


Abstract
Clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR associated protein 9 (Cas9), i.e., CRISPR-Cas9, has been extensively used as a gene-editing technology during recent years. Unlike earlier technologies for gene editing or gene knockdown, such as zinc finger nucleases and RNA interference, CRISPR-Cas9 is comparably easy to use, affordable, and versatile. Recently, CRISPR-Cas9 has been applied in studies of drug absorption, distribution, metabolism, and excretion (ADME) and for ADME model generation. To date, about 50 papers have been published describing in vitro or in vivo CRISPR-Cas9 gene editing of ADME and ADME-related genes. Twenty of these papers describe gene editing of clinically relevant genes, such as ATP-binding cassette drug transporters and cytochrome P450 drug-metabolizing enzymes. With CRISPR-Cas9, the ADME tool box has been substantially expanded. This new technology allows us to develop better and more predictive in vitro and in vivo ADME models and map previously underexplored ADME genes and gene families. In this mini-review, we give an overview of the CRISPR-Cas9 technology and summarize recent applications of CRISPR-Cas9 within the ADME field. We also speculate about future applications of CRISPR-Cas9 in ADME research.
Introduction
Clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR associated protein 9 (Cas9) is an easily adapted gene-editing technology that can be designed to target most genomic sequences. The major difference between CRISPR-Cas9 and previously favored gene-editing systems—such as the zinc finger nucleases (ZFNs) and the transcription activator–like effector nucleases (TALENs)—is its simplicity. CRISPR-Cas9 DNA targeting only requires the design of a short guide RNA molecule compared with the more complex and expensive protein engineering needed for retargeting ZFNs and TALENs (cf. Gaj et al., 2013; Kim, 2016; Editorial, 2017). The availability and affordability of commercial kits and services means that many biomedical research fields are now using CRISPR-Cas9.
In applied genome editing, researchers have traditionally relied heavily on model organisms such as bacteria, yeast, fruit fly, and rodents. In part, this was because these were the only species for which good tools for genetic manipulation were available. The need for faster gene editing led to the development of custom nucleases like ZFNs and TALENs that induce double strand breaks (DSBs) in a predefined region in the gene of interest (Gaj et al., 2013). Only the ZFN technology has been applied within the field of absorption, distribution, metabolism, and excretion (ADME). There, it has been used for selective knockout of drug transporters in C2BBe1 cells (a subclone of Caco-2, which is a widely used model for investigating transporter-mediated intestinal efflux of drugs) (Pratt et al., 2012; Sampson et al., 2015). Similarly, ZFN has been used to knock out endogenous canine transporters in Madin-Darby canine kidney (MDCK) II cells (Gartzke et al., 2015). It has also been used in HepaRG cells, an alternative to primary human hepatocytes, to knock out the nuclear pregnane X receptor (PXR), a receptor that regulates the expression of drug-metabolizing enzymes and drug transporters (Williamson et al., 2016). Despite these successful studies, neither ZFN nor TALEN have become widespread methods within the field of ADME. One reason may be the expensive and demanding protein engineering needed.
CRISPR-Cas9 has made it possible to edit genes in virtually all organisms for a fraction of the cost compared with alternative technologies. Moreover, CRISPR-Cas9 allows modification of the genes of interest at more physiologically relevant levels (i.e., regulated by an indigenous promoter) rather than by overexpression of the gene of interest. In addition to manipulation of individual genes, CRISPR-Cas9 can even be used to eliminate an entire chromosome (Zuo et al., 2017). This has been demonstrated in human pluripotent stem cells with chromosome 21 trisomy and in cancer cells; highlighting potential therapeutic applications for the CRISPR-Cas9 technology. Furthermore, it is possible to study multiple genes simultaneously, thus creating an intricate network for studying or even curing diseases. One example of such simultaneous gene editing is the knockout of 62 porcine endogenous retroviruses in an immortalized porcine cell line (Yang et al., 2015). In a follow-up study, the same technology was used to generate porcine endogenous retrovirus-free piglets, a strategy that would increase safe and effective organs for pig-to-human xenotranplantations (Niu et al., 2017). However, the in vivo application of CRISPR-Cas9 in humans was impeded after a report in 2015, in which CRISPR-Cas9 gene editing in human embryos showed low rates of homology directed repair (HDR) and apparent off-target effects (i.e., alterations in genes unrelated to the target genes) (Liang et al., 2015). Thus, there are crucial safety issues that have to be solved before broader in vivo applications become a reality. Despite this setback, there are ongoing clinical trials involving CRISPR-Cas9 [https://clinicaltrials.gov/ (accessed on May 20, 2018)]. The first of these focuses on T-cells engineering for treatment of metastatic small cell lung cancer. This study started in 2016 and is expected to be completed during 2018. The engineered T-cells were given to the first patient in October 2016 [https://newatlas.com/crispr-gene-editing-first-human-trial/46453/ (accessed on May 20, 2018)], and the results are not yet published.
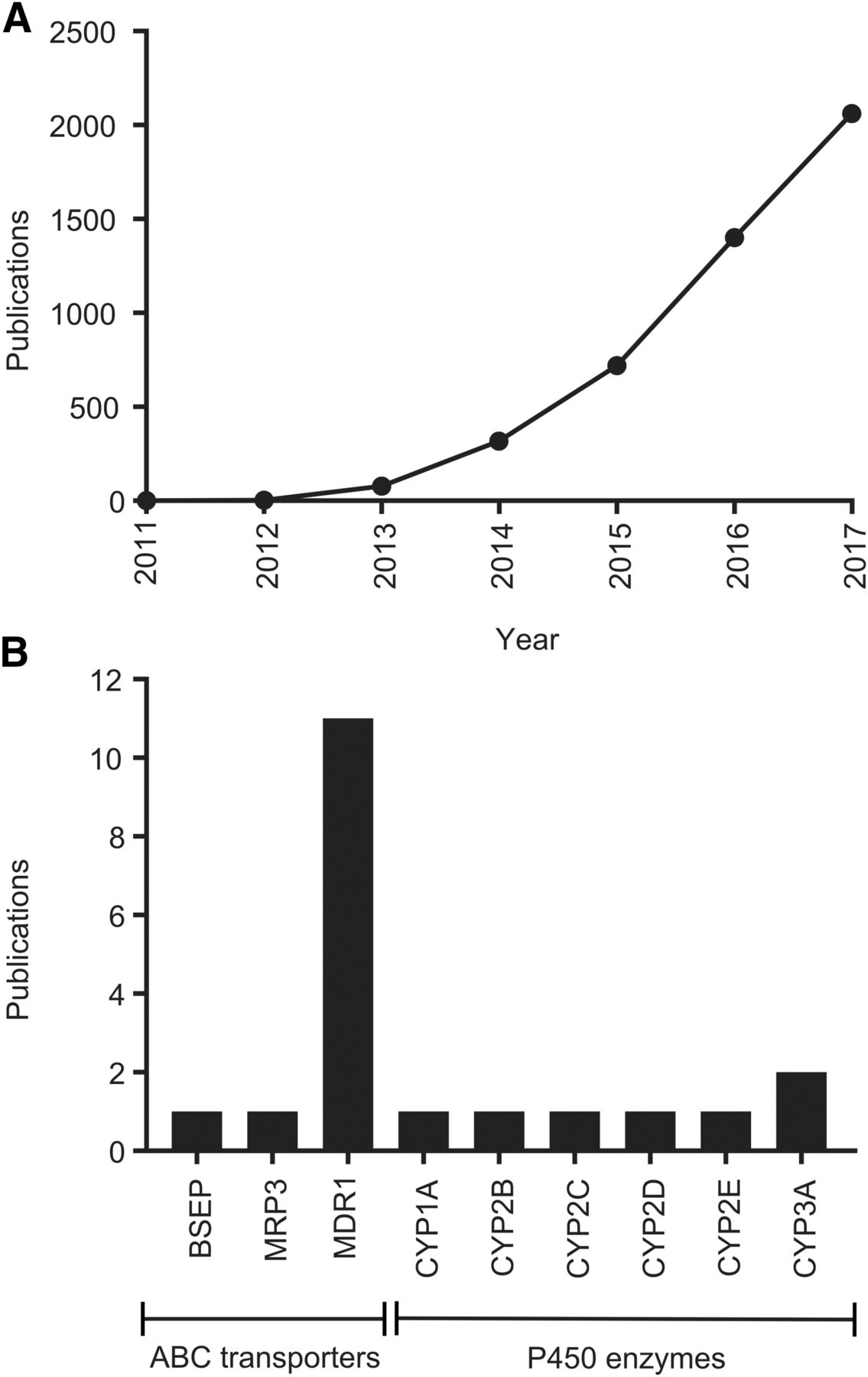
The number of publications regarding CRISPR-Cas9 has increased rapidly (Fig. 1A), from single articles by the originators of the technology in 2011 and 2012, to more than 2000 articles published in 2017. CRISPR-Cas9 is now also being applied within the ADME field (Fig. 1B; Table 1). In this context, it has been mainly used for the generation of in vitro and in vivo knockout models, in parallel with other approaches. Importantly, technologies such as CRISPR-Cas9 also give the opportunity to deorphanize and characterize ADME gene families that are currently underexplored, such as the solute carrier transporter family (César-Razquin et al., 2015). In this mini-review, we give an overview of the application of CRISPR-Cas9 gene editing for ADME studies of drugs and xenobiotics.

(A) Number of publications per year mentioning CRISPR-Cas9. Data were generated by PubMed search using the terms “CRISPR” and “Cas9.” (B) Total number of publications for CRISPR-Cas9 gene editing of ADME genes/proteins. Data were generated by PubMed search using the terms “CRISPR” and “Cas9” and the respective human gene or protein name. All hits were screened and publications were excluded if CRISPR-Cas9 had not been used to edit an ADME gene. If homologous genes from other species had been edited, those publications were included in this compilation. The ADME genes included in the analysis are the solute carrier and ATP-binding cassette (ABC) transporters identified as involved in drug ADME by the International Transporter Consortium (Giacomini et al., 2010) and the most important cytochrome P450 (P450) and uridine diphosphate-glucuronyltransferase enzymes (Evans and Relling, 1999; Williams et al., 2004). The PubMed search was performed May 18, 2018, i.e., articles published after that are not included in this compilation.
Overview of ADME and ADME-related genes edited using CRISPR-Cas9
How Does CRISPR-Cas9 Work?
In nature, CRISPR-Cas9 is an adaptive, acquired immune system, mediated by RNA, which protects eubacteria and archaea from viruses or plasmids (Barrangou et al., 2007). In bacteria, two RNA molecules, together with the endonuclease Cas9, bind to a target sequence. One of the RNA molecules (trans-activating CRISPR RNA) is species conserved and acts as a scaffold that binds Cas9 itself. The other RNA molecule (CRISPR RNA) is variable and confers specificity to a target genome DNA. For gene-editing purposes the technology was simplified and made more efficient by combining both RNAs into a single guide RNA (sgRNA) (Jinek et al., 2012). The sgRNA and Cas9 form a sgRNA-Cas9 complex that binds to its target sequence (Fig. 2, A and B). An additional requirement of the target sequence is the presence of a protospacer adjacent motif (PAM) (Mojica et al., 2009). If the sgRNA hybridize to DNA immediately 5′ to an adjacent PAM domain the Cas9 endonuclease will create a DSB (Fig. 2B). This DSB can be repaired by the mechanism of nonhomologous end joining (NHEJ) (Overballe-Petersen et al., 2013). This, in turn, may result in small deletions or insertions (indels) that disrupt the targeted gene, resulting in loss of its function (Fig. 2C). Alternatively, if there is a donor DNA template to the targeted region the DSB can be repaired by HDR, thereby allowing for precise replacement of nucleotide(s), introduction of mutations, or insertion of sequences in the targeted region (Gong et al., 2005) (Fig. 2C). The frequency of NHEJ indel formation or successful HDR varies, depending on the model cell line/organism and methods used; however, in general HDR is a rarer event than NHEJ indel formation (Mali et al., 2013). For an overview of CRISPR discovery and history, see Lander (2016).
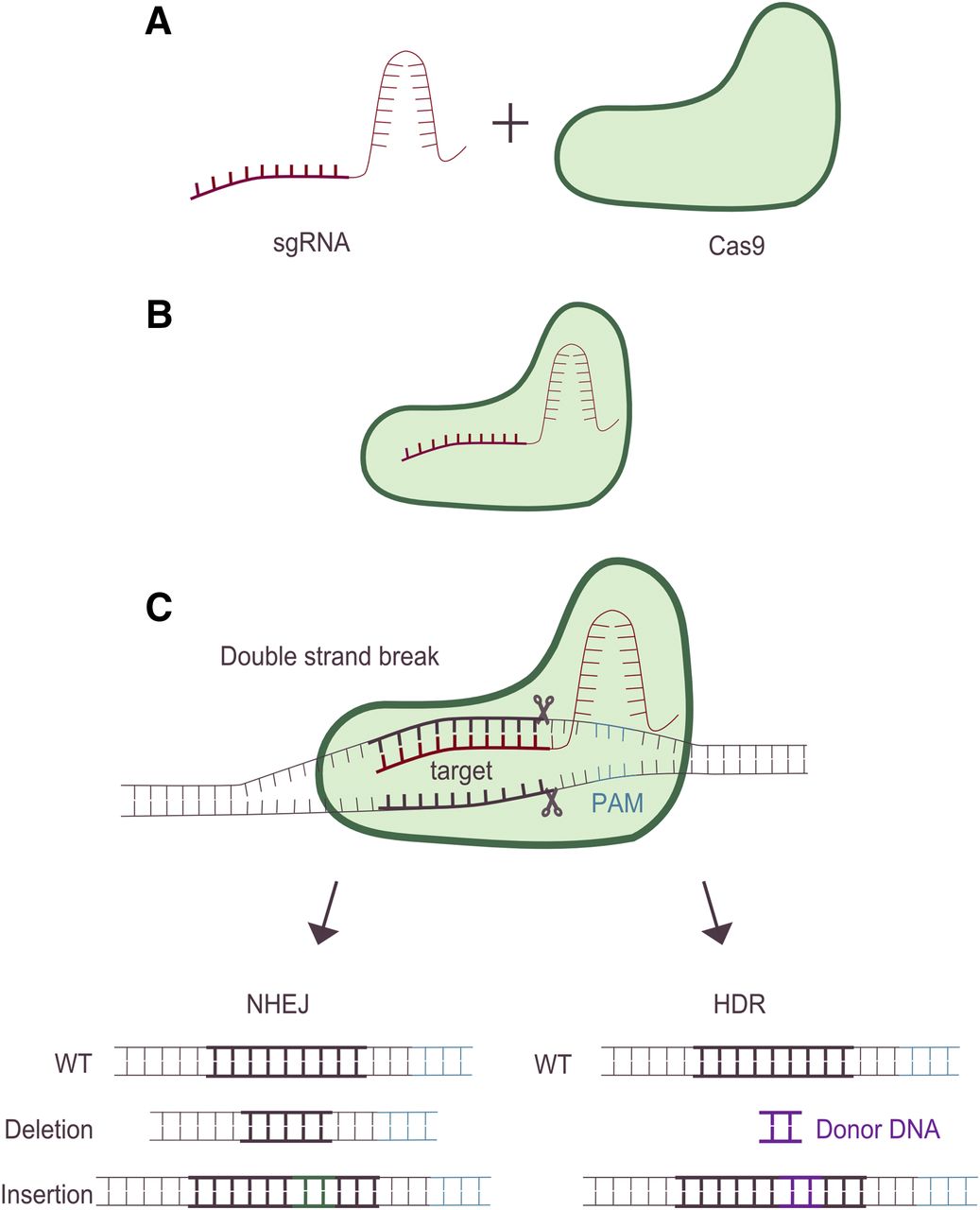
Overview of the CRISPR-Cas9 gene-editing process. For CRISPR-Cas9 gene editing, a sgRNA is designed. This sgRNA consists of two parts, one that is specific for the desired target DNA and one that will interact with the second component in the CRISPR-Cas9 system, the Cas9 endonuclease (A). An important requirement of the target DNA is the presence of a PAM. After formation of a sgRNA-Cas9 complex (B), the complex will bind to the target DNA, and if an adjacent PAM is present Cas9 will cause a DNA DSB three base pairs upstream of the PAM sequence (C). Thereafter, the DSB can be repaired using two different mechanisms, NHEJ or HDR. NHEJ is an error-prone mechanism that will often result in small deletions or insertions (indels) that can lead to a disruption in the targeted locus. Alternatively, if a homology-containing donor-DNA template to the targeted region is present, the DSB can be repaired instead by HDR, thereby allowing for precise replacement, introduction of mutations or insertion of sequences in the targeted region. WT denotes wild type.
The CRISPR-Cas9 system primarily used for gene editing comes from Streptococcus pyogenes. However, Cas9 proteins from other organisms are being explored to increase the flexibility and expansion of application areas (as discussed subsequently). CRISPR-Cas9 has developed from in-house design to predesigned sgRNAs and premade CRISPR kits available from multiple vendors. In addition, software is available, e.g., for sgRNA design, analysis of off-target effects, etc. [e.g., http://crispr.mit.edu/, http://www.e-crisp.org/E-CRISP/, https://crispr.dbcls.jp/, http://www.rgenome.net/cas-offinder/, and http://www.biootools.com/ (accessed on May 23, 2018)]. The commercial availability of kits and software extends the possibilities of the CRISPR-Cas9 technology to researchers within many fields.
Improved CRISPR-Cas9 Technologies.
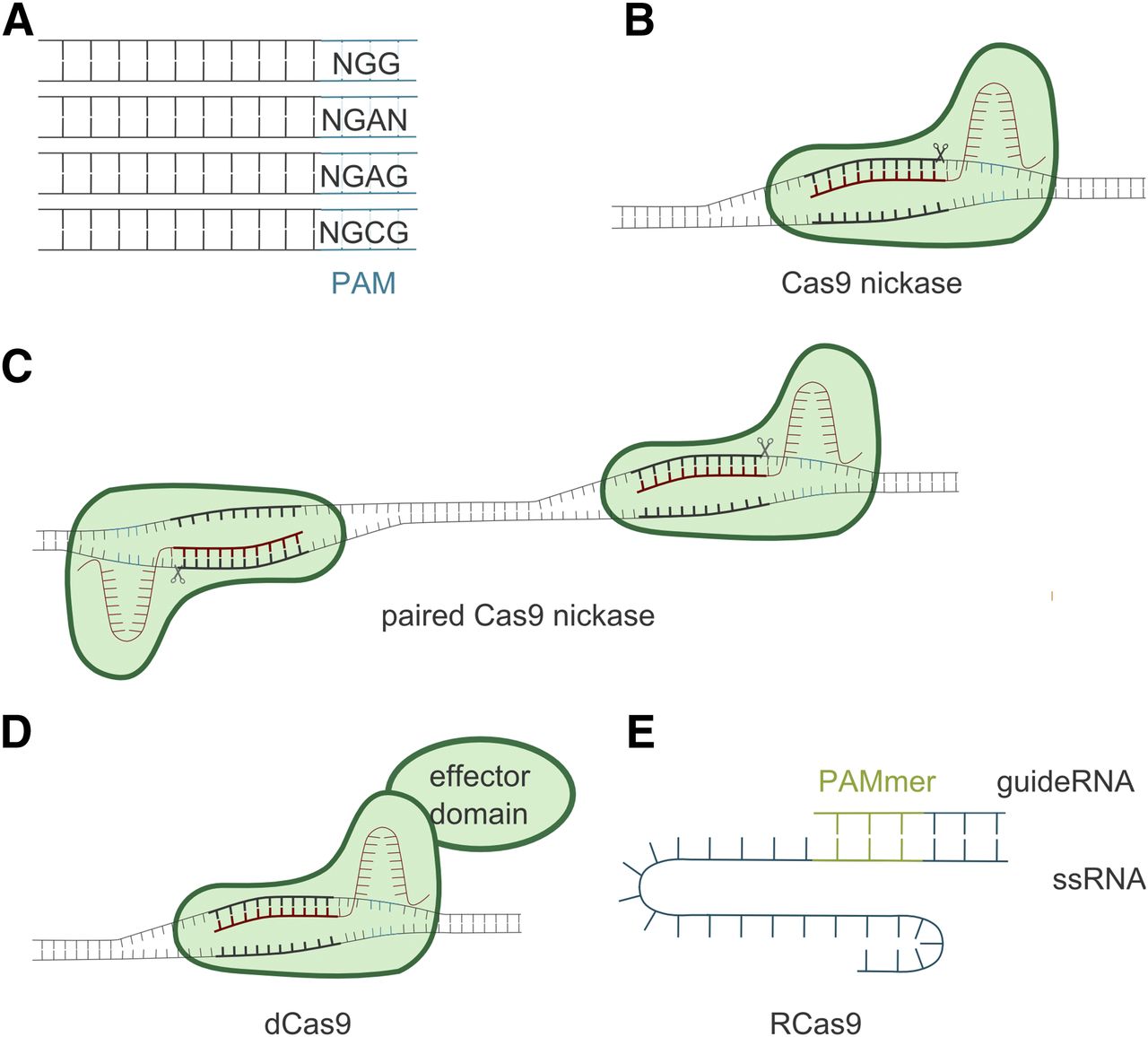
New or improved CRISPR technologies are constantly being developed, offering more opportunities and diverse applications (Sander and Joung, 2014). One limitation of CRISPR-Cas9 is set by the PAM motif—sites must contain the NGG PAM sequence for gene editing to occur (see Fig. 2C). To overcome this limitation, other species, endonucleases, and PAM recognition sequences are being explored, generating even more PAM motif variants (Fig. 3A) (see Nakade et al. (2017), and references therein). Together, these expand the precision and applicability of CRISPR as a gene-editing tool. Recently, a thermostable Cas9 was described with activity up to 70°C. This endonuclease exhibited significant stability in diluted human plasma, potentially providing a new opportunity for therapeutic applications via intravenous administration (Harrington et al., 2017).

The CRISPR-Cas9 technology is constantly being expanded to improve specificity and increase its versatility. Such improvements include more flexible PAM requirements by modification of the Cas9 endonuclease from Stretococcus pyogenes, or by use of endonucleases from other species (A). Another improvement is the engineering of Cas9 into a Cas9 nickase (Cas9D10A) that only cuts one of the DNA strands. This nickase can be used both in single (B) or paired (C) strands, thereby reducing the risk of indels at off-target sites. Another engineered Cas9 is dCas9, which does not cut the DNA, but can be coupled to an effector domain, like a transcriptional activator/repressor, epigenetic modifiers, or fluorescent proteins (D). The Cas9 endonuclease has also been modified to target single-stranded RNA (ssRNA) (RNA-targeting Cas9 or RCas9) instead of double-stranded DNA (E).
One approach to overcome off-target effects is to use mutated Cas9 (Cas9D10A) that introduce a single-strand break (a nick) in the target DNA instead of a DSB (Fig. 3B) (Cong et al., 2013). Hence, two sgRNAs (one on the sense and one on the antisense strand) are needed to produce a DSB and give rise to a 5′ overhang (Fig. 3C). This will minimize the off-target effects, whereas the on-target effects will occur at the same rates as with wild-type Cas9 (Ran et al., 2013).
A modified Cas9, called nuclease deficient Cas9 (dCas9), can be applied for other technical improvements (Gilbert et al., 2013). The dCas9 protein lacks endonuclease function, but it can be further modified by coupling it with a functional protein domain with different activity, e.g., a transcriptional repressor (CRISPR interference) or activator (CRISPR activation) (Lo and Qi, 2017). With the ability of sgRNA to bind to a specific target sequence, the coupled dCas9 can be targeted to the specific gene of interest (Fig. 3D).
Similarly, the dCas9 protein can be coupled to epigenetic modifiers for locus-specific alteration of acetylation or methylation patterns and subsequent suppression/induction of gene expression (cf. Lo and Qi, 2017; Murovec et al., 2017). Other applications of dCas9 include fusion to fluorescent proteins for visualization of specific DNA sequences in living cells (Chen et al., 2013), or fusion to the unspecific endonuclease domain of FokI. Since FokI is only active as a dimer, combining the fused dCas9-FokI with two specific and adjacent sgRNAs increases the specificity of cleavage and reduces the risk for off-target effects (Guilinger et al., 2014).
Cas9 protein has also been modified to target single-stranded RNA (O’Connell et al., 2014). This modified Cas9 is referred to as RNA-targeting Cas9. In this approach, the PAM sequence is mismatched compared with the DNA sequence that allows RNA-exclusive targeting. It is supplied as part of the oligonucleotide that will hybridize to the target RNA (Fig. 3E). Thus far, RNA-targeting Cas9 has been used, e.g., for RNA cleavage, isolation, and tracking in living cells (O’Connell et al., 2014; Nelles et al., 2016). Similarly, a new endonuclease, Cas13, is being used to detect single molecules of RNA or DNA (Shmakov et al., 2017). This endonuclease has been applied in diagnostics to detect single-stranded RNA of Zika and Dengue viruses, as well as mutations in patient liquid samples (Gootenberg et al., 2018).
CRISPR-Cas9 for Generating ADME Models
CRISPR-Cas9, like RNA interference, has been used as a tool for target identification in drug discovery (Taylor and Woodcock, 2015; Xue et al., 2016). Since CRISPR-Cas9 is a relatively new technology, the number of CRISPR-Cas9 studies targeting ADME genes is limited. Nevertheless, by 2013 the first reports of CRISPR-Cas9 gene editing of ATP-binding cassette transporters were presented. In these cases, the gene edited was the CFTR gene (ABCC7) (Schwank et al., 2013) and the white gene (belonging to the ABCG family) regulating eye pigmentation in fruit fly (Kondo and Ueda, 2013), i.e., transporters not considered to be ADME proteins. Two years later, in 2015, the first articles about gene editing of solute carrier transporters were published. These articles described the generation of SLC31A1 and SLC31A2 knockouts, two copper transporters that are also involved in cisplatin and carboplatin transport (Tsai et al., 2015). A year later, in 2016, the first mammalian in vivo knockout ADME models were developed in Sprague-Dawley or Wistar rats. Both models focused on drug-metabolizing cytochrome P450 (P450) enzymes and the genes knocked out were Cyp2e1 (Wang et al., 2016) or the Cyp2d gene cluster (Yoshimi et al., 2016). In the latter publication, a humanized rat model was also generated since the human CYP2D6 gene was knocked in instead of the rodent Cyp2d genes. Since then, CRISPR-Cas9 has been applied in a number of studies focusing on drug-metabolizing enzymes, drug transporters, nuclear receptors, or elements involved in the regulation of ADME genes (for an overview, see Table 1).
Drug-Metabolizing Enzymes
In Vitro.
To our knowledge, only one in vitro P450 model has been established using CRISPR-Cas9. It was based on the human hepatocyte cell line Huh-7 (Dorr et al., 2017). Here, CRISPR-Cas9 editing of the CYP3A5 locus established heterozygous and homozygous in vitro models for the CYP3A5*1 allele. The Huh-7 cell line is homozygous for the CYP3A5*3 allele, an allele with a splice defect resulting in an inactive enzyme, which is also the most prevalent allele in Caucasian populations (Zhou et al., 2017). However, the CYP3A5*1 allele has an allele frequency of up to 56% in non-Caucasian populations, necessitating in vitro models that reflect other genotypes. The gene editing of CYP3A5 was successful and resulted in elevated CYP3A5 mRNA levels and increased metabolism of the two CYP3A5 substrates midazolam and tacrolimus (Dorr et al., 2017).
In Vivo.
Prior to the CRISPR-Cas9 technology, in vivo knockout P450 models, humanized P450 models (where animals express the human P450 ortholog), or chimeric models (where human hepatocytes were transplanted and allowed to repopulate the host liver) were used to study ADME- and toxicity-related problems (Tateno et al., 2004; Cheung and Gonzalez, 2008; Hasegawa et al., 2011; Jiang and Yu, 2012; Kazuki et al., 2013; Scheer et al., 2013). However, the major applications of these models involve, e.g., studies of developmental or physiologic functions, disease mechanisms, validation of target genes, identification of off-targeted effects for drugs and drug candidates, etc.
A number of in vivo CRISPR-Cas9 knockout models have been established for P450 enzymes, including five articles on the generation of mammalian knockout and/or knockin models. These models that are based on Wistar or Sprague-Dawley rats and/or C57BL/6 mice targeted genes coding for enzymes in drug-metabolizing P450 families 2 and 3. In the first model the rat Cyp2d gene locus (containing Cyp2d1-5) was knocked out in Wistar rats and replaced with human CYP2D6 (Yoshimi et al., 2016). Thus, this generated both a Cyp2d knockout rat and a humanized CYP2D6 rat. Unfortunately, no functional characterization was done and to our knowledge no follow-up studies with these in vivo models have been published. In other models Cyp2e1 (Wang et al., 2016) or Cyp2c11 (Wei et al., 2018) were knocked out in Sprague-Dawley rats. The CRISPR-Cas9 technology has also been used to generate a double-knockout rat model in which Cyp3a1 and Cyp3a2 are knocked out (Lu et al., 2017). The Sprague-Dawley knockout models were generated to establish in vivo models for investigating physiologic functions and xenobiotic metabolism by the specific enzyme(s) of interest. In vitro (using liver microsomes from the knockout rats) and in vivo studies using prototypical P450 substrates showed that the metabolic function of the targeted enzyme(s) was indeed impaired in all knockout models. However, all models showed compensatory upregulation or downregulation of other P450s involved in drug metabolism. Moreover, these models also showed other differences, e.g., altered serum concentrations of testosterone (Lu et al., 2017) or alkaline phosphate (Wang et al., 2016) and reduced fertility (Wei et al., 2018). Some of these differences can be attributed to the function of the knocked out gene, e.g., CYP3A and testosterone metabolism, whereas others are unexpected. This suggests that such physiologic alterations need to be taken into consideration when comparing ADME data from the knockout models with data from the wild type.
Besides the rat models, a C57BL/6 mouse model was established, with three Cyp2b genes—Cyp2b9, Cyp2b10 and Cyp2b13—knocked out (Kumar et al., 2017). Interestingly, there were few compensatory changes in expression levels of other P450 enzymes in the triple knockout model. The significant changes that were observed were in female mice for which mRNA expression of Cyp2a4, Cyp2c40, and Cyp3a13 were downregulated.
Drug Transporters
In Vitro.
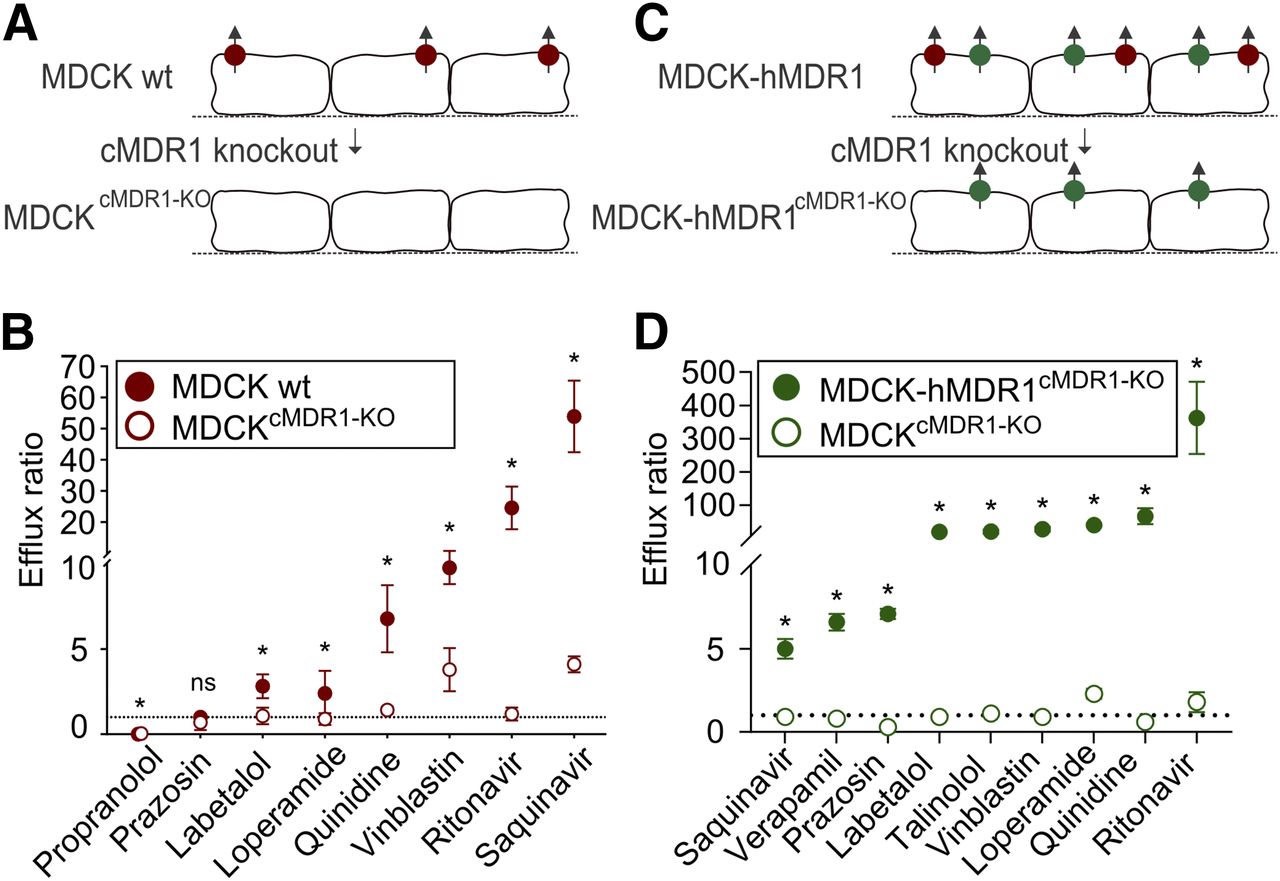
CRISPR-Cas9 gene editing in cell culture models for drug transport has mainly focused on ABCB1/MDR1 (Fig. 1B). To the best of our knowledge, the first paper regarding MDR1 and ADME was published by us in 2016 (Simoff et al., 2016) with a follow up 1 year later (Karlgren et al., 2017). The aim in both studies was to knock out the endogenous canine ABCB1/MDR1 expression in two MDCK cell lines—the wild-type cell line and the cell line already overexpressing the human MDR1 transporter. The monolayer-forming MDCK cell line has been used for decades for heterologous expression of single or multiple drug transporters and subsequent transport studies, and it is favored as a predictive drug efflux model. Unfortunately, due to a large substrate overlap the endogenous background of canine MDR1 interferes with the overexpressed human ABCB1/MDR1 (Kuteykin-Teplyakov et al., 2010; Wang et al., 2013; Karlgren et al., 2017). This makes it difficult to interpret and derive correct kinetic variables for the human homolog. We reasoned that selective knockout of canine ABCB1 would provide an MDCK transport model without interference from the endogenous background. After transfection of the wild-type and overexpressing MDCK cells with plasmids containing sgRNA-Cas9 and clonal selection, two isogenic cell lines were generated with identically disrupted canine ABCB1 genes. Sequencing analysis and liquid chromatography–tandem mass spectrometry–based targeted proteomics verified that the clones were indeed completely lacking canine MDR1 expression. The disruption was homozygous and included four nucleotide deletions that produced frame-shift and premature stop codons in both cell lines. Furthermore, functional analysis showed no efflux of prototypical MDR1 substrates in the knockout (KO) cell line based on wild-type MDCK (MDCKcMDR1-KO) (Fig. 4, A and B). In comparison, the efflux ratios in the original MDCK wild-type cells were significantly higher, indicating that MDCKcMDR1-KO indeed was completely lacking canine MDR1 (Fig. 4B).

Overview of MDCK cell models where the canine ABCB1/MDR1 has been knocked out using CRISPR-Cas9. (A) Canine MDR1 (cMDR1) was knocked out in an MDCK wild-type cell line; after clonal selection, a knockout clone was obtained that completely lacks endogenous cMDR1 expression (MDCKcMDR1-KO). (B) Functional validation using Transwell studies and prototypical MDR1 substrates showed significantly lower efflux ratios for MDCKcMDR1-KO compared with the parental cell line and with most efflux ratios being close to unity (dotted line). (C) cMDR1 was knocked out in an MDCK cell line already overexpressing the human MDR1 transporter (hMDR1). Here, a clone with identical gene editing as MDCKcMDR1-KO and completely lacking cMDR1 expression could be identified (MDCK-hMDR1cMDR1-KO). (D) Functional validation using Transwell studies and prototypical MDR1 substrates showed significantly higher efflux ratios compared with MDCKcMDR1-KO. Furthermore, the hMDR1 activity was retained, indicating no editing of the human ABCB1. Data are presented as mean ± standard deviation for one representative experiment performed in triplicate. ns, not significant, *p < 0.05; using student’s t-test. Graphs (B and D) have been previously published in Simoff et al. (2016) and Karlgren et al. (2017), respectively. These graphs were adapted and republished with kind permission from the publisher.
A knockout cell line, MDCK-hMDR1cMDR1-KO, was also developed for the corresponding MDCK cells overexpressing human MDR1. Significant differences in efflux ratios were seen between MDCKcMDR1-KO and MDCK-hMDR1cMDR1-KO, indicating that the homologous human ABCB1 was largely unaffected by the CRISPR-Cas9 knockout process (Fig. 4, C and D). Recently, we used the MDCKcMDR1-KO cell line, which lacks endogenous canine ABCG2/BCRP activity, for heterologous expression of the human drug transporter BCRP (M. Karlgren et al., manuscript in preparation). Since BCRP and MDR1 share many substrates, the absence of interfering canine MDR1 background is an advantage of this novel model.
Five additional articles published between 2016 and 2018 applied a similar approach to knockout ABCB1/MDR1in various cell lines. These investigated if the knockout of ABCB1 could reverse resistance in human cancer derived cell lines (Ha et al., 2016; Liu et al., 2016; Yang et al., 2016b; Takahashi et al., 2017; Norouzi-Barough et al., 2018). As expected, these studies showed decreased ABCB1/MDR1 mRNA or protein expression levels and increased intracellular accumulation, or increased sensitivity in the MDR1 knockout cells using MDR1 substrates such as rhodamine 123, doxorubicin, and/or carfilzomib. Besides this clinically relevant drug transporter (Giacomini et al., 2010), CRISPR-Cas9 gene editing has also been used in editing of a number of other transporters (see Table 1).
In Vivo.
A number of in vivo transporter models for ADME studies were developed before CRISPR-Cas9. Most of these are in vivo transporter knockout models for studies of drug transport. These models allowed studies of the role of a specific transporter in the presence of all other mechanisms influencing drug disposition in vivo, such as drug uptake, distribution, metabolism, and efflux. For overviews of in vivo knockout transporter models, see Jiang and Yu (2012) and Tang et al. (2013). Prominent examples of the applicability of single and multiple in vivo knockout models for studies of transport mechanisms include ABCB1/MDR1 in the blood-brain barrier (Schinkel et al., 1995) and SLCOs/OATPs in Rotor-type hyperbilirubinemia (van de Steeg et al., 2012), respectively. However, a complicating factor is that knockout of a single transporter protein or several transporter proteins can lead to increased expression of other transporter proteins (e.g., Kuroda et al., 2004; Vlaming et al., 2009). Thus far, in vivo CRISPR-Cas9 gene editing of drug transporters has focused on the ATP-binding cassette transporter family (see Table 1), where it has been used to knock out ABCB1/MDR1 homologs in insects (Denecke et al., 2017; Zuo et al., 2018). In both studies the knockout of ABCB1/MDR1 homologs increased the susceptibility to various insecticides. The ABCB1 homolog was also knocked out in zebrafish. The mutated zebrafish embryos showed a reduced efflux of model substrates and also increased accumulation of cadmium chloride and benzo[a]pyrene (Tian et al., 2017). In another study, abcb11b, the ortholog of human ABCB11/BSEP, was knocked out in zebrafish (Ellis et al., 2018). Like humans with impaired BSEP function, the knockout of abcb11b in zebrafish was associated with, e.g., activation of impaired bile salt excretion and autophagy. See Table 1 for a summary of CRISPR-Cas9 in vivo editing of additional transporters.
Regulation of ADME Genes
The CRISPR-Cas9 technology has been applied to study the regulation of ADME genes. An example is the regulation of the drug transporter ABCC3/MRP3 in the human keratinocyte cell line HaCaT (Takechi et al., 2018). In this study, the authors identified a CpG island, i.e., an area rich in CpG dinucleotides often found near or within the promoter or first exon of a gene, and which may be involved in the epigenetic regulation of genes. This island is located approximately 10 kb upstream of the ABCC3 gene and its methylation status correlates negatively with ABCC3/MRP3 mRNA expression levels in human skin samples. The CRISPR-Cas9-induced deletions in this CpG area reduced ABCC3/MRP3 mRNA expression levels in the HaCaT cell line. This study highlights the possibility of using CRISPR-Cas9 for editing/deletion of nontranscribed DNA targets.
CRISPR-Cas9 has been used for studying expression and function of nuclear receptors involved in P450 regulation (see Table 1). The aryl hydrocarbon receptor is involved in induction of, e.g., CYP1A1. To generate model systems for the study of aryl hydrocarbon receptor signaling and its modulation by xenobiotics, a Luciferase reporter cassette was knocked in at the CYP1A1 transcription start site in induced pluripotent stem cells (iPSCs) using CRISPR-Cas9 (Smith et al., 2016). The authors could monitor aryl hydrocarbon receptor activity by following CYP1A1 expression in these cells. In another study, in human iPSC, CRISPR-Cas9 has been used to tag PXR with the fluorescent protein mCherry (Kim et al., 2018). PXR is involved in the induction of several drug-metabolizing enzymes and transporters (Kandel et al., 2016), but its expression is low in iPSC-derived hepatocytes (Kim et al., 2018). Consequently, this results in low enzyme/transporter levels. The authors reason that the established PXR-mCherry iPSC cell line forms a platform for discovering factors that can increase PXR expression levels during differentiation; however, to date, no such studies have been published. These two articles point to the possibility of following endogenous gene expression with the aid of CRISPR-Cas9. It can be speculated that once established this approach will offer an alternative to other mRNA or protein quantification technologies.
Limitations of CRISPR-Cas9
The CRISPR-Cas9 technology also has its limitations. As mentioned previously, CRISPR-Cas9 gene editing shows apparent off-target effects as well as low efficiency (Liang et al., 2015). DSBs can occur even if a few mismatches exist between the sgRNA and the DNA target. This can result in alterations in genes unrelated to the target gene (Fu et al., 2013). Gene editing at sites with as many as five mismatches has been reported; the rate of such off-target effects can be similar to that of the gene editing at the target site (Fu et al., 2013; Lin et al., 2014).
Potential off-target effects are today often investigated using in silico analysis to avoid target sites with closely resembling sequences. If this is not possible, such off-target sites are investigated for unwanted mutations using polymerase chain reaction and/or DNA sequencing. Nevertheless, it should be noted that a recent report showed that CRISPR-Cas9 editing could also cause single-nucleotide variants in unpredictable off-target sites in vivo in mice (Schaefer et al., 2017). However, that report was retracted during the compilation of this paper since proper genetic background analysis had not been performed (Schaefer et al., 2018). Even so, this shows that—for comprehensive off-target analysis—whole genome sequencing might be the method of choice.
The low efficiency in CRISPR-Cas9 editing is a limitation, especially for in vivo applications. However, even in in vitro studies, e.g., the development of CRISPR-Cas9 knockout models by the NHEJ mechanism, the low efficiency results in the need for extensive screening to find homozygous knockout clones. In our experience, screening of clones enriched using, e.g., green fluorescent protein or antibiotic selection, can result in up to 28% of the screened clones having homozygous deletions (Simoff et al., 2016; Karlgren et al., 2017). This is in line also with other publications reporting similar efficiencies for CRISPR-Cas9 and NHEJ (Moore, 2015; Kosicki et al., 2017), but lower efficiencies for HDR (Mali et al., 2013; Shao et al., 2014). It should be noted that since the gene editing is sequence specific large variation in efficiency is seen for different targets and delivery methods (Kosicki et al., 2017).
The CRISPR-Cas9 technology is also limited by the requirement of a PAM motif in close proximity to the target site. Although endonucleases recognizing alternative PAM sequences are now available (as previously mentioned), this might still constitute a problem when targeting ADME genes that have high sequence identity with other gene family members. In such cases, genomic screening and validation (mRNA/protein expression or activity) might also be more difficult.
After submission of this article, results were published indicating additional problems with CRISPR-Cas9, in particular for its clinical applicability. Haapaniemi et al. (2018) showed that DSBs introduced by CRISPR-Cas9 triggered p53-dependent cell cycle arrest and that this response was independent of the targeted locus. Similarly, Ihry et al. (2018) showed that CRISPR-Cas9-induced DSBs generated a p53-dependent toxic response in human pluripotent stem cells. The targeting of a single locus was sufficient to kill most of the cells (Ihry et al., 2018). This suggests that p53-deficient cells have a growth advantage after CRISPR-Cas9 gene editing. Therefore, if CRISPR-Cas9 is to be used for therapeutic purposes, p53 function needs to be assured to avoid the risk of cancer. Another serious problem, raised by Kosicki et al. (2018), is that CRISPR-Cas9 gene editing may result in large deletions and complex gene rearrangement at the target site. Such alterations are undetectable by the short-range polymerase chain reaction assays commonly used to verify gene modifications. Lesions and crossover events were also detected outside of the targeted locus (Kosicki et al., 2018). Together, these findings highlight the importance of comprehensive genetic analysis to reveal and control unexpected gene modifications prior to a more general clinical use of CRISPR-Cas9.
Future Applications for CRISPR-Cas9 within the ADME Field
Despite the restrictions mentioned, the simplicity, programmability, versatility, and comparably low costs of CRISP-Cas9, together with the rapid methodological development and improvement, indicate a huge potential for a variety of applications in the ADME field. At this point in time, CRISPR-Cas9 has mainly been used to generate in vitro or in vivo knockout models of ADME genes. One major advantage with CRISPR-Cas9 knockout models is the complete elimination of background expression, which will facilitate investigations of clinically relevant drug transporters and drug-metabolizing enzymes. CRISPR-Cas9 knockout models will also be useful in studies of putative ADME genes, such as orphan solute carrier family members (César-Razquin et al., 2015; Lin et al., 2015) and other ADME genes of relevance in drug disposition and drug interactions, as drug targets, and that are associated with human disease. With CRISPR-Cas9, one has the opportunity to not just knock out the gene of interest, but also to study the role of individual nucleotides or amino acids as structural/functional constituents, or to humanize animal ADME genes under a native promoter.
An intriguing opportunity with CRISPR-Cas9 is that noncoding DNA can be targeted. Interindividual variation (expression levels, drug-drug interactions, and pharmacokinetics) of ADME proteins can be due to genetic variation in coding regions or promoter regions, but much of the variation is still unexplained. We know that the DNA encoding proteins, which are fairly well studied, represent only a minor part of the human genome. The regulatory role of noncoding DNA is mainly unexplored. Furthermore, genome-wide association studies have shown that most of the associated sites in human diseases are located in these noncoding DNA regions (Wright and Sanjana, 2016). Similar observations have been reported also for ADME proteins, e.g., regarding variation in ATP-binding cassette transporter expression levels (Matsson et al., 2012). Therefore, Wright and Sanjana (2016) proposed that a future application of CRISPR-Cas9 could be to shed light on this intriguing issue.
While many drugs and drug metabolites interact with both drug transporters and drug-metabolizing enzymes in vivo, transport and metabolism are often studied separately in vitro using recombinant proteins, cell lines overexpressing transporters, or drug-metabolizing enzymes. This is because the contribution of an individual transporter/enzyme can be difficult to determine in more complex in vitro models, such as human hepatocytes. As a consequence, we and others have previously developed in vitro models overexpressing different combinations of transporters and enzymes, in which the contribution of an individual transporter/enzyme can more easily be determined (Crespi et al., 2000; Fahrmayr et al., 2012, 2013; Kwatra et al., 2012; Neve et al., 2013). These models are useful but have drawbacks, including endogenous background expression and time-consuming experimental procedures. With CRISPR-Cas9 it is possible to both completely knock out and knock in genes; hence, with this technology we will be able to develop better and easier-to-use models than the traditional ones. For example, transport-metabolism interplay or drug metabolite interactions could be studied under more controlled conditions using such models.
The use of dCas9 for genome docking and the coupling of dCas9 to an effector domain could provide additional opportunities to generate in vitro models that are more predictive of the in vivo situation. Through dCas9 docking/coupling it could become possible (instead of knocking in or knocking out genes) to modulate expression levels of one or several endogenous genes simultaneously, thereby providing models with more in vivo–like expression profiles.
In summary, we have demonstrated the versatility and applicability of the CRISPR-Cas9 technology. By using examples from more basic research areas, our goal was to stimulate increased use of CRISPR-Cas9 in the ADME field. We predict that the advantages of CRISPR-Cas9 will result in a variety of more advanced and better interpretable in vitro and in vivo models for drug metabolism and disposition.
Acknowledgments
We are grateful to Dr. Janneke Keemink for producing the figures included in this paper.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Karlgren, Simoff, Keiser, Oswald, Artursson.
Footnotes
- Received June 1, 2018.
- Accepted August 3, 2018.
This work is supported by The Swedish Fund for Research without Animal Experiment (Approval Number 0004) to M.Ka.; the Magnus Bergvall Foundation (Approval No. 2305) to M.Ka.; and the Swedish Research Council (Approval No. 1951) to P.A.
Abbreviations
- ADME
- absorption, distribution, metabolism, and excretion
- Cas9
- clustered regularly interspaced short palindromic repeats associated protein 9
- CRISPR
- clustered regularly interspaced short palindromic repeats
- dCas9
- nuclease deficient clustered regularly interspaced short palindromic repeats associated protein 9
- DSB
- double strand break
- HDR
- homology directed repair
- iPSC
- induced pluripotent stem cell
- KO
- knockout
- MDCK
- Madin-Darby canine kidney
- NHEJ
- nonhomologous end joining
- P450
- cytochrome P450
- PAM
- protospacer adjacent motif
- PXR
- pregnane X receptor
- sgRNA
- single guide RNA
- TALEN
- transcription activator-like effector nuclease
- ZFN
- zinc finger nuclease
- Copyright © 2018 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.





{kind=link}
{kind=link}
{kind=link}
{kind=link}