


Abstract
The widespread expression and polyspecificity of the multidrug ABCG2 efflux transporter make it an important determinant of the pharmacokinetics of a variety of substrate drugs. Null ABCG2 expression has been linked to the Junior blood group. Polymorphisms affecting the expression or function of ABCG2 may have clinically important roles in drug disposition and efficacy. The most well-studied single nucleotide polymorphism (SNP), Q141K (421C>A), is shown to decrease ABCG2 expression and activity, resulting in increased total drug exposure and decreased resistance to various substrates. The effect of Q141K can be rationalized by inspection of the ABCG2 structure, and the effects of this SNP on protein processing may make it a target for pharmacological intervention. The V12M SNP (34G>A) appears to improve outcomes in cancer patients treated with tyrosine kinase inhibitors, but the reasons for this are yet to be established, and this residue’s role in the mechanism of the protein is unexplored by current biochemical and structural approaches. Research into the less-common polymorphisms is confined to in vitro studies, with several polymorphisms shown to decrease resistance to anticancer agents such as SN-38 and mitoxantrone. In this review, we present a systematic analysis of the effects of ABCG2 polymorphisms on ABCG2 function and drug pharmacokinetics. Where possible, we use recent structural advances to present a molecular interpretation of the effects of SNPs and indicate where we need further in vitro experiments to fully resolve how SNPs impact ABCG2 function.
Introduction
The ATP-binding cassette (ABC) transporters are a family of membrane-bound protein pumps, which use the hydrolysis of ATP to transport compounds across membranes. They function in a variety of cellular processes such as nutrient uptake, osmotic homeostasis, and protection from xenotoxins. Eukaryotic ABC transporters are possibly exclusively involved in the efflux of molecules from the cytosol; either out of the cell or into organelles within the cell. These molecules include endogenous compounds such as metabolic products and lipids, as well as xenobiotics and xenotoxins, which are of great clinical interest (Mo and Zhang, 2012). Humans possess 48 different ABC transporters, which are categorized into groups ABCA to ABCG (Dean et al., 2001). The three main ABC transporters under study in relation to drug uptake and elimination are ABCB1 (P-glycoprotein), ABCC1 (MRP1), and ABCG2 (breast cancer resistance protein). The ability of these three transporters to efflux multiple, structurally different drugs implicates them in multidrug resistance (MDR) and the efflux of compounds from cells, lowering intracellular concentration and producing relative resistance to a broad variety of drugs (Wong et al., 2014).
The ABC subfamily G isoform 2 (ABCG2) protein is a MDR pump with a wide distribution in the human body, which is found in the small intestine, blood-brain barrier, blood-placenta barrier, liver canalicular membranes, proximal tubule cells of the kidney, and the mammary gland (Horsey et al., 2016). The functions of ABCG2 at its widespread locations are summarized in Table 1, and to illustrate the diversity of its substrate repertoire endogenous and xenobiotic substrates are shown in Fig. 1. The wide distribution and polyspecificity of ABCG2 make it an important determinant of the pharmacokinetics of various substrate drugs (Lee et al., 2015).
An overview of the expression and functions of ABCG2
In addition to these locations, ABCG2 has been found in the adrenal gland, cervix and uterus, bladder, and lungs.
Chemical structures of ABCG2 physiological substrates and pharmacological drug substrates. Structures obtained from PubChem database.
ABCG2 was originally identified in 1998 in placenta and multidrug resistant breast cancer cell lines; hence, it was given the names breast cancer resistance protein and placenta-specific ABC transporter (Allikmets et al., 1998; Doyle et al., 1998; Miyake et al., 1999). Both titles are misnomers: ABCG2 is certainly not restricted to the placenta (Table 1) and clinical correlations between ABCG2 and breast cancer staging, metastasis, and outcome are equivocal (Faneyte et al., 2002; Xiang et al., 2011). On the other hand, the relationship between ABCG2 and the prognosis of hematologic cancers is better developed. Poorer outcomes have been shown in large B-cell lymphoma (Kim et al., 2009b) and acute myeloid leukemia in patients with higher ABCG2 levels (Van den Heuvel-Eibrink et al., 2002; Benderra et al., 2004). ABCG2 also has an established link to survival rates and therapy response in small-cell and non-small-cell lung cancer (see Mo and Zhang, 2012; Horsey et al., 2016).
Along with cancer outcomes, there is a strong and well-researched link between ABCG2 and the development of gout (Woodward et al., 2013). Polymorphisms of ABCG2 (throughout this paper we refer to the gene as ABCG2 and the protein as ABCG2) can result in increased serum urate levels (hyperuricemia); recent analysis of factors correlated with renal urate clearance supports the hypothesis that ABCG2’s role in gout is through decreased gut export of urea (Ichida et al., 2012; Kannangara et al., 2016). Interestingly, recent studies have also hypothesized a link between ABCG2 and the development of Parkinson’s and Alzheimer’s disease. The common polymorphism 421 C>A (Q141K at the amino acid level, described in more detail subsequently) was correlated to a delay in the onset of Parkinson’s (Matsuo et al., 2015) due to an increased concentration of urate in the central nervous system, hypothesized to have a protective effect in the brain (Ascherio et al., 2009). ABCG2 has been found to be upregulated in the brains of Alzheimer’s patients, and 421CC genotypes showed significantly increased susceptibility to Alzheimer’s compared with CA and AA (Xiong et al., 2009; Fehér et al., 2013). At this time, the mechanism of the effects of ABCG2 on Alzheimer’s development appears unclear, although amyloid beta 1–40 peptide transport has been documented for ABCG2 (Do et al., 2012). Additional functions for ABCG2 in stem cell protection (both with respect to cancer stem cells and hematopoietic stem cells) and as a marker of autophagy have been described in the literature, although full appreciation of the importance of ABCG2 in these scenarios remains to be revealed (Zhou et al., 2001; Hirschmann-Jax et al., 2004; Krishnamurthy et al., 2004; Ding et al., 2016).
Many anticancer drugs have been discovered as ABCG2 substrates, such as camptothecin analogs (diflomotecan, irinotecan, and topotecan), tyrosine kinase inhibitors (gefitinib, imatinib, and erlotinib), and other agents (methotrexate and mitoxantrone). Cisplatin, paclitaxel, and vinblastine are not substrates (Stacy et al., 2013) and the anthracyclines doxorubicin and daunorubicin are substrates only of drug-selected ABCG2 variants (Robey et al., 2003). Other xenobiotics that have been discovered as substrates include statins, direct oral anticoagulants, anti-human immunodeficiency virus drugs, antirheumatics, immunosuppressants, and antibiotics (see Fig. 1 for some chemical structures of transported drugs and natural ABCG2 substrates that clearly demonstrates the chemical diversity of its substrates). Alongside traditional anticancer drugs, photodynamic therapy agents such as pheophorbide A and protoporphyrin A have been confirmed as substrates, possibly influencing the effects of photodynamic therapy in some individuals (Robey et al., 2004, 2005; Westover and Li, 2015). Therefore, polymorphisms reducing ABCG2 efflux activity may be of potential importance in contributing to diet-induced phototoxicity, protoporphyria, and other porphyrin-related disorders (Jonker et al., 2002). This exemplifies the importance of understanding how polymorphisms affect ABCG2 function, and for this we will need a detailed description of the structure of the resultant protein.
ABCG2 is coded for by the ABCG2 gene at chromosome 4q22 (Bailey-Dell et al., 2001), and transcription of the gene is regulated by a TATA-less promoter region, containing several specificity protein-1 transcription factor binding sites (Yang et al., 2013). Translation of the mRNA results in the production of a 655 amino acid protein (∼65 kDa). Structurally, ABCG2 is known as a “half transporter” since it contains only one membrane-spanning domain (MSD) and one nucleotide binding domain (NBD) in the polypeptide. The MSD of ABCG2 is found toward the C-terminus and is composed of six membrane-spanning α-helices connected by alternating extracellular and intracellular loops (Fig. 2). The half-transporter structure of ABCG2 means that it must either dimerize or oligomerize before it becomes functional (Mo and Zhang, 2012; Horsey et al., 2016), and although the basis of dimer formation is outside the scope of this review there has been much research in this field (Wong et al., 2016). Recent publications of structural models for ABCG2 (László et al., 2016; Khunweeraphong et al., 2017), and the emergence of cryo-electron microscopy data for the human protein (Taylor et al., 2017; Jackson et al., 2018), mean that we are now close to being able to determine not only how a single nucleotide polymorphism (SNP) in ABCG2 might manifest at the level of protein function but also to relate this to its position in the three-dimensional structure.

Topology map of ABCG2. The membrane topology of ABCG2 is shown with TM helices represented as cylinders and the NBD as an oval. The location of the SNPs is indicated using the single letter amino acid code, as is the location of the N-glycosylation site (N596) and intra- and intermolecular disulphide bonds (C592–C608 and C603–C603).
An important early finding in this regard was the original cloning of two ABCG2 variants from drug resistant cell lines, with a substitution at amino acid 482 from arginine (R) to threonine (T) or glycine (G) (Doyle et al., 1998; Miyake et al., 1999). These R482G/T variants of ABCG2 were shown by the use of fluorescent probes and radioligand binding to have a broadened substrate specificity including anthacyclines (Honjo et al., 2001; Clark et al., 2006). Although it later transpired that the R482 variant sequences were drug-induced mutations (Honjo et al., 2001), it opened the following question. Could natural polymorphisms of ABCG2 also affect its function?
Many single nucleotide polymorphisms have subsequently been identified in ABCG2, including promoter, intronic, and protein coding changes (the latter being displayed in Fig. 2). This review aims to provide a summary of the researched polymorphisms of the ABCG2 gene, the impact of these polymorphisms on ABCG2 expression/function, and the resulting effects on pharmacokinetics.
The Polymorphisms of ABCG2
Thirty-eight ABCG2 polymorphisms were identified in the literature, 21 of which were not included due to lack of significant information. The excluded polymorphisms are (single amino acid notation) S13L, G51C, A149P, R160Q, R163K, Q166E, G268R, G354R, K360X, E366E, C369T, F373C, G406R, T434M, C474T, S476P, P480L, M515R, T542A, F571I, and R575X. The majority of these have minor allele frequencies that are <0.0001 and some even represent somatic mutations; it is likely that most are of questionable clinically significance. Nevertheless, establishing the effects of ABCG2 polymorphisms at as many sites as possible would be beneficial in improving knowledge of the protein’s mechanism. The remaining 24 polymorphisms (see Table 2 for frequency information and Figs. 2 and 3 for their location in the topology and three-dimensional structure of ABCG2) will be described numerically from the N-terminus for convenience.
Allelic variants of ABCG2
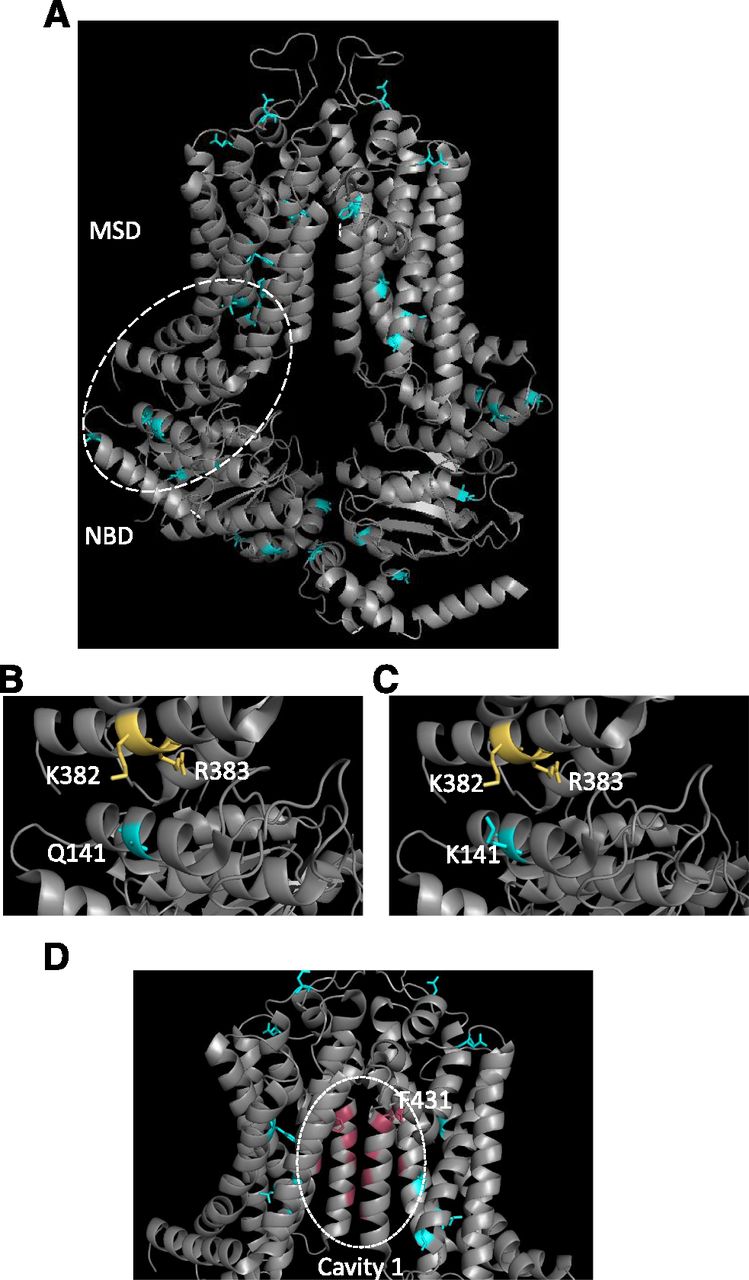
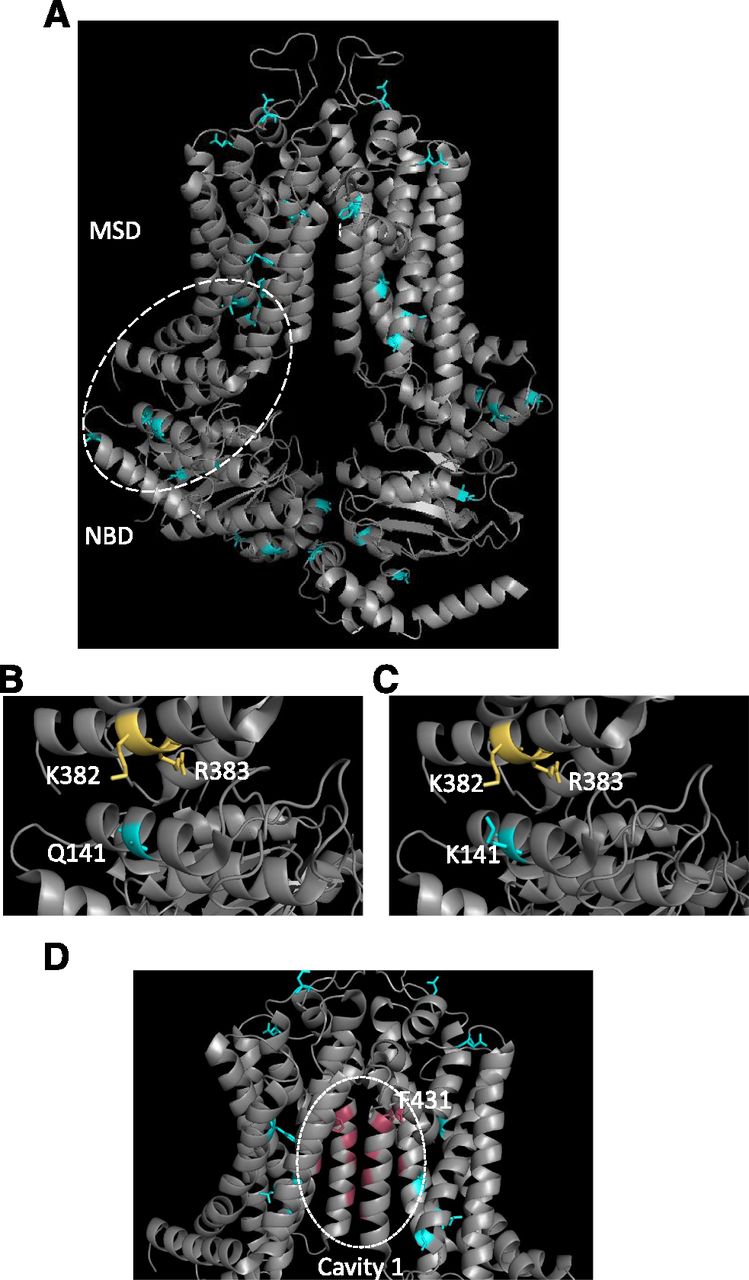
Structural localization of SNPs within ABCG2. (A) The structure of ABCG2 (Taylor et al., 2017) is shown in ribbon format with the location of SNPs indicated by the blue coloring of residues. (B and C) An expanded view of the NBD:MSD interface is shown to highlight the interaction between Q141 and K382/R383 in wild-type ABCG2 (B) that is destabilized by the introduction of the Q141K SNP (C). (D) The ABCG2 drug binding cavity [“cavity 1” in the terminology of Taylor et al. (2017)] is shown with residues in pink proposed to interact with substrates. Of the known SNPs (cyan) only F431 shown in stick format localizes to this cavity.
Polymorphisms in the NBD
The pivotal catalytic role played by the ABC transporter NBDs and our greater understanding of NBD structure and function should enhance our ability to rationalize the effects of NBD polymorphisms on ABCG2 function.
V12M (rs2231137)
V12M (34 G>A) (Table 2) is one of the most common polymorphisms of ABCG2, with a highly variable frequency depending on ethnicity. The polymorphism was found at highest frequencies in Mexican Indians (90%), Pacific Islanders (64%), and South-Eastern Asians (45%), but more rarely in Caucasian (2%–10.3%), African American (8.3%), and Middle Eastern populations (5%) (Zamber et al., 2003; Kobayashi et al., 2005; Poonkuzhali et al., 2008). Residue V12 is located intracellularly, close to the N-terminus of the polypeptide. This region has a substantial influence on the localization of the pump into the plasma membrane, meaning polymorphisms may disrupt the insertion of ABCG2 into the membrane. Structural data currently do not include the N-terminal 30 amino acids; therefore, the exact contribution this region makes to the fold of ABCG2 remains unclear (Taylor et al., 2017). In vitro examination of V12M shows expression system–dependent effects on ABCG2 localization and expression level, which makes it important to verify the effects of polymorphisms in cell lines as close as possible to the in vivo setting (Imai et al., 2002; Kondo et al., 2004; Mizuarai et al., 2004; Morisaki et al., 2005). For V12M, the closest to this comes in a study using Flip-In 293 cell line transfectants, where reduced transport of SN-38 was observed (Tamura et al., 2007a). An in vivo study on liver tissues in a Hispanic population did find that ABCG2 mRNA expression was lower in V12M, possibly as a result of alternative splicing (Poonkuzhali et al., 2008). However, lower ABCG2-V12M protein expression in tissues is yet to be found (Zamber et al., 2003).
If V12M does lower protein expression, this would impact urate transport or cancer chemotherapy. There are conflicting reports regarding the impact on V12M in the development of gout. Several studies have found no significant effect of V12M on urate transport and gout development (Matsuo et al., 2009; Higashino et al., 2017). Despite this, a recent meta-analysis of data concluded that V12M did exert a protective effect against gout. The study found an odds ratio of 0.76 (24% reduction in gout risk) when comparing Czech populations with New Zealand Polynesian and UK Biobank data (Stiburkova et al., 2017). In terms of cancer chemotherapy, there are again conflicting reports. For tyrosine kinase inhibitor (TKI) therapy, overall survival in non-small-cell lung cancer was found to be improved in AA/AG genotypes (Tamura et al., 2012; Chen et al., 2015). Other studies further supported the influence of the V12M SNP on TKI therapy in chronic myeloid leukemia and renal cell metastatic cancer (Kim et al., 2009a; van der Veldt et al., 2011; Tandia et al., 2017); in the latter, the presence of the 34A allele was linked to prolonged progression-free and overall survival in sunitinib therapy using Kaplan-Meier univariate analysis. Furthermore, 34AA/AG genotypes have been linked to improved outcomes in chronic myeloid leukemia mediated by increased toxicity of ctyarabine/anthracycline therapy, with an odds ratio of 8.41 (Hampras et al., 2010). In slight contrast, V12M has been found to show no effect on rituximab plus cyclophosphamide-doxorubicin-vincristine-prednisone therapy response or the side effects of rituximab plus cyclophosphamide-doxorubicin-vincristine-prednisone treatment (Kim et al., 2008); however, doxorubicin is not a good substrate of ABCG2, which at least partially explains this (Mao and Unadkat, 2015). Finally, in terms of cancer, the 34 G>A allele was associated with lower survival rates in pediatric acute lymphoblastic leukemia patients and diffuse large B-cell lymphoma (Hu et al., 2007; Zhai et al., 2012). At this time, the reason for poorer prognosis in V12M for certain cancers remains unclear. Research into other ABCG2 substrates for the V12M SNP is limited, with single reports indicating no effect on disposition for the antiretroviral drugs lamivudine in healthy subjects (Kim et al., 2007) or nelfinavir-receiving patients (Colombo et al., 2005).
Q126X (rs72552713)
The Q126X polymorphism (376 C>T) is a rare polymorphism found most commonly in Japanese populations, with a frequency between 0.9% and 2.4% (Honjo et al., 2002; Imai et al., 2002; Itoda et al., 2003; Kobayashi et al., 2005). Q126X has been investigated in other ethnicities such as Caucasians and African Americans, but in both of these populations the polymorphism was absent (Mizuarai et al., 2004; Kobayashi et al., 2005). The polymorphism results in a premature stop codon, and likely in nonsense-mediated RNA decay and no protein expression in the membrane (Kobayashi et al., 2005). It is, therefore, expected that the influence of this polymorphism on drug pharmacokinetics would be considerable, especially in homozygous carriers (Imai et al., 2002).
Many studies have found a strong connection between the Q126X polymorphism and increased risk of developing gout (Matsuo et al., 2009; Zhou et al., 2014; Li et al., 2015), with Matsuo et al. (2009) showing a significant odds ratio of 5.97. This study also found the Q126X haplotype was present in up to 13.5% of gout patients in a Japanese population (Matsuo et al., 2009). Studies focused on the effects of Q126X on drug pharmacokinetics are rare, most likely due to the low frequency and assumed outcome (i.e., absence of protein in homozygous individuals would clearly have a significant impact on intestinal drug uptake and biliary excretion). A small-scale study has linked the Q126X polymorphism with sulfasalazine (SASP) pharmacokinetics (Gotanda et al., 2015), and effects of Q126X on porphryin transport have also been shown (Tamura et al., 2012). Earlier studies had reported no interaction between Q126X and lamivudine/gefitinib therapy (Kim et al., 2007; Akasaka et al., 2010), suggesting that more studies are needed to fully understand the impact of this polymorphism on drug pharmacokinetics.
Q141K (rs2231142)
Q141K (421 C>A) is probably the most widely studied, common variant of ABCG2, which is found between the Walker A motif and the signature region of the NBD (Woodward et al., 2009). The polymorphism has a highly variable frequency depending on ethnicity. It is found commonly in Japanese (26.6%–35%) and Chinese (34.2%–35%) populations, but more rarely in Caucasian (8.7%–14%), Sub-Saharan (0.9%), and African American (0%–5.3%) populations (Imai et al., 2002; de Jong et al., 2004). Multiple studies have looked at the influence of Q141K on the function of ABCG2, with differing results as to the impacts of the polymorphism. A reduction in overall ABCG2 protein expression has been identified in many studies, both in vitro and in vivo (Imai et al., 2002; Kondo et al., 2004; Kobayashi et al., 2005; Tamura et al., 2007b; Furukawa et al., 2009; Sarankó et al., 2013; Woodward et al., 2013). However, there are studies showing that the mutation also causes reduced ATPase activity, resulting in decreased transport activity (Mizuarai et al., 2004; Morisaki et al., 2005). A plausible structural explanation for this is that when the neutral Q141 is replaced by a positively charged lysine electrostatic repulsion occurs with transmembrane (TM) helix 1a (see Fig. 3, B and C), potentially via an arginine residue in this helix (R383) (Polgar et al., 2009) or the adjacent lysine (K382) (László et al., 2016), leading to an ABCG2 folding defect (Taylor et al., 2017) and degradation by the endoplasmic-reticulum-associated protein degradation system (Furukawa et al., 2009).
Reduced ABCG2 expression in the Q141K variant should result in increased serum-drug concentrations through decreases in gut and urate clearance. The influence of Q141K on ABCG2 has been widely studied due to its connection with gout development, with a number of studies linking Q141K to reduced ATP-dependent urate transport (Woodward et al., 2009, 2013) and increased risk of gout (Cleophas et al., 2017; Higashino et al., 2017). In fact, Q141K is one of the most strongly predictive alleles for high serum urate levels and gout incidence. In addition to increased gout incidence, Q141K was found to be associated with reduced response to the gout treatment allopurinol in a genome-wide association study (Wen et al., 2015). Moreover, Q141K has been linked to poorer outcomes in prostate cancer, possibly due to reduced efflux of folate from the tumor (Sobek et al., 2017), resulting in high folate levels within the tumor that allow cancer cells to proliferate more rapidly. However, the effects of Q141K are mixed since it also reduces efflux of docetaxel in prostate tumors, resulting in improved drug response (Sobek et al., 2017). A wide range of drugs has been investigated with respect to Q141K, as detailed in the following sections.
Camptothecin Analogs.
Camptothecin analogs, i.e., anticancer agents that inhibit topoisomerase I, have shown mixed results in Q141K studies. Diflomotecan showed a 299% plasma level increase in heterozygous Q141K patients compared with wild type (WT) (Sparreboom et al., 2004). Likewise, an in vitro study showed Q141K cells were more susceptible to diflomotecan, with IC50 values 1.2- to 2.3-fold lower compared with wild type (Morisaki et al., 2005). Another camptothecin analog, 9-aminocamptothecin, showed significantly increased area under the curve (AUC) values (i.e., total drug exposure over time) in 421CA genotypes (Zamboni et al., 2006). This means Q141K most likely reduces transport of camptothecins compared with wild type. For other topoisomerase inhibitors, where one might also expect to see increased oral bioavailability in Q141K individuals, the results are less consistent; increased topotecan oral bioavailability was shown by Sparreboom et al. (2005) for 421CA genotypes, but was not found in a more recent study by Li et al. (2013). Q141K was shown to have no significant influence on the disposition of irinotecan and its metabolite SN-38 in individuals in three studies (Han et al., 2007; Jada et al., 2007; Sai et al., 2010), in contrast to an earlier study suggesting a role for Q141K in irinotecan disposition (de Jong et al., 2004). However, a role for ABCG2 in irinotecan pharmacokinetics was supported by in vitro studies of cellular resistance to SN-38 (Tamura et al., 2007b; Jandu et al., 2016). Therefore, more evidence on topotecan and irinotecan pharmacokinetics is needed before a conclusion on these analogs can be made.
Tyrosine Kinase Inhibitors.
Gefitinib pharmacokinetics was the first to be studied of the TKIs, with Li et al. (2007) demonstrating that 421CA genotypes had 1.5-fold increased steady-state gefitinib accumulation compared with wild type. Q141K was also shown by the same group to increase the likelihood of diarrhea after gefitinib treatment, but not skin toxicity (Cusatis et al., 2006). It was expected that erlotinib pharmacokinetics would show similar traits given its similarities to gefitinib. This was confirmed in studies showing a 24% reduction in the clearance of erlotinib in patients with at least 1 Q141K allele (Thomas et al., 2009; Fukudo et al., 2013). For sunitinib, both homozygous and heterozygous genotypes showed an increased dose-adjusted AUC compared with wild type (Mizuno et al., 2012). Q141K showed an association with sunitinib therapy-induced thrombocytopenia, but not with early onset hematoxicity (Low et al., 2016; Kato et al., 2017).
Studies examining imatinib response are more conflicting, although research appears to sway again toward increased accumulation in Q141K carriers. The conflicting results are exemplified in the Gardner et al. (2006) study, which found increased accumulation in vitro but not in 16 heterozygous patients in vivo. Increased sensitivity to imatinib (and dasatinib and nilotinib) in vitro in Q141K-expressing cells was also shown by Skoglund et al. (2014). Several studies have found Q141K showing no influence on imatinib concentration (Yamakawa et al., 2011; Seong et al., 2013; Francis et al., 2015); however, other studies have found 1.4-fold dose-adjusted trough concentration to be higher in Q141K, along with reduced oral clearance and increased drug response (Petain et al., 2008; Takahashi et al., 2010; Au et al., 2014; László et al., 2016), and recent meta-analysis of 23 studies found that Q141K has a significant effect on improved imatinib treatment in chronic myeloid leukemia in Asian populations (Jiang et al., 2017). Currently, no association has been found with ABCG2 polymorphisms and telatinib/danusertib pharmacokinetics (Steeghs et al., 2011a,b). Along with the changes in TKI disposition in ABCG2 polymorphisms, TKIs have been shown to inhibit ABCG2, with a potential role in drug-drug interactions, as discussed by Mao and Unadkat (2015).
Other Anticancer Agents.
It would be expected that loss-of-function polymorphisms such as Q141K would increase methotrexate toxicity since inhibition of ABCG2 has consistently been shown to increase methotrexate AUC (Vlaming et al., 2011; Suthandiram et al., 2014; Gervasini et al., 2017). However, no patient studies have shown this link, with several studies showing no effect of Q141K on plasma concentrations and therapy response (El Mesallamy et al., 2014). The anthracycline daunorubicin was one of the first ABCG2 substrates to be discovered, using MDR breast cancer cell lines. In Q141K-expressing cell lines, daunorubicin and doxorubicin were found to show increased accumulation compared with wild type (Tamura et al., 2007b), but worse outcomes were shown in Q141K acute myeloid leukemia patients treated with idarubicin (Tiribelli et al., 2013). In addition, the presence of the 421C allele was indicative of a weaker treatment response to anthracyclines in a Kurdish population (Ghafouri et al., 2016). The same study showed Q141K produced a weaker response to the taxane paclitaxel (Ghafouri et al., 2016), although Q141K patients were associated with longer progression-free survival when treated with paclitaxel and platinum therapy in a study on ovarian cancer patients (Tian et al., 2012). Another taxane, docetaxel, was shown in a recent study to have reduced resistance in Q141K carriers compared with wild type (Sobek et al., 2017), despite Q141K previously having been shown to have no influence on docetaxel disposition (Baker et al., 2009; Chew et al., 2011). These conflicting findings, particularly for taxanes, reiterate the need for further investigation into the pharmacokinetics of anticancer agents in ABCG2 variants.
Statins.
The effects of Q141K on the family of statins have been extensively researched, with the majority of statins tested (notable exceptions being pitavastatin and pravastatin) (Ieiri et al., 2007; Keskitalo et al., 2009b; Oh et al., 2013; Zhou et al., 2013c) showing altered pharmacokinetics in individuals bearing the C421A allele. Rosuvastatin pharmacokinetics have been consistently shown to be influenced by Q141K, with many studies linking the polymorphism to an increase in plasma AUC. Both the 421CA and AA genotypes show significant increases, with AA individuals showing 135% higher maximum plasma concentration and 140% higher AUC, thus creating drug efficacy at lower doses of rosuvastatin in individuals with the Q141K polymorphism (Zhang et al., 2006; Keskitalo et al., 2009b; Tomlinson et al., 2010; Lee et al., 2013; Zhou et al., 2013a; Birmingham et al., 2015; Liu et al., 2016). In addition, atorvastatin shows a 46% increased plasma AUC in the 421AA genotype, although there is limited evidence as to the effects of CA (Keskitalo et al., 2009b; Birmingham et al., 2015). Simvastatin acid has been shown to have a significantly increased concentration in Q141K patients in studies by Birmingham et al. (2015) and Choi et al. (2015), but evidence for effects on the prodrug simvastatin lactone is more equivocal (Keskitalo et al., 2009a; Birmingham et al., 2015). Research into fluvastatin has also shown varying results. Zhou et al. (2013b) found that neither Q141K nor V12M influenced pharmacokinetics; however, Keskitalo et al. (2009a) found a significant increase in AUC in 421AA individuals. Despite this apparently clear picture of ABCG2:statin interaction there are still ambiguities to resolve. Notably, while Q141K has been shown to result in increased plasma concentration of atorvastatin and rosuvastatin, there was no effect on the elimination half-lives. This means the altered pharmacokinetics is due to tissue-specific ABCG2:statin interactions, i.e., causing increased absorption into the blood rather than changes in biliary excretion (Li and Barton, 2018).
Direct Oral Anticoagulants.
Recent research into anticoagulants has found that dabigatran etexilate, rivaroxaban, edoxaban, and apixaban are substrates of ABCG2 (Gong and Kim, 2013; Gong et al., 2013; Mueck et al., 2013; Zhang et al., 2013; Hodin et al., 2018). It was hypothesized that these drugs may be influenced by loss-of-function polymorphisms (e.g., Q141K) and drug-drug interactions, possibly risking patient safety and impacting drug action. Apixaban was recently shown to have a 1.5-fold higher clearance rate in WT or heterozygous genotype compared with the 421AA genotype (Ueshima et al., 2017), supporting the need for further studies to determine whether patients with C421A alleles are at greater risk of bleeding. This is an important field of potential study, especially due to the significant risks in under- or overdosing of anticoagulants, and the lack of antidotes for some direct oral anticoagulants.
Anti-Human Immunodeficiency Virus Medications.
Several anti-human immunodeficiency virus medications have been shown to act as ABCG2 substrates. Disposition of both lamivudine and nelfinavir seem to be unaffected by Q141K (Colombo et al., 2005; Kim et al., 2007). On the other hand, dolutegravir showed a significantly increased peak plasma concentration in the 421AA genotype compared with CC and CA. It was hypothesized this was due to reduced ABCG2 expression in the small intestine, which resulted in increased dolutegravir absorption (Tsuchiya et al., 2017). Increased drug concentration in Q141K patients has also been found for raltegravir (Tsuchiya et al., 2016).
Disease-Modifying Antirheumatic Drugs.
Leflunomide, used in the treatment of rheumatoid arthritis, produces the active metabolite teriflunomide/A771726. A 70% increase in maximal serum concentration and 40% increase in AUC of teriflunomide have been shown in Q141K as a result of reduced ABCG2 activity (Kim et al., 2011; Wiese et al., 2012). The effects of Q141K on SASP have been extensively researched, but data remain conflicting. Studies by Urquhart et al. (2008), Ieiri et al. (2008), and Gotanda et al. (2015) all showed a significant increase in the AUC of SASP for CA/AA genotypes, but this was not found in the Adkison et al. (2010) study. The increase in drug concentration may be caused by increased SASP absorption in the small intestine or decreased clearance in the liver (Ieiri et al., 2008).
Other Drugs.
A significant effect on the in vitro accumulation of diabetic medication glyburide/glibenclamide in cells expressing Q141K has been shown by Pollex et al. (2010), with possible importance in the placenta during pregnancy. Increased glyburide accumulation in Q141K-expressing cells was also found by Bircsak et al. (2016). Q141K has also recently been shown to influence the exposure of clozapine, an antipsychotic medication (Bircsak et al., 2016). The dispositions of the immunosuppressants tacrolimus and mizoribine have shown no change in Q141K (Fukao et al., 2011; Ogasawara et al., 2013), although mycophenolic acid showed more mixed results (Miura et al., 2008; Geng et al., 2012). There are also multiple drugs that have been studied with respect to Q141K, which have thus far shown no evidence of pharmacokinetic effects. No effects have been found thus far for the angiotensin II inhibitors telmisartan and olmesartan (Yamada et al., 2011; Kim et al., 2012; Chen et al., 2013) or for nitrofurantoin, an antibiotic used in bladder infections (Adkison et al., 2008; Huang et al., 2012). However, the wide variety of drugs potentially influenced by Q141K highlights the importance of testing new drugs for interactions with this specific polymorphism.
“Rescuing” the Mis-Trafficked Q141K Isoform.
The pharmacological modulation of misfolded ABC transporters has been intensively studied in the case of ABCC7/CFTR (De Boeck et al., 2014; Vauthier et al., 2017). The F508del variant of CFTR, which is found in approximately 70% of cystic fibrosis patients, is located in a structurally equivalent position to Q141K, which has prompted significant research into the use of pharmacological chaperones to increase Q141K protein expression at the cell membrane. A number of small molecules were discovered by Woodward et al. (2013) to increase the expression of Q141K ABCG2 within the membrane including 4-phenybutyrate (a histone deacetylase inhibitor) and the CFTR corrector VRT-325. In the case of VRT-325, it was found to restore function (uric acid transport) of Q141K-expressing cells, showing its potential benefits in gout treatment. Other histone deacetylase inhibitors, such as romidepsin, have been shown to rescue Q141K ABCG2, causing restored ABCG2 expression in the membrane and increased ABCG2 function (Basseville et al., 2012). The potential for ABCG2 modulation in gout therapy is in its infancy but there is promise in this area.
Less Well-Studied SNPs in the NBD
The T153M (458 C>T; rs753759474) polymorphism was shown to cause decreased ABCG2 expression, resulting in decreased resistance to SN-38 (irinotecan), due to reduced efflux activity (Mizuarai et al., 2004; Yoshioka et al., 2007; Stiburkova et al., 2017). A recent study linked T153M to gout induction, along with the nearby polymorphism R147W. It was hypothesized that these effects may be due to disruption in ATP binding (Stiburkova et al., 2017); however, no in vitro examination of the effect of this SNP on ATP binding has been performed. Indeed, examination of the structure of ABCG2 indicates that both T153M and R147W are positioned close to the NBD:TM domain interface (Fig. 3B). Similar to Q141K, these residues may either disturb the folding of the protein or the allosteric communication of ATP hydrolysis with drug binding and transport. Furthermore, in vitro examination of these SNPs is certainly warranted since it may shed light on the mechanism of the transporter.
Close to the Walker B catalytic glutamate residue (E211 in human ABCG2) there are two SNPs, I206L (rs12721643) and F208S (rs1061018) (Zamber et al., 2003). Their location in the Walker B region means that impacts on ATP binding are expected, particularly for the drastic change from Phe to Ser in the F208S variant. In vitro experiments on cells expressing the I206L variant indicated that there was significantly lower ABCG2 expression compared with wild type (Vethanayagam et al., 2005), which is paralleled by other mutations in neighboring residues (Cui et al., 2001)). F208S (623 T>C) expression is also reduced, and furthermore the mutated protein is not localized to the cell surface membrane. Concomitantly, F208S was found to show no resistance to SN-38 (irinotecan) and mitoxantrone in studies by Yoshioka et al. (2007) and Tamura (2007b), and is defective in porphyrin transport. This makes it likely to be a loss-of-function polymorphism, a view reinforced by transport studies of TKI in cells expressing this variant (Skoglund et al., 2014). For both I206L and F208S, to date, there are no studies regarding the effects of this SNP in patients.
Polymorphism rs3116448, which results in S248P (742 T>C), shows impaired efflux function and defective porphyrin transport compared with wild-type protein (Tamura et al., 2006; Deppe et al., 2014), despite showing similar expression at the plasma membrane. S248P has been shown to reduce drug resistance for SN-38 and mitoxantrone compared with wild type (Tamura et al., 2007b). This is a result of impaired transport, which will result in an increased concentration within target cells. Structurally, S248 is located C-terminally to a conserved NBD motif—the histidine switch region (Mao and Unadkat, 2015)—which is known to be critical in ATP hydrolysis; therefore, a dramatic effect on protein function is expected here.
Finally, in a loop toward the end of the NBD, the SNP P269S (805 C>T) is located close to the linker region; it is found in Korean and Vietnamese populations at a frequency of 0.2% (Lee et al., 2007). The activity of P269S ABCG2 was decreased modestly compared with wild type, despite similar protein expression levels, for a range of drugs including prazosin, estrone-3-sulfate, and mitoxantrone (Kondo et al., 2004; Lee et al., 2007; Higashino et al., 2017). Similarly, there is no effect of this SNP on urate transport, suggesting P269S is not clinically relevant in gout; the clinical significance of this SNP on pharmacokinetics remains to be determined.
Polymorphisms in the Linker Region
The recent structural advances for ABCG2 now mean that interpretations of linker region SNPs will be more informed (Taylor et al., 2017). Only one SNP in the linker region of ABCG2 has been described in any detail. D296H (886 G>C) was originally identified by Polgar et al. (2008), and was shown to reduce ABCG2 expression and increase the toxicity of several TKIs (Skoglund et al., 2014). This can be rationalized by inspection of the structure of ABCG2, which shows D296 to be located at an NBD:NBD interaction site. It is feasible that disruption of the NBD:NBD interface by this SNP could have dramatic effects on the transporter’s ability to hydrolyze ATP, although this remains to be seen.
Polymorphisms in the MSD
The membrane spanning region of ABCG2, like other ABC transporters, is the site at which transported substrates interact (see Fig. 3D for the proposed drug binding site in ABCG2). The structural basis for multidrug transport by ABCG2 is not fully understood but is believed to be underpinned by multiple pharmacologically distinct and potentially spatially distinct drug interaction sites (Clark et al., 2006; Cox et al., 2018). A loop between two of the helices that comprise the MSD is also the location of the single N-linked glycosylation site in the transporter, full maturation of which is necessary for ABCG2 stability (Nakagawa et al., 2009). It might, therefore, be expected that mutations in the MSD would impact drug specificity as well as protein stability.
F431L.
F431L (1291 T>C) is found within the TM2 segment of the MSD. Studies have found that ABCG2 expression at the cell surface membrane was not affected by the polymorphism, although there was loss of function (Yoshioka et al., 2007; Deppe et al., 2014; Sjöstedt et al., 2017). This loss of function results in impaired transport of porphyrins (Tamura et al., 2007b), decreased resistance to SN-38 and mitoxantrone compared with wild type (Tamura et al., 2007a), and differences in response to TKIs and ABCG2-specific inhibitors (Yoshioka et al., 2007; Kawahara et al., 2010). This may result in an increased risk of hypersensitivity to certain anticancer agents, especially methotrexate, where transport is completely abolished. Yoshioka et al. (2007) hypothesized the F431 residue may be important in substrate recognition, and inspection of the structure of ABCG2 (Fig. 3D) shows the location of this residue close to a proposed drug-binding pocket (Taylor et al., 2017); the recent determination of the structure of ABCG2 with a bound inhibitor confirms the importance of this residue in transporter pharmacology (Jackson et al., 2018).
S441N (rs758900849).
The S441N polymorphism (1322 G>A), also in TM2, has been shown to cause instability in the protein, presumably by disrupting the TM helical fold, despite the relatively conservative nature of the substitution. This results in enhanced proteasomal degradation of the S441N variant protein, and a lower overall protein expression level within the plasma membrane (Nakagawa et al., 2008). The lower protein expression in S441N appears to completely eliminate the transport function of ABCG2 in multiple studies (Tamura et al., 2006, 2007a,b; Sjöstedt et al., 2017) in various different cell-based systems; these describe reductions in and abolition of transport of urate, porphyrins, SN-38, and mitoxantrone. How this translates to effects on ABCG2 in individuals with either GA or AA haplotypes is unclear. Notably, the polymorphism S441N is close to numerous residues identified in structural and functional work on ABCG2 as being important for drug binding and transport. It lies adjacent to S440, shown to be critical in the transport of mitoxantrone and pheophorbide A (Cox et al., 2018), and is located at a site where multiple structural and molecular docking studies predict drugs may bind (László et al., 2016; Taylor et al., 2017; Cox et al., 2018).
F489L (rs192169063).
F489L (1465 T>C) is found in the TM3 segment of the MSD, producing ABCG2 with impaired functioning, but with similar expression levels in the membrane (Sjöstedt et al., 2017). This impaired transport has been found to be around 10% of wild type, with F489L ABCG2 shown to be unable to transport methotrexate (Tamura et al., 2006). Although not the exact residue change seen in the SNP, it is interesting that Cox et al. (2018) have also expressed an F489A variant, which showed a contribution to a drug binding site in molecular docking studies and reduced drug export function (reduced pheophorbide A transport), but with no effect on protein expression. In contrast, a study by Deppe et al (2014) found that F498L reduced levels of ABCG2, which was reversed by proteasome inhibition, suggesting a protein processing defect.
N590Y (rs34264773).
N590Y (1768 A>T) is located in the extracellular loop between TM5c and TM6a and was found at a frequency of 1% in a Caucasian population (Zamber et al., 2003). Results on this polymorphism are limited to in vitro studies that do not show strong agreement. Vethanayagam et al. (2005) found that N590Y increased the expression of ABCG2 within the membrane, but reduced drug transport and resistance to mitoxantrone and topotecan to just 30% of wild type. This was not supported by Yoshioka et al. (2007), who showed comparable expression to wild type and no change in SN-38 pharmacokinetics. Research on the pharmacokinetics of F489L and N590Y is limited and conflicting, thus no reliable judgement can be made on their effects. It is interesting to note that the side chain of N590 is in very close proximity to N596, which is the site of glycosylation in ABCG2; therefore, an effect of this SNP on processing and maturation of the transporter would not be unexpected.
D620N (rs34783571).
The D620N polymorphism (1858 G>A) is also located at the very extracellular end of the TM6a helix and its side chain interacts with the polar head group region of the lipid bilayer (Taylor et al., 2017). The impact of the polymorphism on protein expression again shows contradictory results. Vethanayagam et al. (2005) showed 240% increased ABCG2 expression compared with wild type, similar to N590Y. This study also showed that the mutated pump had 50% of the efflux activity of the wild-type pump, with a concomitant 50% decrease in methotrexate resistance, although there was no effect on mitoxantrone or topotecan resistance (Vethanayagam et al., 2005). On the other hand, another study found a decrease in ABCG2 expression (Yoshioka et al., 2007) and reduced resistance to SN-38 (Yoshioka et al., 2007), while a further report indicated no effect on efflux activity (Morisaki et al., 2005). Reduced excretion of urate in the kidney may result in a possibly damaging effect of D620N in gout (Stiburkova et al., 2017), although sample sizes are not currently high enough to provide firm evidence of this.
Polymorphisms in Noncoding Regions
Mutations in the introns may affect ABCG2 expression and function via effects on mRNA splicing or RNA stability, although the exact mechanisms of many such polymorphisms remain unclear. Both 16702 C>T and 12283 T>C have been shown to increase ABCG2 expression in liver samples (Poonkuzhali et al., 2008), with 16702 C>T showing some association with skin toxicity and rash in erlotinib therapy. These effects of 16702 C>T are likely caused by the gain of a GATA4 transcription factor binding site (Rudin et al., 2008). Conversely, 1143 C>T was shown to lower ABCG2 expression in the same study with 1143CT/TT carriers showing an increased concentration response to erlotinib (Poonkuzhali et al., 2008; Rudin et al., 2008), and CT carriers have shown a minor link to sorafenib response (Tandia et al., 2017). Another study looked at the effects of intronic polymorphisms on methotrexate therapy. Two polymorphisms were identified with an enhanced response: 5566 C>T in intron 2 and 3531 G>C in intron 9. The 18271 G>A (rs1564481) polymorphism was shown to correlate with the pharmacokinetics of, and improved response to, therapy with flavopiridol, a CDK9 inhibitor used in the treatment of acute myeloid leukemia (Ni et al., 2010). No effect on flavopiridol pharmacokinetics was found for other intron polymorphisms, 152 G>A, 10130 A>G, 14952 G>T, and 18271 G>A.
Promoter mutations may affect expression via effects on the transcription of the ABCG2 gene. The −15622 C>T polymorphism has been shown to lower ABCG2 protein expression in tissue (Poonkuzhali et al., 2008). Both heterozygous and homozygous haplotypes result in 40%–50% increases in maximum concentration in patients treated with erlotinib, along with increased incidence of side effects such as diarrhea (Rudin et al., 2008). An increased risk of side effects was also shown in gefitinib therapy, although Lemos et al. (2011) did not find any link between the TT genotype and therapy outcomes. Conversely, the −15994 C>T (rs7699188) polymorphism significantly increases ABCG2 expression. These effects are possibly due to the gain of a hepatocyte nuclear factor 4 binding site, a transcription factor that may be involved in ABCG2 expression. The increased expression as a result of this polymorphism has been shown to increase the clearance of oral imatinib in vivo (Poonkuzhali et al., 2008).
Junior Blood Group
As well as its influence on pharmacokinetics, ABCG2 has recently been explained as having a key role in the Junior (Jr) blood group system, which is especially common in Asian populations. Several millions of people are predicted to not express ABCG2, producing the Jr(a−) blood group, because the Jr antigen is within the ABCG2 protein (Saison et al., 2012; Zelinski et al., 2012). Antibodies to the Jr(a−) blood group may result in blood transfusion reactions, but do not commonly cause severe hemolytic disease of the newborn, despite some reports of this (Zelinski et al., 2012). The Jr(a−) blood group is induced by ABCG2 null alleles, which lead to insertion of a premature stop codon, such as the Q126X (376 C>T) polymorphism (Honjo et al., 2002; Zelinski et al., 2012). These individuals have no ABCG2 expression on the surface of their erythrocytes. Homozygous Q126X was detected in 80% of Jr(a−) blood samples in a recent study in a Japanese population (Tanaka et al., 2018). Saison et al. (2012) also suggested that the R236X polymorphism (706 C>T) may be the basis of the Jr(a−) blood type in Romani Gypsy diaspora communities of Southwestern Europe, and a homozygous R246X (736 C>T) variant has been found, resulting in Jr(a−) erythrocytes in two Caucasian individuals. Heterozygous R246X was shown to lower ABCG2 expression approximately 50% in vivo during flow cytometry analysis of erythrocytes (Kasza et al., 2012). Along with these studies, it is predicted that the E334X variant also produces the Jr(a−) blood group (Zelinski et al., 2012). Recently, an increasing catalog of alleles has been linked to the Jr blood group, with studies reporting R113X, G262X, Q531X, and S340del as mutations causing Jr(a−) (Zelinski et al., 2012). It is as yet unknown whether individuals with the Jr(a−) blood group have altered pharmacokinetics of drugs, although they would be expected to have similar pharmacokinetics to those detailed in the previous section on Q126X. An interesting line of research would be to investigate the differences in drug disposition and treatment outcomes in populations with/without the Jr(a−) group.
Perspectives for Future Research
The effects of the Q141K polymorphism are well researched, with a variety of substrate drugs being shown to have altered pharmacokinetics. These effects are likely caused by the decreased ABCG2 expression in the membrane in the various ABCG2 sites, a result of increased instability and degradation of this isoform. The influence of Q141K on pharmacokinetics may be a result of increased oral availability (e.g., due to reduced excretion in the small intestine), decreased efflux of drugs from the target cells, or decreased transport across barriers (e.g., the blood-brain barrier).
Summarizing the effects of the less-common polymorphisms in the NBD and polymorphisms in the MSD (see Table 3) shows varied conclusions, with polymorphisms found to increase, decrease, or have no change on levels of ABCG2 expression in vitro; however, there is limited research into their effects on drug therapy. More research into the in vivo effects of these polymorphisms on ABCG2 expression and drug efficacy is required before any definitive conclusions can be drawn. An ideal outcome of research into the ABCG2 polymorphisms would be to establish a grouping system for polymorphisms based on their characteristics of protein expression, efflux activity, and substrate-specific effects. One grouping method was presented previously, based upon protein expression, transport activity, drug resistance, and prazosin-stimulated ATPase activity (Tamura et al., 2007a). Based on the accumulation of limited research into rarer polymorphisms, we propose an updated six-group system based on ABCG2 expression and effects on activity or resistance (Table 4). However, to substantiate and validate this system requires much more work on the rarer SNPs.
Summary of the effects of less-well studied ABCG2 SNPs on expression and function
Potential categorization of ABCG2 SNPs
Where will such information come from? One novel method involves the use of antibody-based flow cytometry analysis to quantify the levels of ABCG2 in erythrocytes via a blood sample, coupled with genomic analysis of the DNA in the same sample. The use of blood samples could be a well-tolerated method of analyzing ABCG2 levels since it is noninvasive and quantifiable with good anti-ABCG2 antibodies. This method has been used to describe significant decreases in protein expression in heterozygous Q141K and R236X carriers (Kasza et al., 2012). This proved the method could be successful in showing the varying tissue levels of ABCG2 for differing genotypes. An expansion of this method to discover the in vivo expression levels of the many other natural polymorphisms detailed in this review would be an ideal next step in research, since it would remove the more subjective interpretation of cell line studies. Complementary functional studies in cell lines do have an important role to play as long as changes in transport are correlated with the ABCG2 expression level to ensure that an accurate picture of a SNP’s effect on drug transport is obtained. One additional question that is not often discussed in the literature is the effect of heterozygous alleles in the final assembled ABCG2 dimer. Specifically, in a heterozygous individual there is a possibility that following translation and trafficking ABCG2 dimers are present in three forms: WT-SNP, SNP-SNP, or WT-WT. Given that translation occurs through the action of polyribosomes on the same mRNA, it seems feasible that SNP-SNP ABCG2 dimers and WT-WT dimers would occur rather than “mixed dimers,” but this remains to be determined. Finally, better structural data now set the scene for molecular docking approaches that should be able to rationalize effects in patients and enable better predictions regarding possible drug interactions with this important MDR pump.
Acknowledgments
We thank other members of the Kerr laboratory for informative discussions, in particular, Megan Cox and Aaron Horsey.
Authorship Contributions
Performed data analysis: Heyes, Kapoor, Kerr.
Wrote or contributed to the writing of the manuscript: Heyes, Kapoor, Kerr.
Footnotes
- Received June 12, 2018.
- Accepted September 20, 2018.
Abbreviations
- ABC
- ATP-binding cassette
- AUC
- area under the curve
- Jr
- Junior
- MDR
- multidrug resistance
- MSD
- membrane-spanning domain
- NBD
- nucleotide binding domain
- SASP
- sulfasalazine
- SNP
- single nucleotide polymorphism
- TKI
- tyrosine kinase inhibitor
- TM
- transmembrane
- WT
- wild type
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}