; breed not reported) by application of the dermal product to the back of the pig. Cryopreserved excised Göttingen minipig skin (male; 5 months; 9.3 kg) was cut from the back of the minipig and dermatomed to a thickness of 400–600 µm. Cryopreserved excised human skin was obtained from female Caucasian human subjects (during abdominal plastic surgery). The human and pig skin permeabilities of the test drugs generally showed similar time–course profiles, with the pig having about a 2-fold higher permeability. Using the in vitro pig permeability data and convoluting with the human systemic drug disposition data, the human PK following dermal application were reasonably well predicted. The observed AUC and maximum plasma concentration values were about within a 2-fold range of predictions. Time of occurrence of maximum plasma concentration varied, though, suggesting that the delayed appearance of the drugs in the circulation may not be adequately captured by the in vitro permeability studies. This thorough study, with some limitations, reported the following information: in vitro permeability in pig skin; in vitro permeability in human skin; in vivo PK following dermal application in pigs; in vivo PK following dermal application in humans; and in vivo systemic PK in humans (literature data). What is missing is the systemic PK profile in pigs and an analysis of the in vitro and in vivo relationship in pigs. It was not clear how the BA was obtained without systemic PK in the pigs, although an estimate might have been obtained from analysis of the residual drug in the dermally applied formulation. The more complete study design would include an i.v. arm in the same pigs, together with the thorough data analysis, as noted: i.v. PK in the pig; in vivo estimation of absorption in pigs (e.g., by deconvolution); evaluation of correlations between in vitro and in vivo data in both pigs and humans; and quantification of the IVIVC in pigs and prediction of human in vivo transdermal absorption with and without consideration of the correlation in pigs. The IVIVC established in pigs will lend more confidence in predicting human PK. If the human PK predictions turn out to be substantially different from those based on the in vitro human permeability, there is a reference in pigs to evaluate whether the in vitro system is appropriate for prediction purposes. Completeness in the IVIVC data and methodology for both pig and human is important. Ultimately, if experimental protocols can be standardized (such as pig breed, dermatome methodology, storage of skin, Franz cell setup, etc.) and generally agreed upon, and the in vitro data are found to have adequate correlation to in vivo results, then human skin studies need not be done, that is, prediction of human PK directly based on in vitro pig permeability, as demonstrated by Yamamoto et al. (2017).
Yoshimatsu et al. (2017) conducted in vitro and in vivo studies on six drugs (ketoprofen, flurbiprofen, diclofenac, buprenorphine, fentanyl, and lidocaine) in mice, rats, minipigs, and humans [diclofenac and lidocaine data from the Yamamoto et al. (2017) study]. The study protocols were basically the same as in Yamamoto et al. (2017). The results indicated considerably lower in vitro and in vivo permeability in pigs compared with the other species, including humans. For example, the ratios of the fractions permeated in vitro at 24 hours between humans and pigs for the six drugs varied from 4.58 to 34.5, whereas the differences observed in the Yamamoto et al. (2017) study were all within about 2-fold. It is difficult to explain the much lower permeability in pigs noted in this study, especially since two of the drugs were the same in both studies.
In-House Experience with Pig Transdermal Models.
In our experience, in vitro pig and human skin generally showed similar permeabilities, as has been reported by others (e.g., Rohatagi et al., 1997). Skin was dermatomed to a thickness of 400–600 µm from the back of Göttingen minipigs. The Franz cell was used to determine in vitro permeability. An example design of IVIV studies and analysis is shown in Fig. 3. Three minipigs were used in a crossover design and three dose groups (i.v., patch formulations A and B). This design generates, in the same animal, systemic and absorption PK, allowing estimation of in vivo absorption for the individual, controlling the interindividual differences in systemic disposition and increasing the power of the analysis. The two patch formulations were made to give different release rates and thus to provide different permeabilities, allowing the development of an IVIVC for one formulation and external validation with data from the other formulation. In addition, a minipig skin bank was established in-house. As long as the two formulations were tested with the same pig skin (Pig N in Fig. 3), the effect of any differences in absorption between the in vitro (Pig 1) and in vivo (Pig N) results that are due to differences in the skin behavior between the two pigs may be cancelled in terms of predicting the in vivo PK for Formulation B. Note, the above crossover design with N = 3 and the IVIVC analysis method is not intended for meeting the criteria for IVIVC per the Food and Drug Administration’s guidance (1997) on the development, evaluation, and applications of IVIVC for extended-release oral dosage forms. Rather, the cost-efficient IVIVC study design and analysis, as illustrated (and the similarity in in vitro permeability between the two species), will achieve the following: provide confidence in predicting human transdermal PK based on in vitro permeability with human skin under the same experimental conditions as in minipigs; facilitate an optimal study design in humans (for example, by examining the time course and PK profile in minipigs, which may demonstrate the delay in PK exposure, skin retention of drugs, etc.); and allow a reasonable definition of the product profile at an early stage of drug development.
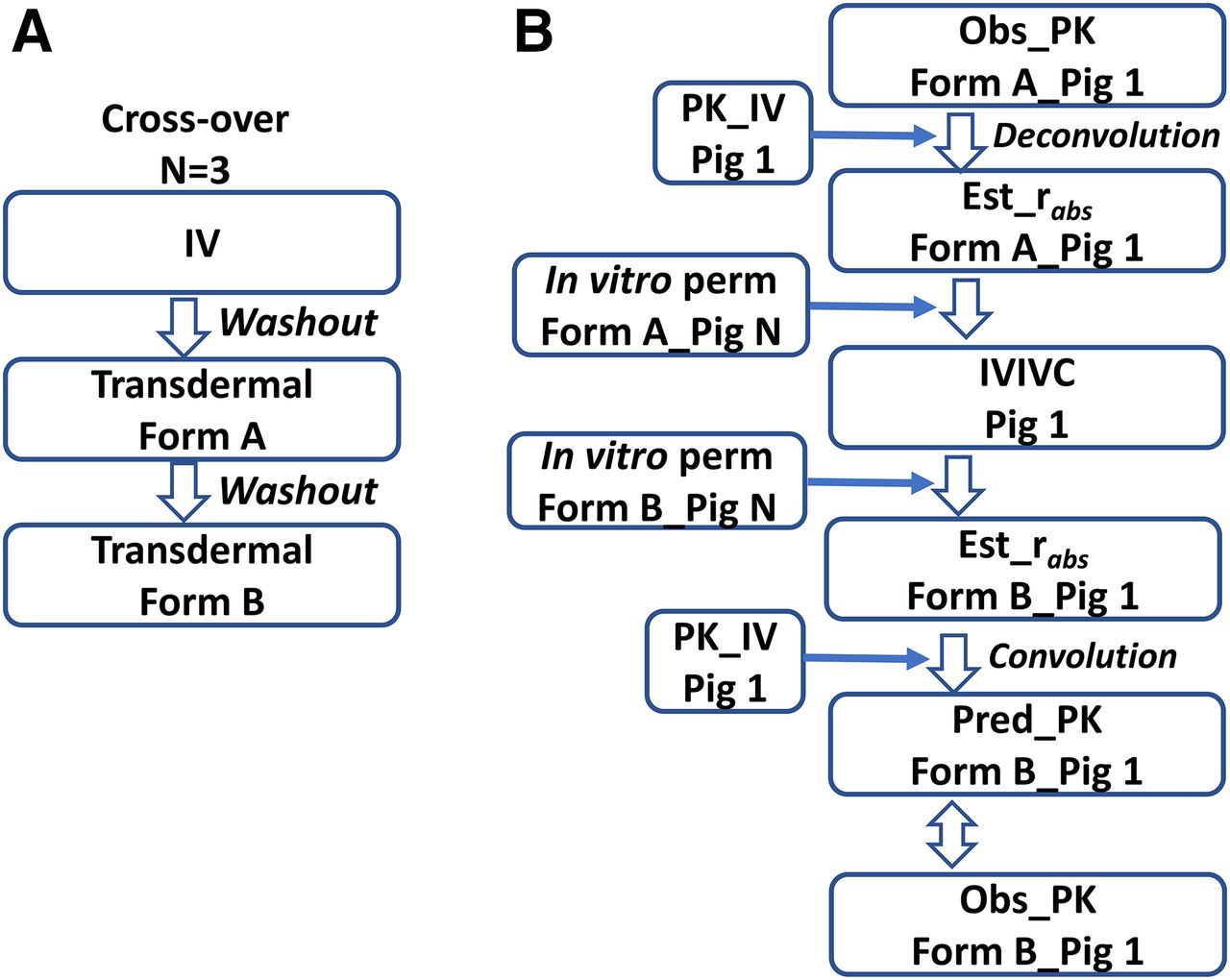
(A) An example IVIVC study design in three Göttingen minipigs with a sequence of IV, Formulation A, and Formulation B, with a 1-week washout in between dosing the groups. (B) The IVIVC analysis for one pig (Pig 1). The i.v. PK in Pig 1 is modeled, and impulse response function is obtained. Deconvolution is conducted to estimate the in vivo absorption rate (rabs) of Formulation A. The correlation, based on Formulation A, is modeled between in vivo rabs in Pig 1 and in vitro permeability in Pig N from the minipig skin bank. The in vitro permeability in Pig N for Formulation B, thus, is plugged into the correlation model to estimate rabs in Pig 1 of Formulation B. The next step is to use the convolution method to predict the in vivo transdermal PK of Formulation B based on the estimated rabs in Pig 1 and the impulse response function from the i.v. PK. The last step is to evaluate the performance of the predicted PK vs. the observed PK with Formulation B. Reproduced with permission of the copyright owner (Saguaro Technical Press).
As we have noted in previous sections, there needs to be standardization of methods and experimental protocols. We cannot explain some of the dramatic disparities noted in the literature; however, effort needs to be expended to attend to and resolve those differences. Studies need to be well designed, and data analyses need to be robust and complete, as we tried to exemplify in the above in-house example, to realize the full potential value of an IVIVC.
Summary and Conclusions
The swine has played an important role in biomedical research, and current literature suggests a greater awareness of that role, which is expected to increase markedly with time. We have attempted to review in this work those biologic aspects of the pig and its similarities to humans that make it an attractive animal model in drug discovery and development. Needless to say, there are always limitations to any animal model used to make predictions in humans; as noted, these are models of, not duplicates of, humans. What does the future hold? We fully expect that use of minipigs will gain greater momentum in the immediate future. As we have noted, it is important that standardization be applied across studies (and organizations) to optimize experimental results and minimize variability. Among those considerations are the following: selection of the minipig breed, type of diet, and frequency of feeding, etc. An area of research that does not seem to have been explored is pharmacodynamics (PD). The minipig can be instrumented to provide PD measurements and to offer PK/PD model development. We look forward to those developments. The authors indeed believe that there is a “pig in the poke.”
Acknowledgments
We gratefully acknowledge the time and effort of Jennifer Martin, University of Arizona Health Sciences Library (librarian par excellence), in conducting a maxi-literature search for this mini-review. We thank Da Zhang for sharing the oral bioavailability data in dogs and humans and Hock Tan and Wenfei Liang for providing the GDB in-house experience on transdermal in vitro and in vivo studies in humans and pigs.
Authorship Contributions
Participated in research design: Tang, Mayersohn.
Performed data analysis: Tang, Mayersohn.
Wrote or contributed to the writing of the manuscript: Tang, Mayersohn.
Footnotes
- Received June 22, 2018.
- Accepted August 20, 2018.
Abbreviations
- AAFE
- absolute average fold error
- AOX
- aldehyde oxidase
- AUC
- area under the plasma concentration–time curve
- BA
- bioavailability
- BCS
- Biopharmaceutical Classification System
- CL
- clearance
- CLint
- intrinsic CL
- CYP
- cytochrome
- F
- fraction of the dose absorbed systemically
- fup
- fraction unbound in plasma
- GE
- gastric emptying
- GI
- gastrointestinal
- GIT
- GI tract
- IVIVC
- in vitro–in vivo correlation
- IVIVE
- in vitro–in vivo extrapolation
- MMC
- migrating myoelectric complex
- NAT
- N-acetyltransferase
- NCE
- new chemical entity
- PBPK
- physiologically-based PK
- PD
- pharmacodynamics
- PK
- pharmacokinetic
- TPGS
- d-α-Tocopheryl polyethylene glycol 1000 succinate
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics
References
In this issue





{kind=link}
{kind=link}
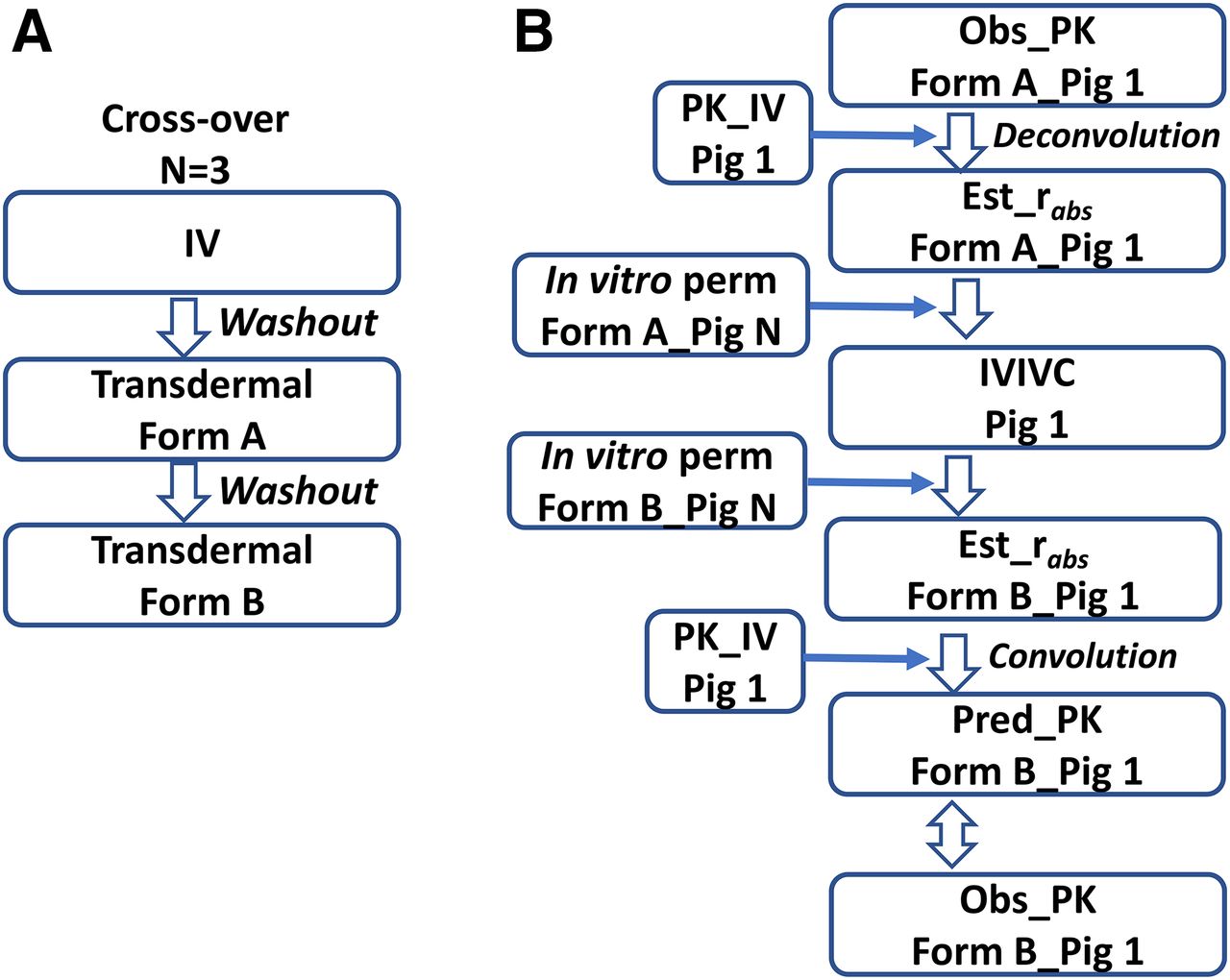{kind=link}
Jump to section
- Article
- Abstract
- Introduction and Historical Perspective
- Physiologic and Anatomic Considerations
- Gastric Emptying and Intestinal Transit
- Hepatic and GIT Enzymatic Metabolism and Transporters
- Oral Absorption and BA
- In Vitro and In Vivo Porcine Prediction of In Vivo Human CL
- PBPK Models
- Evaluation of Formulations
- Dermal Absorption
- Summary and Conclusions
- Acknowledgments
- Authorship Contributions
- Footnotes
- Abbreviations
- References
- Figures & Data
- Info & Metrics
- eLetters
- PDF + SI