


Abstract
Imetelstat, a 13-base oligonucleotide (5′-TAGGGTTAGACAA-3′), is a potent, investigational telomerase inhibitor in clinical development for the treatment of hematologic myeloid malignancies. Modifications to imetelstat oligonucleotide chemistry include an N3′–P5′ thio-phosphoramidate backbone linkage to improve biologic stability and the addition of a palmitoyl tail at the 5′-position to enhance cellular membrane permeability. Other oligonucleotides have been previously shown to have in vitro test-system–dependent outcomes when potent cytochrome P450 inhibition in human liver microsomes (HLM) is observed, but such inhibition is not observed in cryopreserved human hepatocytes (CHH). Outcomes in CHH are consistent with clinical reports in which no interactions were reported. In the present study, imetelstat was evaluated for in vitro inhibition of eight P450 enzymes, namely CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4 in CHH (0.5 million cells/ml). Assays were performed using validated conditions, including short substrate times (10 minutes), and at the approximate substrate Km concentration. Imetelstat was found to have little to no inhibition of all P450 isoforms evaluated, with inhibitor concentration that causes 50% inhibition (IC50) values >100 μM. Maximum percent inhibition values for each P450 isoform at 100 μM imetelstat were <20% except for CYP2C8 activity, which was inhibited by 49%. Using a static mechanistic model, the predicted change in area under the curve of a victim drug coadministered with imetelstat was 1.04-fold, projecting no relevant clinical interaction. Overall, the results from this in vitro study suggest that clinical use of imetelstat is unlikely to affect the pharmacokinetics of concomitant therapies that undergo cytochrome P450-mediated metabolism.
Introduction
Oligonucleotide based therapeutics are an attractive area for pharmaceutical development due to the ability to provide target specificity with base-pair sequence, especially in the areas of cancer treatment, genetic disorders, infectious diseases, and metabolic disorders (Lundin et al., 2015; Sharma and Watts, 2015; Aartsma-Rus, 2016). Usually, these molecules function by having an anti-sense base-pair sequence complementary to a target mRNA sequence, leading to downregulation of mRNA translation and protein expression, and are thus also known as anti-sense oligonucleotides (ASOs). Chemical modification of oligonucleotides has been adopted as a strategy to enhance target interaction and biologic stability, yielding a general improvement in pharmacokinetic and pharmacodynamic properties of these therapeutics. This includes sulfurization of the phosphodiester bond, methoxy or methoxyethyl group addition to sugar moieties, locked/unlocked nucleic acids, and internucleotide linkage modifications to form peptide nucleic acids (Prakash, 2011; Deleavey and Damha, 2012; Sharma and Watts, 2015; Chen et al., 2018). Among the many oligonucleotide biomolecules investigated thus far, only fomivirsen, pegaptanib, mipomersen, eteplirsen, nusinersen, and defibrotide sodium have received U.S. Food and Drug Administration (FDA) approval (Stein and Castanotto, 2017).
Imetelstat (13-mer oligonucleotide N3′–P5′ thio-phosphoramidate with a covalently linked C16 [palmitoyl] lipid moiety at the 5′ end) is in clinical development as a treatment option for hematologic myeloid malignancies (Röth et al., 2010; Baerlocher et al., 2015; Chiappori et al., 2015; Tefferi et al., 2015; Dillen et al., 2017). Imetelstat is a high-affinity, active-site telomerase inhibitor with specificity for the RNA template region of telomerase, directly competing with telomere binding. Therefore, its mode of action is similar to a conventional small-molecule competitive enzyme inhibitor rather than like an ASO (knockdown of target) (Herbert et al., 2005). Activation of telomerase is thought to be important for tumor progression by enabling cancer cells to maintain short telomeres, gain immortality, and evade apoptosis (Lichtsteiner et al., 1999). The mechanism of action by imetelstat results in potent inhibition of telomerase and leads to progressive telomere shortening (Herbert et al., 2005; Marian et al., 2010). Structurally (see Fig. 1), the thio-phosphoramidate linkage within imetelstat confers high resistance to nuclease digestion in blood and tissues, while the properties of the 5′-palmitoyl chain result in improved cellular permeability and consequently strong tissue penetration and retention (Herbert et al., 2005).
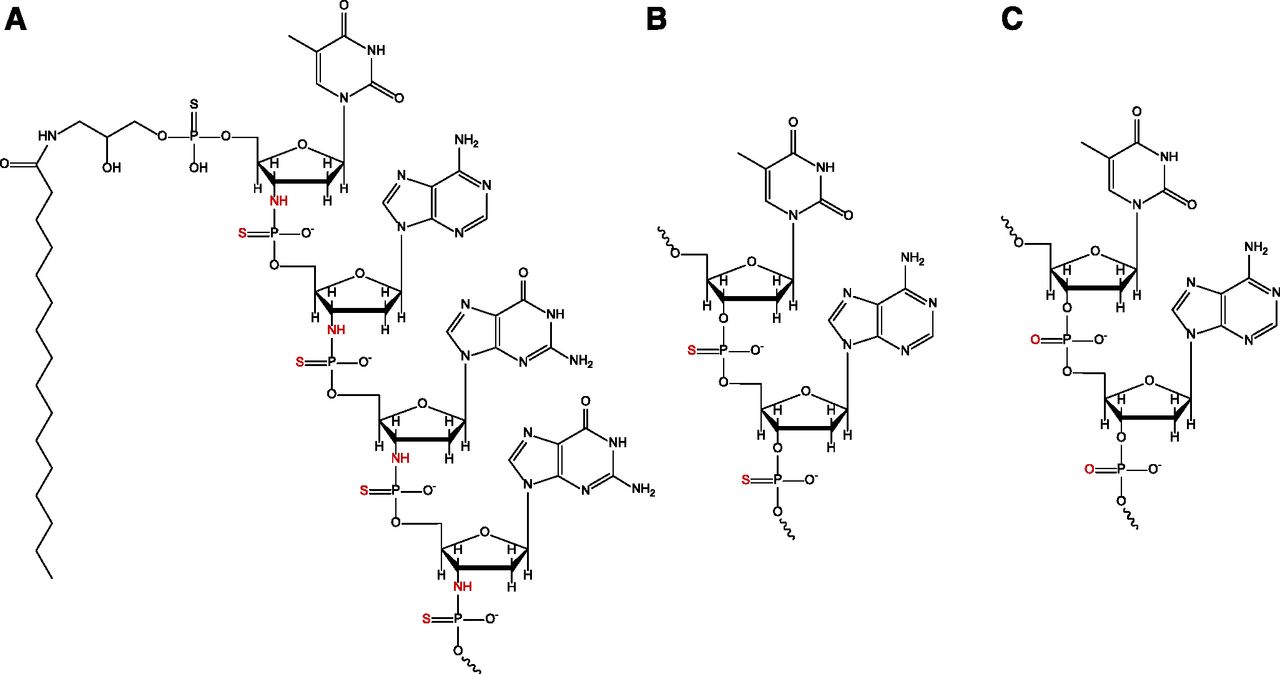
Truncated structures of (A) imetelstat, (B) phosphorothioate, and (C) phosphodiester oligonucleotide linkages. The thio-phosphoramidate, phosphorothioate, and phosphodiester linkages on each respective molecule are highlighted in red.
The potential for oligonucleotide-based molecules to act as perpetrators of drug-drug interactions (DDIs) has not been reported extensively in the literature. However, of the clinical interaction studies conducted with victim drugs subject to clearance by cytochrome P450, glucuronidation, renal elimination, or nucleoside kinases, none has reported a clinically relevant DDI by therapeutic oligonucleotides (Adjei et al., 2003; Villalona-Calero et al., 2004; Geary et al., 2006; Yu et al., 2009; Li et al., 2014). Industry regulatory guidances from the FDA (2017 draft—https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM581965.pdf) and the European Medicines Agency (EMA; 2013 final— http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf) recommend in vitro P450 inhibition studies for small-molecule drugs in appropriate test systems (such as human liver microsomes or cryopreserved human hepatocytes) to ascertain the perpetrator DDI potential of a drug. Recent studies on P450 inhibition in vitro suggest that cryopreserved human hepatocytes (CHH) are a more clinically relevant test system for the prediction of DDIs by oligonucleotides, and that studies conducted in human liver microsomes may result in artifactual results (Kazmi et al., 2018). This is supported by another recent in vitro report in which no P450 inhibition by oligonucleotides in CHH was reported (Shemesh et al., 2017). In the present study, the in vitro P450 inhibitory potential of imetelstat was evaluated in CHHs to determine the risk for P450-mediated DDIs with concomitantly administered medications.
Materials and Methods
Chemicals and Reagents.
Imetelstat, a 13-base oligonucleotide (5′-TAGGGTTAGACAA-3′) N3′–P5′ thio-phosphoramidate containing a palmitoyl tail at the 5′-position (5′-palmitoyl-TAGGGTTAGACAA-NH2-3′) was provided by Geron Corporation (Menlo Park, CA). Krebs-Henseleit buffer (KHB), calcium chloride, phenacetin, bupropion, tolbutamide, dextromethorphan, chlorzoxazone, midazolam, α-naphthoflavone, ticlopidine, sulfaphenazole, quinidine, 4-methylpyrazole, ketoconazole, amodiaquine, montelukast, and acetaminophen were all purchased from MilliporeSigma (St. Louis, MO). Sodium bicarbonate and HEPES were purchased from Life Technologies/Thermo Fisher Scientific (Carlsbad, CA). N-3-benzylphenobarbital was obtained internally from Janssen Research & Development (Beerse, Belgium); S-(+)-mephenytoin was purchased from Corning Life Sciences (Corning, NY); metabolite reference standards including hydroxytolbutamide, (+/−)-4′-hydroxymephenytoin, dextrorphan, 6-hydroxychlorzoxazone, 1′-hydroxymidazolam, hydroxybupropion, and N-desethylamodiaquine were purchased from Ultrafine Chemicals and BD Gentest, both formerly part of BD Biosciences and now part of Corning Discovery Labware, Inc. (Woburn, MA). Hepatocyte Thawing Media was purchased from Bioreclamation IVT (Baltimore, MD). The Strata Impact protein precipitation 96-well plates were purchased from Phenomenex Corporation (Torrance, CA). All other reagents and solvents were of high-performance liquid chromatography grade or equivalent analytical standards.
Test Systems.
Pooled cryopreserved human hepatocytes, lot PDI [cryopreserved human hepatocytes (CHH), n = 20, mixed gender] were purchased from Bioreclamation IVT. Hepatocyte incubations were performed in oxygen-sparged KHB supplemented with calcium chloride, sodium bicarbonate and HEPES. Buffer pH was adjusted to 7.4 ± 0.1.
In Vitro P450 Inhibition.
Imetelstat (0.1–100 μM) was evaluated for its potential to reversibly inhibit eight P450 enzymes, namely CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4 in CHH at the approximate substrate Km concentration (determined in house with the same lot of CHH) as shown in Table 1. Triplicate incubations for each concentration were conducted with an automated liquid handling system (Biomek; Beckman Coulter, Indianapolis, IN) in 100-μl mixtures at 0.5 million cells/ml in KHB. After serial dilution in KHB, imetelstat, or experimental controls (single concentrations), and the probe substrates were aliquoted equally (10 μl each) into the incubation vessels. The final organic content was ≤0.05% for imetelstat and ≤0.15% for experimental controls (with the exception of ≤0.85% for montelukast) in each incubated sample (considering the substrate and the inhibitor). Reactions were initiated with the addition of 80 μl CHH to the incubation vessels and incubations were conducted at 37°C with 95% humidity, 5/95% CO2/air, and 350 rpm shaking for 10 minutes. Reactions were quenched after 10 minutes by the addition of 200 μl of acetonitrile containing an appropriate mixture of internal standards. After incubation, the samples were transferred to a 2-ml Strata Impact protein precipitation 96-well plate (Phenomenex) containing 300 μl of acetonitrile. The samples were filtered on a vacuum manifold, the filtrate was evaporated to dryness under nitrogen, and then reconstituted in 250 μl mobile phase (1:1, methanol/water containing 0.1% acetic acid) for further liquid chromatography/tandem mass spectrometry analysis using a cocktail method (see Table 1). Early screening studies for P450 inhibition by imetelstat in human liver microsomes (HLM) were conducted by In Vitro Technologies Inc. (Baltimore, MD) and Cerep (Redmond, WA) as described in Supplemental Table 1.
Experimental conditions for the measurement of P450 activity in pooled cryopreserved human hepatocytes
Analytical Methods.
Samples were analyzed by liquid chromatography/tandem mass spectrometry on a Shimadzu Nexera liquid chromatography system (binary LC-30AD pumps with SIL-30ACMP autosampler (Shimadzu Scientific Instruments, Columbia, MD)), coupled to a SCIEX API 4000 triple-quadrupole mass spectrometer (AB SCIEX, Foster City, CA) operated in multiple reaction monitoring and electrospray ionization scan modes. For analysis of metabolites, a gradient elution method comprising 0.1% acetic acid in water (A) and methanol with 0.1% acetic acid (B) ramping from 6% to 90% over 9 minutes (vs. 4 minutes for 6-hydroxychlorzoxazone, negative electrospray ionization mode) was applied to a Zorbax Eclipse Plus Phenyl Hexyl column (2.1 × 100 mm, 1.8 μm; Agilent Technologies, Santa Clara, CA) and column temperature was maintained at 50°C.
Data Analyses.
The acquired data were processed using Analyst version 1.6.2 (AB SCIEX) and the area ratios of metabolite and internal standard peaks were exported to Microsoft Excel 2010 (Microsoft, Redmond, WA). Nonlinear regression and inhibitor concentration that causes 50% inhibition (IC50) curve fitting analysis were performed using SigmaPlot, version 12.5 (Systat Software, Inc., Chicago, IL). Inhibition was measured as the percent reduction of probe substrate metabolite formed in the presence of imetelstat compared with the amount formed in the absence of test compound (maximal activity). Area ratios (AR) of the metabolite peak to the internal standard peak were used in the calculations, using eq. 1: Residual activity (i.e., maximal activity – % inhibition) was plotted as a function of inhibitor concentration (numerical data in Supplemental Table 2). Prediction of hepatocyte fraction unbound (fuinc) was determined by calculation of a predicted imetelstat logP with MarvinSketch version 16.1.11 (ChemAxon, Cambridge, MA) followed by application of the fuinc equation described previously by Kilford et al. (2008).
Residual activity (i.e., maximal activity – % inhibition) was plotted as a function of inhibitor concentration (numerical data in Supplemental Table 2). Prediction of hepatocyte fraction unbound (fuinc) was determined by calculation of a predicted imetelstat logP with MarvinSketch version 16.1.11 (ChemAxon, Cambridge, MA) followed by application of the fuinc equation described previously by Kilford et al. (2008).
Imetelstat inhibition data were analyzed with basic and mechanistic static models for the assessment of DDI risk as described by the current FDA and EMA guidance documents. The static mechanistic model describing hepatic-only DDI risk is shown in eq. 2: where A = reversible inhibition or (1/(1 + [Ih]/Ki)); B = time-dependent inhibition (TDI) parameters; and C = induction parameters. Because imetelstat is dosed intravenously, gut parameters were excluded. B was assumed to equal 1 due to a lack of TDI by imetelstat (Supplemental Fig. 1). Induction parameters were not considered (i.e., C = 1) per the current FDA and EMA guidance recommendations on assessment of inhibition only DDI potential. Ih = hepatic inlet concentration = fraction unbound in plasma (fuplasma) × [Cmax + [{fraction absorbed (Fa) × absorption rate constant (ka) × dose}/hepatic blood flow (Qh)]/blood to plasma ratio (Rb)].
where A = reversible inhibition or (1/(1 + [Ih]/Ki)); B = time-dependent inhibition (TDI) parameters; and C = induction parameters. Because imetelstat is dosed intravenously, gut parameters were excluded. B was assumed to equal 1 due to a lack of TDI by imetelstat (Supplemental Fig. 1). Induction parameters were not considered (i.e., C = 1) per the current FDA and EMA guidance recommendations on assessment of inhibition only DDI potential. Ih = hepatic inlet concentration = fraction unbound in plasma (fuplasma) × [Cmax + [{fraction absorbed (Fa) × absorption rate constant (ka) × dose}/hepatic blood flow (Qh)]/blood to plasma ratio (Rb)].
Results
Inhibition of P450 Enzymes by Imetelstat in Cryopreserved Human Hepatocytes.
Imetelstat (0.1–100 μM) was evaluated as a potential direct inhibitor of P450 isoforms in CHH (0.5 million cells/ml) at the approximate substrate Km concentration (see Table 1) as described in Materials and Methods. As shown in Fig. 2 and Table 2, imetelstat had little to no inhibitory effect on CYP1A2 (phenacetin), CYP2B6 (bupropion), CYP2C8 (amodiaquine), CYP2C9 (tolbutamide), CYP2C19 (S-mephenytoin), CYP2D6 (dextromethorphan), CYP2E1 (chlorzoxazone), and CYP3A4 (midazolam) with IC50 values all >100 μM. The maximum percent inhibition at the highest concentration of imetelstat tested (100 μM) was less than 20% for all P450 isoforms, except for CYP2C8, where the maximum inhibition was 49%. However, a modest inhibitory concentration dependency for CYP2C8 was observed, with inhibition reaching a plateau between 30 and 100 μM imetelstat. In contrast, all positive control inhibitors showed >80% inhibition of their respective P450 isoforms, as expected (see Table 2). Furthermore, a lack of CYP2C8 TDI by imetelstat was observed with an IC50 shift assay (Supplemental Fig. 1).
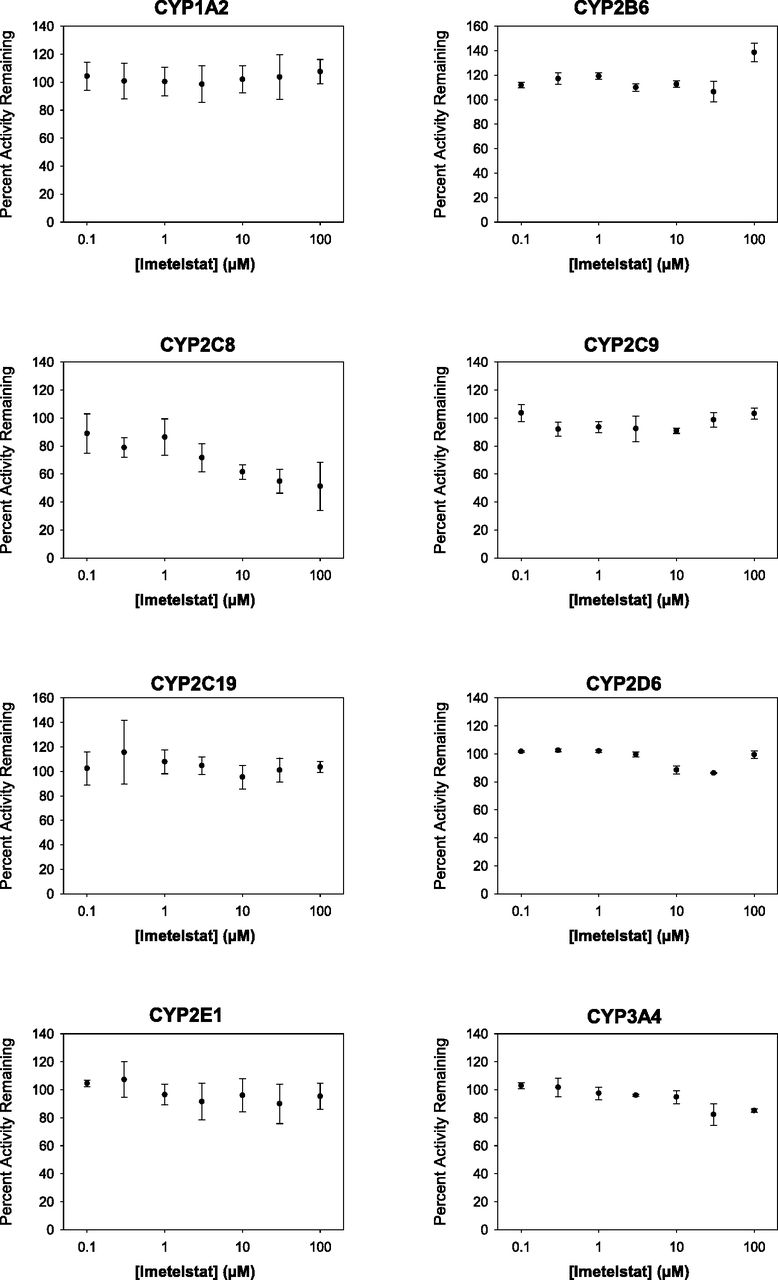
IC50 plots of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4 inhibition by imetelstat in cryopreserved human hepatocytes. As described in Materials and Methods and Table 1, the inhibition of P450 enzymes was assessed at 0.1–100 μM imetelstat (n = 3) in CHH at 0.5 million cells/ml for 10 minutes followed by liquid chromatography/tandem mass spectrometry analysis. Curve-fitting was not performed when IC50 values were projected to be >100 μM.
IC50 and maximum percent inhibition values for the inhibition of P450 isoforms by imetelstat and positive control inhibitors in pooled cryopreserved human hepatocytes
Discussion
For small-molecule drugs, regulatory agencies such as the FDA and EMA recommend in vitro evaluation of the drug-drug interaction (DDI) potential of a new molecular entity as part of the submission package toward drug approval. This is typically done through in vitro assessment of the inhibition of drug-metabolizing enzymes, namely P450 enzymes, in validated test-systems such as HLM or cryopreserved human hepatocytes (CHH). In the case of oligonucleotide drugs, although these biomolecules as a class differ in structure and composition from conventional small-molecule drugs, assessment of the DDI potential of oligonucleotides is typically evaluated in a manner similar to the FDA and EMA DDI study recommendations outlined for small-molecule drugs. Thus, in the present study the in vitro potential for imetelstat to inhibit P450 enzymes, namely CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4 was evaluated in CHH.
As shown in Fig. 2 and Table 2, imetelstat was found not to be an in vitro inhibitor of any P450 enzymes tested in CHH with IC50 values greater than 100 μM. Furthermore, the maximum percent inhibition at highest imetelstat concentration (100 μM) was minimal (<20%) for all P450 isoforms, except for CYP2C8 activity (49% inhibition), where modest concentration dependency was observed. However, it is unclear why the inhibition of CYP2C8 plateaued between 30 and 100 μM (45%–49% inhibition), as this is typically observed when inhibitor solubility is an issue; a phenomenon not observed with imetelstat (high aqueous solubility). Overall, these findings are consistent with other in vitro reports that oligonucleotides as a class do not cause significant inhibition of P450 enzymes in CHH (Shemesh et al., 2017; Kazmi et al., 2018). The recent report by Kazmi and colleagues (2018) suggests that oligonucleotide molecules as a class may cause differential test-system–dependent inhibition in HLM and CHH based on the chemical properties of the oligonucleotide backbone (phosphodiester vs. phosphorothioate linkage), and that inhibition profiles for oligonucleotides in CHH are more clinically relevant. In the case of imetelstat, a thio-phosphoramidate linkage is present (see Fig. 1) along with a 5′-palmitoyl chain for the improvement of biologic stability and tissue uptake (Herbert et al., 2005). Liver uptake of imetelstat has been previously observed in mice (Herbert et al., 2005), consistent with in house preclinical toxicology studies where liver distribution of imetelstat was seen in rodents and primates, confined largely to Kupffer cells (data not shown). This accumulation into tissue macrophages (often observed as basophilic granules) has been shown to be a class effect of oligonucleotides, especially with high doses (Frazier, 2015). In screening studies, potent P450 inhibition by imetelstat was observed in HLM for CYP2C8 (IC50 value of 0.7 μM) with IC50 values for other P450 enzymes spanning 1.8–26 μM (see Supplemental Table 1). The P450 inhibition observed in HLM, particularly of CYP2C8, was completely attenuated in the present study conducted with CHH, suggesting test-system–dependent inhibition, possibly due to the thio-phosphoramidate linkage of imetelstat.
With respect to predictions of a clinical DDI, the static basic model approach, as recommended by the EMA and FDA, was first used where R1 = 1 + Imax,u/Ki, with R1 representing the predicted area under the curve (AUC) ratio in the presence or absence of inhibitor, Imax,u representing maximal free plasma concentration of the inhibitor, and Ki representing the unbound in vitro inhibition constant. Imetelstat Cmax concentration with a 2-hour intravenous infusion dosage of 9.4 mg/kg was approximately 30 μM (136 μg/ml). The in vitro Ki value for P450 inhibition can be predicted reliably from the IC50 value by using the equation IC50/2 = Ki (Haupt et al., 2015). Nonspecific binding of imetelstat to CHH was expected to be negligible (fuinc = 1) based on physicochemical properties (hydrophilicity [predicted logP = −3.19] and charge status), and application of the predicted hepatocyte binding equation described by Kilford et al. (2008). In the case of CYP2C8 (the P450 enzyme with the greatest maximal inhibition by imetelstat), the Ki was estimated to be 50 μM based on an approximate IC50 value of 100 μM in CHH. Considering the plasma protein binding of imetelstat (at least 92.7%; Janssen internal data), the R1 value was calculated as 1.04, which is above the EMA and FDA cutoff criteria of R1 ≥1.02. The basic static models recommended by the EMA and FDA are designed to be stringent in nature by including a 50-fold safety margin for I/Ki (I = 1, Ki = 50; or I/Ki = 0.02). Should an assessment with the basic static model fail to pass DDI cutoff criteria, the EMA and FDA recommend assessment of DDI prediction with a static mechanistic model or a dynamic model, which incorporate much more detailed drug disposition parameters than the basic static model (Fahmi et al., 2009). For the static mechanistic model, these parameters include the effect of hepatic reversible inhibition, time-dependent inhibition (TDI), enzyme induction, and hepatic inlet concentration of a perpetrator drug; as well as fractional metabolism (fm) of a victim drug. However, the EMA and FDA recommend, in their most recent DDI guidelines, that inhibitory potential and induction potential with the static mechanistic model be considered separately and not simultaneously, as was originally proposed by Fahmi and colleagues (2009). Thus, predictions of inhibitory perpetrator DDI potential with this model are to be based on an assessment of direct and time-dependent inhibition only (as shown in eq. 2). Furthermore, use of the static mechanistic approach described by the EMA and FDA in their respective DDI guidances requires an estimate of hepatic inlet concentration (Ih) that is obtained by including absorption, dose, blood flow, and blood-partitioning parameters, in addition to plasma Cmax and plasma protein-binding parameters (Ito et al., 1998). However, in the case of imetelstat, because it is delivered intravenously, oral absorption parameters are not applicable and the hepatic inlet concentration is equivalent to the unbound systemic plasma concentration (fuplasmaXCmax= Ih = 2.19 μM). Imetelstat is not expected to be a time-dependent inhibitor of CYP2C8 (Supplemental Fig. 1), and assuming a worst-case scenario of a victim drug with 100% systemic clearance through one P450 pathway (fm = 1), application of these parameters to the static mechanistic model yields a prediction of 1.04-fold change in the AUC of a victim drug that is a substrate of CYP2C8. This is well below the EMA and FDA AUC ratio cutoff criteria of ≥1.25 and indicates that imetelstat would likely not be a perpetrator of a clinical DDI.
The predicted lack of clinical DDI for imetelstat is consistent with the reported lack of clinical interactions with other oligonucleotides. Clinical pharmacokinetic interaction studies with simvastatin (CYP3A4-mediated clearance), warfarin (CYP2C9/3A4/1A2-mediated clearance), rosiglitazone (CYP2C8/2C9-mediated clearance), glipizide (CYP2C9/2C8-mediated clearance), ezetimibe (glucuronidation-mediated clearance), metformin (renal transporter-mediated clearance), cisplatin (renal and copper transporter-mediated clearance), and gemcitabine (nucleoside kinase-mediated clearance) have all shown no significant pharmacokinetic interaction with therapeutic oligonucleotides (Adjei et al., 2003; Villalona-Calero et al., 2004; Geary et al., 2006; Yu et al., 2009; Li et al., 2014). Furthermore, an in vivo rat preclinical interaction study with imetelstat coadministered with paclitaxel, which is metabolized by CYP3A1/2 in rat (Vaclavikova et al., 2004), showed no significant pharmacokinetic interaction (Janssen internal data), supporting the lack of reported DDIs by oligonucleotide-based therapeutics.
In conclusion, the findings presented in this study indicate that imetelstat is not a significant in vitro inhibitor of P450 enzymes in CHH, and the in vitro–to–in vivo extrapolation (IVIVE) of DDI potential using the EMA- and FDA-recommended static mechanistic model also projects that imetelstat is not likely to be a clinically significant P450 inhibitor, and thus consequently unlikely to affect the pharmacokinetics of concomitant pharmacotherapies that are metabolized by P450s.
Acknowledgments
Medical writing assistance was provided by Ashwini Patil (Tata Consultancy Services, Mumbai, India) and additional editorial support was provided by Namit Ghildyal (Janssen Research & Development, LLC). We would like to thank Shannon Dallas and Avijit Ghosh for helpful discussions and manuscript review.
Authorship Contributions
Participated in research design: Kazmi, Sensenhauser, Greway.
Conducted experiments: Kazmi, Sensenhauser.
Performed data analysis: Kazmi, Sensenhauser.
Wrote or contributed to the writing of the manuscript: Kazmi, Sensenhauser, Greway.
Footnotes
- Received August 28, 2018.
- Accepted October 29, 2018.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- ASO
- anti-sense oligonucleotides
- AUC
- area under the curve
- CHH
- cryopreserved human hepatocytes
- DDI
- drug-drug interaction
- EMA
- European Medicines Agency
- FDA
- U.S. Food and Drug Administration
- HLM
- human liver microsomes
- IC50
- inhibitor concentration that causes 50% inhibition
- KHB
- Krebs-Henseleit buffer
- P450
- cytochrome P450
- TDI
- time-dependent inhibition
- Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}