


Abstract
Lesinurad [Zurampic; 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)], a selective inhibitor of uric acid reabsorption transporters approved for the treatment of gout, is a racemate of two atropisomers. The objective of this investigation was to evaluate the stereoselectivity of metabolism, the inhibitory potency on kidney uric acid reabsorption transporters (URAT1 and OAT4), and the clinical pharmacokinetics of the lesinurad atropisomers. Incubations with human liver microsomes (HLM), recombinant CYP2C9, and recombinant CYP3A4 were carried out to characterize the stereoselective formation of three metabolites: M3 (hydroxylation), M4 (a dihydrodiol metabolite), and M6 (S-dealkylation). The formation of M3 in HLM with atropisomer 1 was approximately twice as much as that with atropisomer 2, whereas formation of M4 with atropisomer 1 was 8- to 12-fold greater than that with atropisomer 2. There were no significant differences in the plasma protein binding among lesinurad and the atropisomers. Following oral administration of 400 mg lesinurad once daily for 14 days to healthy human volunteers, the systemic exposure (Cmax at steady state and area under the concentration-time curve from time zero to the time of dosing interval) of atropisomer 1 was approximately 30% lower than that of atropisomer 2, whereas renal clearance was similar. In vitro cell-based assays using HEK293 stable cells expressing URAT1 and OAT4 demonstrated that atropisomer 2 was approximately 4-fold more potent against URAT1 than atropisomer 1 and equally active against OAT4. In conclusion, lesinurad atropisomers showed stereoselectivity in clinical pharmacokinetics, metabolism, and inhibitory potency against URAT1.
Introduction
Lesinurad acetic acid [formerly known as RDEA594; 2-(5-bromo-4-(4-cyclopropylnaphthalen-1-yl)-4H-1,2,4-triazol-3-ylthio)], is a selective uric acid reabsorption inhibitor (Tan et al., 2011; Miner et al., 2016). The US Food and Drug Administration approved the use of lesinurad in combination with a xanthine oxidase inhibitor for the chronic treatment of hyperuricemia associated with gout (Zurampic as a single agent tablet was approved in 2015 and Duzallo as a fixed dose combination product of lesinurad and allopurinol was approved in 2017).
Absorption, metabolism, and excretion of lesinurad were evaluated in human healthy volunteers and have been reported separately (Shah et al., 2018). After oral administration, lesinurad is rapidly and completely absorbed from the gastrointestinal tract with an absolute bioavailability of approximately 100% (Shah et al., 2018). Metabolites accounted for 64.1% of the dose excreted (feces and urine) and unchanged lesinurad accounted for 31.3%, including a minor extent in the feces (1.5%) (Shah et al., 2018). CYP2C9 was the major enzyme responsible for the oxidative metabolism of lesinurad, and the fraction of lesinurad metabolized by this enzyme was 47.3% based on the recovery of oxidative metabolites in the urine and feces from a human mass balance study (Shah et al., 2018). Among the metabolites excreted in the urine, M3 and M4 accounted for approximately 33% of the dose (Shah et al., 2018). M4 is formed via an epoxide intermediate (namely, M3c), which is subsequently hydrolyzed by microsomal epoxide hydrolase to M4 (Shah et al., 2018).
Lesinurad has a sterically hindered biaryl axis due to bulky substituents on the triazole ring, which lead to hindered rotation about the axis, resulting in distinct atropisomers that can be separated by chiral chromatography. Therapeutic agents that possess chiral axial centers are much less common than chiral compounds with a stereogenic center. Besides lesinurad, other drugs with a chiral axial center on the market are gossypol and telenzepine, as well as a drug under investigation by Bristol-Myers Squibb (BMS) (BMS-986142) (Eveleigh et al., 1989; Watterson et al., 2016; Yang et al., 2018). BMS-986142 is a reversible inhibitor of Bruton’s tyrosine kinase, which is being evaluated in clinical trials for the treatment of autoimmune disorders such as lupus and rheumatoid arthritis. The two atropisomeric centers in BMS-986142 are rotationally locked to provide a single, stable atropisomer that produces enhanced potency and selectivity and reduces safety liabilities (Watterson et al., 2016). A significant challenge in the development of a pure atropisomeric drug is the isolation of each atropisomer via chromatographic separation in which one-half of the final material is discarded, unlike a carbon chiral center where the enantiomeric product can be controlled by chemical synthesis (LaPlante et al., 2011a,b).
Previously, Wang et al. (2017) published a study on the characterization of lesinurad atropisomers by elucidating the absolute configuration of lesinurad ester via X-ray crystallography for the (+) and (−) assignment; the ester bonds were subsequently hydrolyzed to obtain (+) and (−) lesinurad as solutions. The authors further characterized lesinurad metabolism (in vitro studies) and animal pharmacokinetics (PK), in which lesinurad atropisomers were stable in rat and human plasma at 37°C up to 2 hours. In addition, the two atropisomers showed similar permeability and physiochemical properties but exhibited species-specific differences in PK in rats and monkey (Wang et al., 2017).
The objectives of this investigation were to further elucidate the chemical properties of lesinurad atropisomers, characterize in vitro stereoselective metabolism based on metabolite formation and stereoselective inhibitory effects against uric acid reabsorption transporters (URAT1 and OAT4), and assess human PK differences following oral administration of multiple doses of lesinurad given once daily to healthy male subjects.
Materials and Methods
Materials
Lesinurad, its two atropisomers (atropisomer 1 and 2), and deuterated racemate lesinurad ([D6]lesinurad) were supplied by Ardea Biosciences, Inc. (San Diego, CA). All other chemicals were commercially available including acetonitrile, high-performance liquid chromatography (HPLC) or liquid chromatography/mass spectrometry grade (Fisher Scientific, Rockford, IL); 0.1% formic acid in acetonitrile and 0.1% formic acid in water, HPLC spectrophotometry grade (Honeywell International Inc., Muskegon, MI); dimethylsulfoxide (DMSO) (Fisher Scientific); 7-hydroxycoumarin (Sigma-Aldrich, St Louis, MO); formic acid (Fisher Scientific); magnesium chloride (MgCl2) solution (1 M) (Sigma-Aldrich); β-nicotinamide adenine dinucleotide phosphate, reduced form (NADPH) (Sigma-Aldrich); potassium phosphate buffer powder (Sigma-Aldrich); methyl tert-butyl ether (Sigma-Aldrich); valpromide (Sigma-Aldrich); and water, HPLC grade (EMD Millipore, Burlington, MA).
Human liver microsomes (HLM) (ultrapooled, mixed gender), recombinant human cytochrome P450 isoforms CYP2C9, and recombinant CYP3A4 (supersomes) were purchased from Corning Life Sciences (Corning, NY). Pooled human plasma was purchased from Bioreclamation IVT (Westbury, NY).
Analytical Method for Separation of Atropisomers
Analysis of atropisomers was carried out using normal phase chiral chromatography consisting of an amylose-based chiral column (Daicel CHIRALPAK IA-3µm 4.6 × 100 mm; West Chester, PA) and an Agilent (Santa Clara, CA) 1100 pump system connected to a UV diode array detector set at 226 nm. Separation of the atropisomers was achieved using an isocratic elution with mobile phase methyl tert-butyl ether and 0.1% trifluoroacetic acid at 35°C. The run time was 10 minutes and the retention times of lesinurad atropisomer 1 and atropisomer 2 were 2.7 and 4.2 minutes, respectively. The naming convention of lesinurad atropisomers in this paper was based on the order of elution as noted previously.
Interconversion of Atropisomers
Individual atropisomers of lesinurad were exposed to a variety of conditions to ascertain whether they readily interconvert.
Thermal Challenge.
The atropisomers were challenged in both organic and aqueous media to evaluate interconversion. Solutions of individual atropisomers at a concentration of 0.25 mg/ml in acetonitrile were held at 60°C for 4 days. Individual atropisomers of lesinurad were dissolved in dilute sodium bicarbonate solution. The resulting solutions were exposed to room temperature, 60°C, and 100°C for 4 days.
Biologic Condition.
Individual atropisomers were evaluated for interconversion in biologically relevant conditions in the presence of buffer or human serum. First, 2 µl of each lesinurad atropisomer solution prepared in DMSO at 40, 4.0, or 0.4 mM was mixed with 1998 µl of Tris buffer (50 mM, pH 7.4) or human serum to make 40, 4.0, or 0.4 μM incubation solutions, respectively. The prepared solutions were incubated at 37°C for predetermined time intervals. Aliquots (100 µl) of each incubation solution were taken immediately after mixing or at predetermined time points (0.5, 1, 4, 8, and 24 hours) and mixed with 300 µl of chilled acetonitrile. For serum samples, the precipitated proteins were removed by centrifugation at 3300g for 15 minutes at 4°C, and the supernatant fractions (Tris and serum) were transferred to 1.5-ml centrifuge tubes and concentrated to dryness using a DNA120 SpeedVac (Thermo Electron Corporation, Milford, MA) on the high concentration setting. Acetonitrile (100 μl) was added to the tube, and the tube was sonicated for 5 minutes. The tube was then centrifuged for 2 minutes, and the resulting supernatant was transferred to HPLC vials for analysis.
Effect of Excess of One Atropisomer on the Ratio of Atropisomers.
To investigate the atropisomer ratio in the presence of excess of one atropisomer, a sample of 10 g of racemate lesinurad was spiked with 0.5 g of atropisomer 1. This blend was dissolved and then reprocessed using the standard crystallization conditions as follows. Racemic lesinurad and pure atropisomer 1 (5% or 10%) were dissolved in a solution of 2.60 g of 50% aqueous NaOH in 53 ml of water (1.05 Eq) at ambient temperature, and a clear solution was confirmed. The solution was warmed to 35°C and ethyl acetate (116 ml, 11 volumes) was added. Under rapid stirring, 48% HBr solution was added at 35°C until a pH of 2.5 was achieved. The layers were separated and the ethyl acetate solution of the product was subjected to the exact final processing steps of the current manufacturing technique. The solution was slowly distilled under reduced pressure (60 mm Hg) from a 300 ml jacketed flask and at a jacket temperature of approximately 20°C until a volume of 79 ml (7.5 volumes) was achieved after 1 hour and 37 minutes. The mixture was then warmed to 38°C and stirred for 4.5 hours, during which time crystallization was observed. Next, the suspension was distilled slowly under reduced pressure (75 mm Hg) and at a jacket temperature of approximately 20°C until a final volume of 42 ml (four volumes) was achieved after 45 minutes. The mixture was again warmed to 38°C and heptane (10.5 ml, one volume) was slowly added. The mixture was allowed to stir for 1 hour at this temperature and then cooled slowly to 8°C and stirred for 2 hours at this temperature. The suspension was drained from the reactor and immediately filtered through a medium-porosity glass frit. The initial mother liquor was used twice to rinse the reactor walls, and this addition material was also filtered. The filter cake was washed once with a chilled (5–10°C) mixture of ethyl acetate (7.7 ml) and heptane (7 ml). The filter cake was suction dried and then dried at 50°C under high vacuum overnight to afford 8.84 g (88% yield) of white, crystalline solid.
Plasma Protein Binding Assay
In vitro binding of lesinurad atropisomer 1, atropisomer 2, and lesinurad racemate to human plasma protein was evaluated by equilibrium dialysis using a rapid equilibrium dialysis device (Thermo Scientific Waltham, MA). Stock solutions of lesinurad atropisomer 1, atropisomer 2, and lesinurad racemate at 1.0, 10.0, and 50.0 mM in DMSO were spiked into control human plasma at 1:1000 to make the fortified plasma samples at 1.0, 10.0, and 50.0 µM, respectively. The Teflon base and the fortified plasma samples were separately preincubated at 37°C for 10 minutes. The rapid equilibrium dialysis device inserts were then added to the base, and 500 µl of phosphate buffer solution (pH 7.4) was added to the buffer chamber and 300 µl of fortified plasma samples was added into the sample chamber (indicated by the red ring on the insert). All protein binding determinations were conducted in triplicate. The equilibrium dialysis apparatus was covered with sealing tape and incubated in a shaking water bath at 37°C and 300g for 20 hours. After incubation, aliquots (150 μl) of plasma sample or phosphate buffer (pH 7.4) were removed from each chamber and mixed with equal volume of control phosphate buffer (pH 7.4) or blank plasma. A 50-µl aliquot of these samples was extracted with 150 µl of acetonitrile containing 20.0 ng/ml of [D6]lesinurad as the internal standard. After mixing, the precipitated proteins were removed by centrifugation. A 100-µl aliquot of the supernatant was mixed with an equal volume of water and then analyzed by high-performance liquid chromatography–tandem mass spectrometry (LC-MS/MS) as described previously (Shah et al., 2018).
In Vitro Metabolism Studies to Characterized Lesinurad Atropisomers
Stereoselective Metabolism of Lesinurad Atropisomers by Recombinant Human CYP2C9, Recombinant CYP3A4, and HLM.
Time and protein linearity were established and incubation conditions were optimized for evaluation of the metabolism of lesinurad atropisomers. Human liver microsomes (0.5 mg/ml), CYP2C9 supersomes (100 pmol/ml), or CYP3A4 supersomes (100 pmol/ml) were incubated in duplicate at 37°C in 0.2 ml (final volume) of incubation mixtures containing potassium phosphate buffer (100 mM, pH 7.4), MgCl2 (3 mM), NADPH (1 mM), and lesinurad atropisomer (1, 10, or 50 µM) at the final concentrations indicated. Stock solutions of atropisomers were prepared with DMSO at 100 mM. The final concentration of DMSO in the incubation was at or less than 0.05%. Reactions were initiated by the addition of NADPH solution and terminated at a predetermined time point (30 minutes) by the addition of 300 µl of stop reagent (chilled acetonitrile containing 0.1 µM of 7-hydroxycoumarin as an internal standard). After incubation, microsomal proteins were precipitated by centrifugation at 3300g for 15 minutes at 4°C. Supernatant fractions were analyzed by LC-MS/MS.
Incubations of Lesinurad Atropisomers with Human Liver Microsomes in the Presence of Microsomal Epoxide Hydrolase Inhibitor Valpromide.
Human liver microsomes (0.5 mg/ml) were incubated in duplicate at 37°C in 0.2 ml (final volume) of incubation mixtures containing potassium phosphate buffer (100 mM, pH 7.4), MgCl2 (3 mM), NADPH (1 mM), lesinurad atropisomer (1, 10, or 50 µM), and valpromide (0, 50, or 100 µM) at the final concentrations indicated. Reactions were initiated by the addition of NADPH solution and terminated at a predetermined time point (30 minutes) by the addition of 300 µl of stop reagent (chilled acetonitrile containing 0.1 µM of 7-hydroxycoumarin as an internal standard). After incubation, microsomal proteins were precipitated by centrifugation at 3300g for 15 minutes at 4°C. Supernatant fractions were analyzed by LC-MS/MS.
LC-MS/MS Methods for Metabolite Monitoring
An API4000 triple quadrupole mass spectrometer (AB Sciex, Framingham, MA) equipped with an electrospray ionization ion source was used and operated in positive TurboIonSpray mode. Selected reaction monitoring was used to monitor the precursor → product ion transitions of the mass-to-charge ratios: 404 → 386 (lesinurad), 420.2 → 402 (M3c), 438 → 402 (M4), 346 → 152.3 (M6), and 163 → 107 (7-hydroxycoumarin). Separation of lesinurad (achiral) and metabolites was achieved by a reversed phase C8 column (Luna C8 (2), 5 μm, 100 Å, 150 × 4.6 mm; Phenomenex, Torrance, CA). Mobile phases A and B were 0.1% formic acid in water and 0.1% formic acid in acetonitrile, respectively. The mobile phase elution flow rate was 0.9 ml/min with a linear elution gradient profile as follows: 0 to 1 minutes, 10% B; 1–10 minutes, 10%–90% B; 10–15.9 minutes, 90% B; 15.9–16 minutes, 90%–10% B; and 16–20 minutes, 10% B.
In Vitro Assays to Evaluate the Inhibitory Effect against Uric Acid Reabsorption Transporters
Stable Cell-Line Uptake Assays.
The generation of HEK293-hourURAT1 and HEK293-OAT4, both uric acid reabsorption transporter stable cell lines, has been described by Miner et al. (2016). Briefly, HEK293-hourURAT1 or HEK293-OAT4 were seeded onto 96-well poly-D-lysine-coated tissue culture plates at a density of 1.25 × 105 cells per well and grown at 37°C overnight. The next day, the cell culture was washed once with wash buffer (125 mM sodium gluconate, 25 mM HEPES, pH 7.4). URAT1 and OAT4 activity assays were conducted in assay buffer consisting of 25 mM 2-(N-morpholino)ethanesulfonic acid pH 5.5 [from a 1-M solution of 2-(N-morpholino)ethanesulfonic acid Sigma-Aldrich adjusted to pH 5.5 with sodium hydroxide], 125 mM sodium gluconate, 4.8 mM potassium gluconate, 1.2 mM KH2PO4, 1.2 mM MgSO4, 1.3 mM calcium gluconate, and 5.6 mM glucose. Lesinurad racemate, atropisomer 1, or atropisomer 2 wase serially diluted into assay buffer (concentration ranged from 0 to 200 μM) and preincubated with the cells for 5 minutes at room temperature in a volume of 30 μl. URAT1-expressing cells were incubated with 500 µM 14C-uric acid for 10 minutes and OAT4-expressing cells were incubated with 75 µM 6-carboxyfluorescein (Life Technologies, Inc., Carlsbad, CA) for 10 minutes. For 14C-uric acid, cells were washed three times in 25 mM of 2-MES acid (pH 5.5/125 mM sodium gluconate), and solubilized in Ultima Gold (Perkin Elmer, Waltham, MA) prior to liquid scintillation counting. For 6-carboxyfluorescein, cells were lysed by adding 150 µl of 1 N NaOH to each well and incubating at 37°C for 1 hour. Plates were then read on an Analyst HT plate reader (Molecular Devices, San Jose, CA). Each treatment was measured in triplicate.
Evaluation of the Stereoselective Pharmacokinetics of the Lesinurad Atropisomers
Study Design and Sample Collection.
A specific clinical study to evaluate the stereoselective PK of lesinurad atopisomers was not conducted; clinical samples (plasma and urine) from a previously conducted clinical study were used for this assessment (Shen et al., 2017). The clinical study is described as follows: A multiple-dose, open-label study conducted at a single center in 11 healthy male subjects (four Caucasian, six African American, and one Hispanic) with a median age of 31 years (range, 20–52 years) and median weight of 83 kg (range, 59–100 kg). The study was conducted in accordance with the Declaration of Helsinki, approved by an institutional review board, and all subjects were provided voluntary informed consent. All subjects received lesinurad 400 mg oral tablet on day 1 and then were dosed daily from day 7 to day 14. Plasma samples were collected at the following time points in relation to dosing of lesinurad on days 1 and 14: predose (within 30 minutes before dosing) and at 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 10, 12, and 24 hours postdose. Urine (total catch) samples were collected on day 14 at the following time intervals: 0–6, 6–12, and 12–24 hours postdose. The plasma and urine samples used for the atropisomer PK assessment were stored under the validated storage conditions and analyzed within the time period at which the long-term stability of lesinurad in plasma and urine was established (Shen et al., 2017). For the analysis presented herein, day 14 plasma samples were analyzed.
Pharmacokinetic Analysis.
The Cmax, Cmin, and time of occurrence of maximum observed concentration were calculated from individual plasma concentration-time profiles using noncompartmental analysis in Phoenix WinNonlin (version 6.3; Certara, Princeton, NJ). The PK parameters calculated included the area under the concentration-time curve from time zero to the time of dosing interval, the half-life under steady state, the amount excreted in urine from time zero to 24 hours, renal clearance (CLr), and the fraction of urinary excretion from zero to 24 hours.
Plasma and Urine Sample Preparation and LC-MS/MS Method.
For quantitation of lesinurad atropisomers in plasma, an aliquot (70 µl) of plasma from each sample was extracted with 500 µl of acetonitrile containing 400 ng/ml [D6]lesinurad as the internal standard. After precipitated proteins were removed by centrifugation, 500 µl of the supernatant was completely dried down under nitrogen stream and reconstituted with 300 µl of methanol. A 10 μl aliquot of the extract was injected onto a chiral HPLC column (Chiralpak AS-3R, 4.6 × 50 mm, 3 μm; Chiral Technologies West Chester, PA) equipped with an Agilent 1100 HPLC pump system and running isocratic mobile phase consisting of 50% water with 0.1% formic acid (v/v) and 50% acetonitrile with 0.1% formic acid (v/v) at 0.9 ml/min. The column effluent entered the mass spectrometer and was analyzed by selected reaction monitoring as described in the achiral method (Shah et al., 2018). The calibration curves for atropisomers 1 and 2 were linear (1/x2 weighting) over the concentration range of 2.50–1000 ng/ml with a lower limit of quantitation of 2.50 ng/ml for each atropisomer.
Urine samples (25.0 µl) were extracted with 200 µl of acetonitrile containing 200 ng/ml [D6]lesinurad. Following mixing and centrifugation, 55.0 µl of the supernatant was taken, completely dried down under nitrogen stream, and reconstituted with 300 µl of methanol. The reconstituted solution was then injected onto the HPLC column and analyzed by LC-MS/MS as described previously for the plasma extracts. The calibration curves for atropisomers 1 and 2 were linear (1/x2 weighting) over the concentration range of 2.50–1000 ng/ml with a lower limit of quantitation of 2.50 ng/ml for each atropisomer.
Results
Interconversion Stability.
Atropisomerism was observed for lesinurad due to restricted rotation along the C1′–N1 bond as a result of steric hindrance between substituents on each of the aryl moieties (Fig. 1). Lesinurad atropisomers were exposed to a variety of conditions in an attempt to cause interconversion. No conversion of the atropisomers was observed for samples held at room temperature, 60°C in either organic (acetonitrile) or aqueous media (dilute sodium bicarbonate), or exposed to biologic conditions (Tris buffer or human serum at 37°C for 24 hours). Only minimal interconversion (0.2%) was observed for samples held at 100°C for 4 days. The failure to see significant (i.e., >1%) interconversion of the isolated atropisomers of lesinurad over 4 days in aqueous solution at 100°C indicates that the half-life, based on first-order kinetics, for the interconversion of the atropisomers is in excess of 275 days at 100°C (and longer at room temperature or 37°C).

Lesinurad atropisomer stereochemistry.
The single-crystal X-ray structure of lesinurad (Fig. 2) showed that the individual atropisomers alternate within the lattice such that each S-atropisomer is associated with an R-atropisomer and vice versa. This resulted in a 50:50 mixture of atropisomers within the crystal and implied that even a nonracemic mixture of atropisomers in the solution state would result in a 50:50 mixture of crystallized lesinurad. The introduction of either 5% or 10% excess of pure atropisomer to lesinurad resulted in a crystalline product that was still an approximately 50:50 mixture of atropisomers (no enrichment observed), after processing through the crystallization steps.

Lesinurad crystal structure.
Effect of Excess of One Atropisomer on the Ratio of Atropisomers.
Analysis of the solids by chiral HPLC indicated a 50.2:49.8 mixture of atropisomer 1 and atropisomer 2 with either 5% or 10% excess of atropisomer 1. Analysis of a mother liquor sample (clear, light yellow solution) indicated a 70.1:29.9 mixture of atropisomer 1 and atropisomer 2, which indicated that the 5% excess spike of atropisomer 1 was retained in the filtrate or 87.9:12.1 mixture of atropisomer 1 and atropisomer 2 and the 10% excess spike of atropisomer 1 was primarily retained in the filtrate. This result indicates that the lesinurad crystal structure acts as a control in providing a 50:50 mixture of atropisomers.
Plasma Protein Binding.
The percentages of atropisomer 1 binding to plasma proteins in pooled human plasma at 1, 10, and 50 μM were 99.0 (±0.03), 97.8 (±0.10), and 98.7 (±0.08), respectively. The percentages of atropisomer 2 binding to plasma proteins in pooled human plasma at 1, 10, and 50 μM were 98.7 (±0.16), 97.3 (±0.22), and 98.0 (±0.98), respectively. The percentages of lesinurad racemate binding to plasma protein in pooled human plasma at 1, 10, and 50 μM were 98.5 (±0.43), 97.9 (±0.06), and 98.2 (±0.49), respectively. The mean (±S.D.) percentages of atropisomer 1, atropismer 2, and lesinurad racemate binding to human plasma proteins at the three concentration levels were 98.5 (±0.57), 98.0 (±0.76), and 98.2 (±0.43), respectively. There are no significant differences in the plasma protein binding between the two atropisomers or between either one of the lesinurad atropisomers and lesinurad racemate based on t test analysis (P > 0.05).
In Vitro Metabolism of Lesinurad Atropisomers.
The structural elucidation of lesinurad metabolites has been previously described by Shah et al. (2018). The metabolites formed from lesinurad atropisomers are depicted in Fig. 3, which represents an abbreviated scheme of that reported in Shah et al. (2018). Metabolite profiles of lesinurad atropisomers in human liver microsomes are presented in Fig. 4. The formation of M6 was linear up to 50 μM; however, in contrast, the formation of M3 and M4 was not linear, indicating saturable Michaelis-Menten kinetics at high concentrations.

Pathway of oxidative metabolism of lesinurad in human liver microsomes.

Formation of metabolites M3, M3c, M4, and M6 in human liver microsomes. Data are expressed as a mean ± S.D. from four independent experiments (each experiment was conducted in duplicate).
The formation of M3, M3c, and M4 (based on the peak area ratio of analyte to internal standard) in human liver microsomal incubation was greater with atropisomer 1 than with atropisomer 2. The formation of M3 from atropisomer 1 was approximately 2-fold greater than from atropisomer 2, while formation of M3c and M4 was 8- to 12-fold greater from atropisomer 1 than atropisomer 2 over the concentration range from 10 to 50 μM. The formation of M6 favored atropisomer 2 at all concentrations evaluated. The formation of M3c in human liver microsomes with atropisomer 1 was greatly increased in the presence of the microsomal epoxide hydrolase inhibitor, valpromide, while M4 formation in HLM from atropisomer 1 was inhibited by up to 29% (Fig. 5).

M4 and M3c formation in human liver microsomes in the presence of microsomal epoxide hydrolase inhibitor valpromide. Data are expressed as a mean ± S.D. from three or four independent experiments for 100 μM inhibitor and no inhibitor, respectively (each experiment was conducted in duplicate).
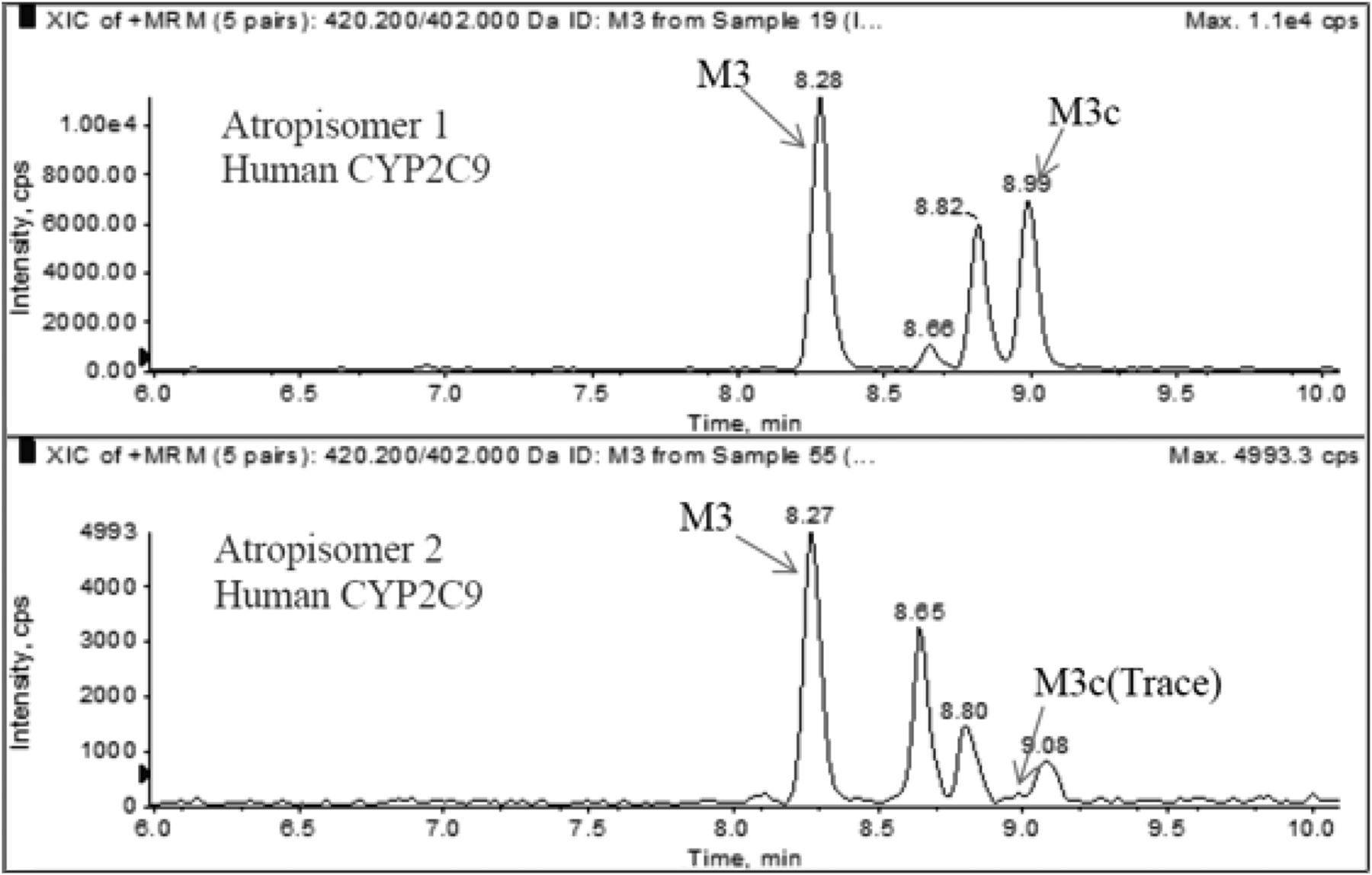
The stereoselective metabolism of lesinurad atropisomers to M3, M3c, and M6 was confirmed with recombinant human CYP2C9 and CYP3A4. M3c was readily formed in CYP2C9 incubation with atropisomer 1, and only a trace level of M3c was observed in CYP2C9 incubation with atropisomer 2 (Fig. 6). M3 formed by recombinant CYP2C9 following incubation with atropisomer 1 was approximately 2-fold greater than that observed with atropisomer 2. M6 formation by recombinant CYP3A4 was approximately 5-fold higher in incubation with atropisomer 2 than with atropisomer 1.

Representative chromatograms of M3c and M3 formed in incubations with recombinant CYP2C9 and lesinurad atropisomer.
Inhibition of URAT1 and OAT4 Transporters.
The half-maximal inhibitory concentration (IC50) of lesinurad atropisomers to inhibit URAT1 or OAT4 was measured by the decrease in 14C-labeled uric acid or 6-carboxyfluorescein uptake, respectively, with increasing concentrations from 0 to 200 µM in URAT1 or OAT4-overexpressing HEK293 cells. The IC50 value is calculated using a median of percent inhibitions from triplicate evaluations for each concentration using the sigmoidal dose-response model in XLFIT (IDBS, Alameda, CA). The average IC50 and S.E.M. values for each atropisomer were calculated for each transporter. The IC50 values for inhibition of URAT1 by atropisomers 1 and 2 were 17.3 ± 3.2 and 4.3 ± 1.1 µM, respectively. The IC50 values for inhibition of OAT4 by atropisomers 1 and 2 were 3.7 ± 0.27 and 4.4 ± 0.40 µM, respectively. Atropisomer 2 has approximately 4-fold greater inhibitory potency against URAT1 than atropisomer 1 and the two are equally active against OAT4. The results for positive control inhibitors benzbromarone for URAT1 and verinurad for OAT4 were consistent with published data (Miner et al., 2016; Tan et al., 2017).
Human Pharmacokinetics.
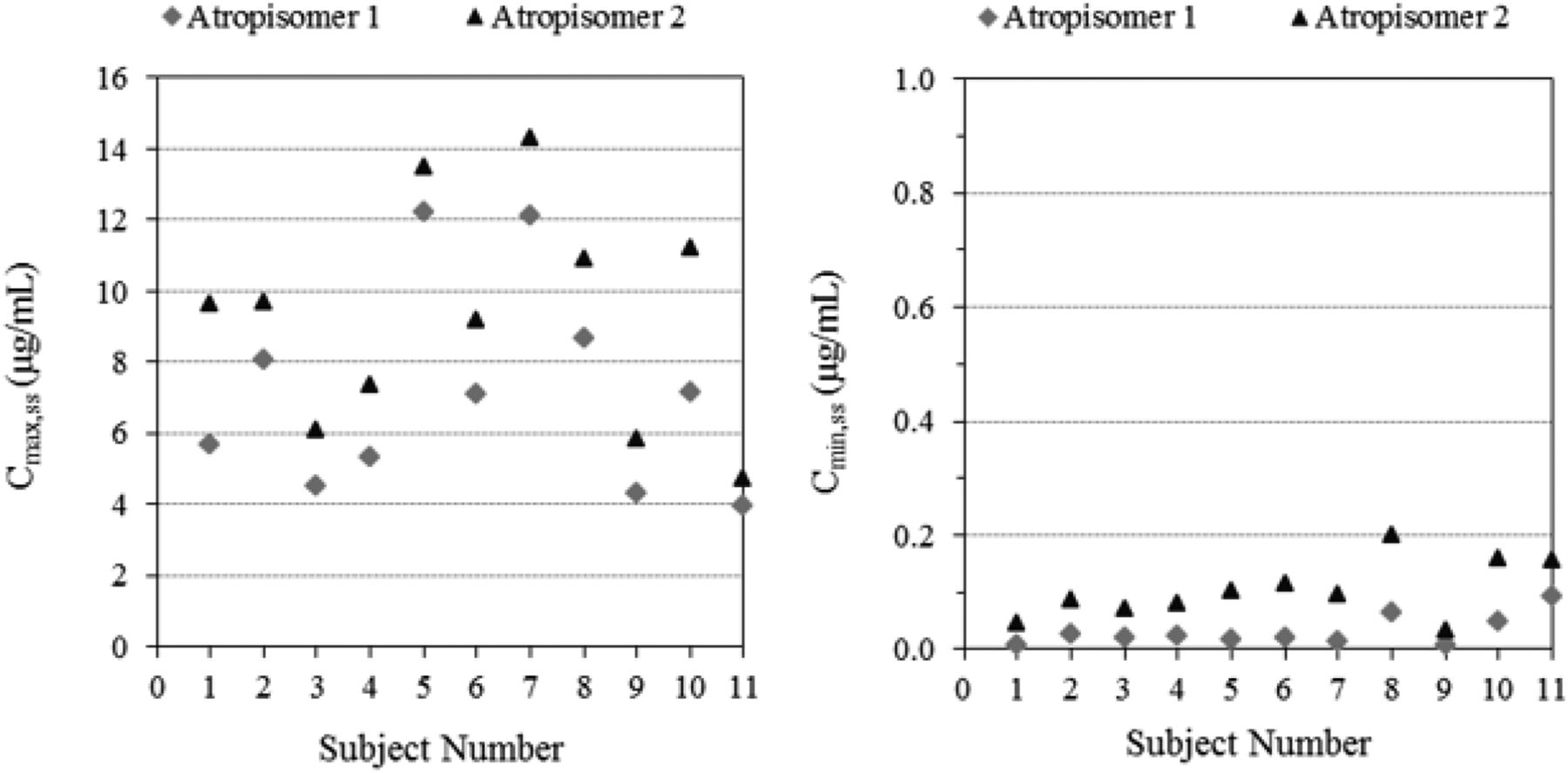
The combined concentration-time profile of the two atropisomers was nearly superimposable with the racemic profile. At each time point, the concentrations of atropisomers 1 and 2 were less than that of lesinurad racemate and the concentration of atropisomer 2 was slightly higher than that of atropisomer 1 (Fig. 7). The plasma PK parameters and urinary excretion results for lesinurad and its atropisomers are summarized in Table 1. The individual plasma maximum observed concentration at steady state (Cmax,ss) and the minimum observed concentration at steady state (Cmin,ss) for the atropisomers are presented in Fig. 8, in which all subjects showed that Cmax,ss and Cmin,ss were lower for atropisomer 1 than for atropisomer 2. The mean ratios of atropisomer 1 to atropisomer 2 were 0.76, 0.25, and 0.78 for Cmax,ss, Cmin,ss, and the area under the concentration-time curve from time zero to the time of dosing interval, respectively (Table 2). The Cmax,ss values for atropisomers 1 and 2 were not statistically different (P value = 0.1100); however, the Cmin,ss ratio was different (P value = 0.0009), which is consistent with the stereoselective metabolism dominated by CYP2C9. The urinary excretion of lesinurad atropisomers 1 and 2 was determined on day 14 (Table 2). The urinary PK results showed that the ratios of atropisomer 1 to atropisomer 2 were 0.65 for the amount excreted in urine from time zero to 24 hours and 0.84 for CLr. Urinary excretion showed that a greater amount of atropisomer 2 was excreted unchanged in the urine (fraction excreted in urine of 51% or 101 mg/200 mg dose) compared with atropisomer 1 (fraction excreted in urine of 33% or 65.9 mg/200 mg dose). It should be noted that the renal excretion of unchanged drug (fraction excreted in urine) was calculated based on an atropisomer dose equal to one-half of the administered 400 mg racemate in a 50:50 mixture of two atropisomers.

Mean plasma concentration-time profiles of lesinurad and lesinurad atropisomers 1 and 2.
Lesinurad atropisomer plasma pharmacokinetic parameters (geometric mean and 95% confidence interval)
The P values were determined using a paired t test.

Individual maximum (Cmax,ss) and minimum (Cmin,ss) plasma concentrations of atropisomers 1 and 2 at steady state.
Lesinurad atropisomer urine pharmacokinetic parameters (geometric mean and 95% confidence interval)
The P values were determined using a paired t test. The fraction excreted in urine and CLr values for each atropisomer were calculated using one-half of the racemate dose based on the fact that the racemate exists as a 50:50 mixture of two atropisomers and absorption of lesinurad was assumed to be nonstereoselective.
Discussion
Lesinurad is comprised of a 50:50 mixture of two atropisomers due to the restricted rotation of substituents on the aryl moieties along the C–N bond. The atropisomers strongly prefer a 50:50 mix in crystalline form and this ratio is resistant to alteration due to high temperatures and excess of one atropisomer over the other. The interconversion was minimal with a half-life estimated to be 275 days at 100°C.
Chiral drugs with axial chirality compared with those containing a stereogenic chiral center are less well characterized since fewer therapeutic agents exhibit this chemical trait. Many therapeutic agents are marketed as a pure enantiomer to afford greater efficacy and/or to minimize toxicities (LaPlante et al., 2011b). The racemization of atropisomers due to limitation on bond rotation is time dependent with vastly different half-lives ranging from minutes to years depending on the extent of steric hindrance, electronic influences, temperature, solvents, and conditions (Yang et al., 2018). Many compounds may have rotational hindrance around a single bond, but these are generally very short lived and not detected. Consequently, it is important to evaluate the thermal stability of chirality to ascertain restricted rotation (Hasegawa et al., 2017). Investigation of the kinetics of atropisomer interconversion using the isolated individual atropisomers of lesinurad revealed that the S- and R-atropisomers prefer a locked configuration and do not readily interconvert. The atropisomerism of lesinurad is caused by the addition of the bromine, and the lesinurad intermediate without this moiety can freely rotate (data not shown). A wide variety of conditions were tested to enhance interconversion but none led to alterations in the equilibrium, indicating a high-energy rotational barrier for conversion with a resulting half-life of 275 days at elevated temperature. Despite the stability of lesinurad atropisomers, the main challenge in commercializing only one lesinurad isomer would be the cost to perform a chiral separation in which half of the product would be discarded, unlike a stereogenic chirality in which the production of desired form is directed by chemistry synthesis (Wang et al., 2017). Thus, the cost of producing a product that requires chiral separation is likely one reason for the limited number of therapeutics with axial chirality on the market.
The lesinurad atropisomers were not individually crystallizable, thus absolute configuration was not assigned and our notation was based on its elution from a chiral column. Wang et al. (2017) determined the optical rotation based on the ester moiety of lesinurad and assigned −/+, where the more metabolic stable atropisomer (by HLM evaluation) was the (+) atropisomer, which corresponded to atropisomer 2 in our work (Wang et al., 2017).
Lesinurad is highly permeable with complete absorption after oral administration since bioavailability is 100% (Shah et al., 2018), indicating that absorption of lesinurad atropisomers was not stereoselective. Stereoselective metabolism was observed with each atropisomer in vitro based on a semiquantitative approach using peak area ratios, and stereoselective pharmacokinetics was also observed following oral administration of racemic lesinurad. The Cmax ratio (0.760) of atropisomer 1/atropismer 2 at steady state was approximately 3-fold that of the Ctrough ratio (0.250), which is consistent with the fact that Cmax is affected mostly by nonstereoselective absorption and Ctrough is affected by stereoselective metabolism. For stereogenic drugs, stereoselective metabolism is often observed, resulting in different kinetic parameters (Vmax and Km) for each enantiomer (Yamazaki and Shimada, 1997; Fjordside et al., 1999; Scordo et al., 2005; Rulcova et al., 2010). Stereoselective metabolism was also observed for lesinurad atropisomers in which CYP2C9 preferentially metabolized atropisomer 1, while CYP3A4 preferred atropisomer 2. The stereoselective metabolism by CYP2C9 implies that only one-half of the dose of lesinurad undergoes formation of the M3c epoxide metabolite intermediate such that the exposure to this metabolite is primarily associated with atropisomer 1. In vivo, due to the stereoselective metabolism of lesinurad by CYP2C9, atropisomer 1 had lower systemic exposure (Cmax,ss, Cmin,ss, and area under the plasma concentration-time curve from time zero to 24 hours) than atropisomer 2. This was also consistent with the greater amounts of atropisomer 1–related dihydrodiol metabolite (M4) excreted in the urine (data not shown). While both lesinurad atropisomers inhibited URAT1, atropisomer 2 was 4-fold more potent than atropisomer 1. Overall, stereoselective metabolism and efficacy to URAT1 were observed with the atropisomers in which the more potent inhibitor underwent less active metabolism, prolonging its activity to lower uric acid.
Inhibition of in vivo CYP2C9 metabolism may preferentially increase plasma concentrations of the less active URAT1 inhibitor atropisomer 1. This expectation was consistent with results of a fluconazole-lesinurad drug interaction study (ZURAMPIC, 2015) in which lesinurad plasma exposure, as measured by a nonchiral assay, increased approximately 56% in the presence of fluconazole (CYP2C9 inhibitor); however, there was no discernible effect of the increased lesinurad exposure on serum uric acid lowering (URAT1 inhibition). Similarly, in an identified genetic poor metabolizer (CYP2C9 genotype *3/*3; lesinurad in combination with febuxostat in gout patients), plasma lesinurad exposure (nonchiral assay) was approximately doubled compared with the mean plasma exposure in the remainder of the group; however, the urate pharmacodynamic response in the poor metabolizer subject was not obviously different from the other subjects. Thus, inhibition of CYP2C9 by a coadministered inhibitor or genetic variants will likely have limited effect on urate pharmacodynamic response to lesinurad, since alterations in metabolism affected the less potent lesinurad atropisomer.
The CLr of lesinurad was not highly stereoselective (the CLr ratio of atropisomer 1 to atropisomer 2 was 0.84) since the ratio fell within the 0.8–1.25 bioequivalence criterion. Although a somewhat larger fraction of unchanged atropisomer 2 was excreted into urine, this was attributed to higher plasma concentration and exposure of atropisomer 2. Renal impairment has been reported to decrease metabolic enzyme activity, including CYP2C9, as a result of elevated uremic proteins (Dreisbach and Lertora, 2008; Rowland Yeo et al., 2011). The effect of uremic proteins on metabolic enzyme activity may be expected to preferentially affect atropisomer 1 since it is more prone to metabolism than atropisomer 2. When considering the effects of both reduced kidney function (slightly larger effect on atropisomer 2) and uremic proteins (slightly larger effect on atropisomer 1), it is likely that both atropisomers may be elevated in plasma with a decrease in urinary excretion in patients with mild-to-moderate renal impairment. Given the opposite directional effects, it is anticipated that the atropisomer ratios in plasma and urine are not likely to be markedly different in patients with renal impairment compared with patients with normal renal function.
Characterizing the pharmacokinetics of a drug with atropisomeric properties spans the full spectrum from high to low certainty. A compound like lesinurad with very slow interconversion enables proper and accurate PK assessment of each atropisomer or accurate characterization as racemate. Each lesinurad atropisomer behaves as a separate entity since there is no interconversion in vivo or postsample collection to alter accurate concentration assessment. On the other hand, a compound with rapid interconversion that can occur in vivo and postsampling, as in the case of BMS-207940, makes it difficult to develop as a single atropisomer in clinical development. Specifically, BMS-207940 showed a low-energy barrier for interconversion in which its atropisomer was able to convert in powder form, in plasma, and at lower concentrations (Zhou et al., 2004). Interestingly, BMS-207940 was developed as a racemate (50:50) and was studied in phase I and II trials with an average terminal half-life ∼15 hours following oral administration of single and multiple doses (Zhou et al., 2004). It is unclear if BMS-207940 had equal potency toward its target. Zhou et al. (2004) have suggested that accurate PK assessment of atropisomers is also possible when interconversion is rapid such that atropisomers are indistinguishable in vivo; thereby the chirality degenerates and both atropisomers behave as a single entity. Thus, difficulties in characterizing PK arises when interconversion is neither highly rapid nor extremely slow but lies in between like the BMS compound such that assessment of the atropisomer is constantly changing.
In summary, lesinurad atropisomers showed stereoselective metabolism resulting in exposure as well as urinary excretion differences. The individual atropisomers of lesinurad exhibited stereospecific pharmacologic activity toward URAT1 but not OAT4 in modulating uric acid uptake into stable cell lines expressing these transporters. Thus, lesinurad atropisomers showed stereoselective pharmacologic activity to URAT1. The lack of interconversion of the atropisomers facilitated the assessment of the two separate entities that contributed to lesinurad’s efficacy toward URAT1 and pharmacokinetics. As such, separation of the individual atropisomers via either chiral chromatography or selective crystallization is not necessary since lesinurad has preference to coexist as a 50:50 mixture. Developing lesinurad as an individual atropisomeric drug would be costly and impractical, thus racemic lesinurad remains the ideal pharmaceutical form of the tablet product.
Authorship Contributions
Participated in research design: Yang, Zhou, Shen, Wilson, Renner, Miner, Girardet, Lee.
Conducted experiments: Yang, Zhou, Shen, Renner, Miner.
Performed data analysis: Yang, Zhou, Shen, Renner, Miner, Lee.
Wrote or contributed to the writing of the manuscript: Yang, Renner, Girardet, Lee.
Footnotes
- Received January 16, 2018.
- Accepted November 2, 2018.
Abbreviations
- BMS
- Bristol-Myers Squibb
- CLr
- renal clearance
- Cmax,ss
- maximum observed concentration at steady state
- Cmin,ss
- minimum observed concentration at steady state
- DMSO
- dimethylsulfoxide
- HLM
- human liver microsomes
- HPLC
- high-performance liquid chromatography
- LC-MS/MS
- liquid chromatography–tandem mass spectrometry
- PK
- pharmacokinetics
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}