


Abstract
Anacetrapib is an inhibitor of cholesteryl ester transfer protein (CETP), associated with reduction in LDL cholesterol and increase in HDL cholesterol in hypercholesterolemic patients. Anacetrapib was not taken forward into filing/registration as a new drug for coronary artery diease, despite the observation of a ∼9% reduction in cardiovascular risk in a large phase III cardiovascular outcomes trial (REVEAL). Anacetrapib displayed no adverse effects throughout extensive preclinical safety evaluation, and no major safety signals were observed in clinical trials studying anacetrapib, including REVEAL. However, anacetrapib demonstrated a long terminal half-life in all species, thought to be due, in part, to distribution into adipose tissue. We sought to understand the dependence of anacetrapib’s long half-life on adipose tissue and to explore potential mechanisms that might contribute to the phenomenon. In mice, anacetrapib localized primarily to the lipid droplet of adipocytes in white adipose tissue; in vitro, anacetrapib entry into cultured human adipocytes depended on the presence of a mature adipocyte and lipid droplet but did not require active transport. In vivo, the entry of anacetrapib into adipose tissue did not require lipase activity, as the distribution of anacetrapib into adipose was-not affected by systemic lipase inhibition using poloaxamer-407, a systemic lipase inhibitor. The data from these studies support the notion that the entry of anacetrapib into adipose tissue/lipid droplets does not require active transport, nor does it require mobilization or entry of fat into adipose via lipolysis.
Introduction
Agents that inhibit cholesteryl ester transfer protein (CETP) prevent the exchange of neutral lipid between high-density, low-density, and very-low-density lipoproteins (HDL, LDL, VLDL, respectively). The resultant effects of CETP inhibitors on the distribution of cholesterol among these lipoproteins, notably increases in HDL cholesterol and decreases in LDL cholesterol, led to their development as drugs that could potentially reduce cardiovascular risk. Across several clinical studies, anacetrapib, a potent inhibitor of CETP, reduced LDL cholesterol and raised HDL cholesterol in normal healthy volunteers and in hypercholesterolemic patients at high risk of cardiovascular disease on a background of statin therapy. In a large phase 3 cardiovascular outcomes trial (Randomized EValuation of the Effects of Anacetrapib through Lipid-modification, REVEAL), treatment of patients with atherosclerotic vascular disease with anacetrapib for a mean duration of 4 years led to a relative reduction of ∼9% compared with placebo in the primary composite outcome of heart attack, coronary revascularization procedure, or death from coronary heart disease, an effect that appeared to be driven by a reduction in non-HDL cholesterol (Bowman et al., 2017). Despite this reduction in cardiovascular risk, however, a decision was made based on a comprehensive evaluation of the clinical profile of anacetrapib to not proceed with regulatory filings (Merck & Co. press release, October 11, 2017).
Anacetrapib displays a long terminal half-life in preclinical species and in humans (Gotto et al., 2014; Hartmann et al., 2016). In an extension of a phase 3 clinical safety study (Determining the EFficacy and Tolerability of Cholesteryl ester transfer protein INhibition with AnacEtrapib, DEFINE), where patients were treated with anacetrapib for 18 months, low levels of anacetrapib in the range of approximately 50 nM were detectable in plasma 2.5–4.0 years after cessation of dosing (Gotto et al., 2014). We previously reported that in mice treated with anacetrapib for 6 weeks anacetrapib levels increased in white and brown adipose tissue, and during 35 weeks after cessation of dosing levels of anacetrapib in white adipose tissue remained relatively unchanged (Hartmann et al., 2016). Given the lipophilicity of anacetrapib (LogD 7.1), one possibility for the long elimination phase from plasma is that adipose tissue serves as a large depot in the body from which anacetrapib can slowly exit to the plasma compartment, but the mechanisms behind the preferential distribution of anacetrapib into adipose have not been characterized.
Our study furthers our understanding of the mechanisms behind anacetrapib distribution into adipose. We used mice to examine the entry and subcellular localization of anacetrapib into adipose in vivo, and we used human cultured adipocytes to assess active transport mechanisms of entry in vitro. We report that anacetrapib distributes into the lipid droplet of adipocytes through a mechanism that does not involve active transport or dependence on lipase-dependent mobilization of lipid.
Materials and Methods
Animals.
All testing protocols described herein were performed in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the U.S. National Institutes of Health, were approved by the Merck Research Laboratories Institutional Animals Care and Use Committee in Rahway, NJ, and adhered to the Public Health Service policy on humane care and use of laboratory animals. Mice were maintained in a 12-hour light/dark cycle with free access to food and water, in group housing conditions in a temperature-controlled environment (22°C). Male wild-type (WT) C57BL/6 mice, which were used for adipose and plasma pharmacokinetics and adipocyte localization experiments, were obtained from Taconic Farms (Germantown, NY). The mice were maintained on regular chow (7012, 5% dietary fat; 3.75 kcal/g; Teklad, Madison, WI). The mice were 12–16 weeks of age at the time the experiments were performed.
Anacetrapib Pharmacokinetics.
To understand the acute (24 hour) versus longer-term time-course of anacetrapib deposition into adipose tissue, we treated male C57BL/6 mice (N = 33) with anacetrapib (100 mg/kg per day, oral gavage, dosed as a suspension in 0.5% methycellulose) for 28 days. We euthanized n = 3 mice (CO2 asphyxiation followed by cardiac puncture) and collected blood/tissues at each of the following time points: 0.5, 1, 2, 6, and 24 hours after a single dose; days 3, 7, 10, 14, 21, and 28 during once-daily dosing (collection occurred 24 hours after the previous day’s dose). Anacetrapib levels in plasma and adipose tissue were determined as described in Hartmann et al. (2016).
Anacetrapib Localization in Mouse Adipose Tissue.
To determine the subcellular localization of anacetrapib, radiolabeled anacetrapib was prepared by the Radiolabeled Synthesis Group at Merck Research Laboratories in Rahway, NJ. This was a preparation of [1-14C-propane]-2-yl-anacetrapib, with the 14C group on the isopropyl moiety as previously described in Kuethe et al. (2013) with specific activity 0.08932 mCi/mg; 0.3204 mCi/ml; and high-performance liquid chromatography purity: 14C content = 99.64%. This is referred to hereafter as [14C]-anacetrapib.
For dosing of radiolabeled anacetrapib in mice, cold anacetrapib was initially prepared in Imwitor:Tween-80 (1:1, w:w) in a starting concentration of 10 mg/ml, and sonicated. To make enough solution for 10 mice, 0.44 ml of [14C]-anacetrapib in ethyl alcohol was added slowly to 0.4 ml of cold anacetrapib solution under continuous stirring. This preparation was then slowly (under continuous stirring) diluted with 1.16 ml of water (final cold anacetrapib concentration was 2 mg/ml, [14C]-anacetrapib was 70 μCi/ml), vortexed, and sonicated for 5 minutes. A stable emulsion was obtained. Mice were dosed via oral gavage at 5 ml/kg. Mice (average weight of 28 g) received a total dose of 10 μCi/mouse.
Twenty-four hours after dosing, the mice were euthanized by CO2 asphyxiation. Epididymal white adipose tissue (eWAT) was collected and pooled (n = 8 mice). Adipocytes were isolated, and subcellular fractionation was performed as described by Yu et al. (2000) and Bourez et al. (2012). Briefly, adipose tissue was minced and digested with collagenase (1 mg/ml in Krebs-Henseleit buffer supplemented with 2% bovine serum albumin [BSA]) for 1 hour at 37°C.
The resulting suspension was then filtered through a 100 μm mesh cell strainer and subjected to a series of washes. The floating fat layer containing the isolated adipocytes was collected and resuspended in disruption buffer (25 mM Tris-HCl, 100 mM KCl, 1 mM EDTA, 5 mM EGTA, pH 7.4, supplemented with protease inhibitors). Cells were disrupted by nitrogen cavitation at 450 psi for 15 minutes at 4°C, after which the cavitate was collected dropwise and transferred to a polyallomer sealable tube.
Nuclei were pelleted by spinning at 1500g for 10 minutes. The top fat layer was removed via a tube slicer and the infranatant was collected away from the nuclei pellet. The fat layer and the infranatant were each mixed with an equal volume of disruption buffer containing 1.08 M sucrose and then overlaid sequentially with 2 ml each of 270 mM sucrose buffer, 135 mM sucrose buffer and Top Solution (25 mM Tris-HCl, 1 mM EDTA, 1 mM EGTA, pH7.4).
After centrifugation for 1 hour at 150,000g in a SW 41 Ti rotor (Beckman Coulter, Brea, CA), eight 1.5-ml fractions were collected from top to bottom. The resulting microsomal pellet as well as the nuclei pellet collected earlier were washed and resuspended in 1.5 ml of top solution by sonication, 1.0 ml of which was added to Ultima Gold scintillation cocktail (PerkinElmer, Waltham, MA) and counted on a scintillation counter.
Immunoblotting.
Protein from cellular fractions was concentrated and collected from spin columns (Amicon Centrifugal Filter Device; Millipore, Billerica, MA) using the manufacturer’s instructions. Proteins were isolated from buoyant lipid fractions using methods described by Brasaemle and Wolins (2016), and the resultant pellet was resuspended in 2× SDS sample buffer and incubated at 60°C for 4–6 hours in a sonicating water bath. Five micrograms of protein per fraction were run under denaturing conditions on 12% Tris-glycine gels. After transfer to nitrocellulose membranes, blots were blocked with Odyssey Blocking Buffer (LI-COR, Lincoln, NE) for 1 hour at room temperature. Membranes were probed with primary antibodies against either perilipin (Fitzgerald Industries International, Acton, MA) or GLUT4 (Abcam, Cambridge, MA) diluted in blocking buffer with 0.2% Tween 20. Blots were washed in phosphate-buffered saline (PBS) with 0.1% Tween 20 three times for 5 minutes before incubation with the appropriate IRDye secondary antibody (LI-COR) diluted in blocking buffer with 0.2% Tween 20 and 0.1% SDS. After another round of washes, the protein signals were visualized, and the intensities of the bands were measured using Odyssey Imager (LI-COR). Intensity readings were converted to percentage of total intensity in graphical representations.
Cell Culture Experiments.
Human preadipocytes (hPADs, pooled as a mix of unknown gender) were obtained from Cell Applications (San Diego, CA). Preadipocytes were thawed from frozen stocks (received frozen stocks at passage 5–10) and plated into 24-well plates for differentiation at ∼90,000 cells/well. The cells were cultured in growth medium, which contained Dulbecco’s modified Eagle’s medium (DMEM)/Ham’s F-12 medium, 3.3 nM biotin, 1.7 mM pantothenate, and 10% fetal bovine serum. Adipocyte differentiation was initiated by placing the cells in Quick Differentiation Medium, composed of DMEM/Ham’s F-12 medium, 3.3 mM biotin, 1.7 mM pantothenate, 0.01 mg/ml transferrin, 20 nM insulin, 100 nM cortisol, 0.2 mM T3, 25 mM dexamethasome, 250 μM 3-isobutyl-1-methylxanthine (IBMX), and 2 μM rosiglitazone. After 4 days of differentiation induction, the cells were cultured in 3FC medium, which contains DMEM/Ham’s F-12 medium, 3.3 mM biotin, 1.7 mM pantothenate, 0.01 mg/ml transferrin, 20 mM insulin, 100 mM cortisol, and 0.2 mM T3 for up to an additional 10 days.
The cells were typically studied between 10 and 14 days after differentiation with 70%–90% fully differentiated. For experiments involving ATP depletion, on the day of the experiment the cells were pretreated for 1-hour with either control medium (serum-free culture medium containing 0.2% BSA), or serum- and glucose-free medium supplemented with 0.2% BSA and ATP depletion agents (1 μM rotenone plus 1 μM antimycin A, and 10 mM 2-deoxy-d-glucose) (Wick et al., 1957; Palmer et al., 1968; Giudicelli et al., 1977; Bashan et al., 1993; Kim et al., 1999). The concentrations of ATP-depletion agents were determined as the minimum concentrations, in combination, that blocked radiolabeled adenosine uptake (as discussed later) in adipocytes without impacting cell viability during the treatment and uptake protocol.
After ATP depletion, [2,8-3H]-adenosine from American Radiolabeled Chemicals (St. Louis, MO) and [14C]-labeled anacetrapib were used to investigate transporter-mediated substrate uptake. For adenosine uptake, a final concentration of 2 μCi/ml of [2,8-3H]-adenosine mixed with 10 μM of nonlabeled adenosine was used for uptake experiments. A specific adenosine transporter inhibitor, S-(4-nitrobenzyl)-6-thioinosine (NBMPR; Sigma-Aldrich, St. Louis, MO) (Aronow et al., 1985), at 0.1 μM was used as a positive control for adenosine uptake blockade.
A final concentration of 1.44 μCi/ml of [14C]-anacetrapib mixed with 10 μM of nonlabeled anacetrapib was used for anacetrapib uptake experiments. In these experiments, cells were treated with radiolabeled anacetrapib (triplicate replicate wells in a 24-well plate), the medium was collected, and the cells were washed 3 times with PBS followed by lysis with 0.5 ml of 0.1 N NaOH. The lysate and medium were stored in −20°C until ready for analysis.
Three hundred μl of medium and lysate were counted on a liquid scintillation counter in 3 ml of Ultima Gold (PerkinElmer). In some experiments, the uptake of radiolabeled anacetrapib was compared among the mature adipocytes, preadipocytes, and a nonadipocyte cell line (CHO-K1; ThermoFisher, cultured in DMEM/Ham’s F-12 medium + 10% fetal bovine serum).
Effects of Lipase Inhibition on Anacetrapib Plasma and Adipose Pharmacokinetics In Vivo.
To examine the role of lipase in the localization of anacetrapib to adipose tissue, male C57BL/6 mice were dosed with either PBS or poloxamer 407 (P-407) to inhibit hepatic, endothelial, and lipoprotein-associated lipases (Wasan et al., 2003; Millar et al., 2005; Johnston, 2010) (1000 mg/kg, intraperitoneal injection) 1 hour before they were orally dosed with anacetrapib at 100 mg/kg. We euthanized n = 4 mice as described earlier, and plasma and epididymal white adipose tissue were collected for measurement of anacetrapib concentration in plasma and tissue, and measurement of plasma lipids. Plasma triglyceride and total cholesterol were measured using triglyceride and total cholesterol kits from Wako Diagnostics (Mountain View, CA), according to the manufacturer’s instructions.
Statistical Analysis.
For experiments involving two factors, two-way analysis of variance followed by Sidak multiple comparison tests was performed (GraphPad Software, San Diego, CA). For individual pairwise comparisons, Student’s t test was performed (GraphPad Software).
Chemicals and Compounds.
Reagents were obtained from commercial sources as indicated. Anacetrapib was synthesized and prepared in Medicinal Chemistry Department at Merck & Co., Inc. (Rahway, NJ). radiolabeled anacetrapib synthesis is described above (page 2).
Results
Anacetrapib Localizes to the Lipid Droplet of Adipocytes.
After oral dosing of anacetrapib in mice (100 mg/kg), the levels of anacetrapib reached a maximum of approximately 10 μM in white adipose tissue and 4 μM in plasma after a single dose (Fig. 1A), but dropped to approximately 3.0 and 0.1 μM by 24 hours, respectively. During 28 days of dosing, the levels of anacetrapib in white adipose tissue remained at approximately 2 μM, and the plasma levels remained between 0.1 and 0.2 μM (Fig. 1B).

Pharmacokinetics of anacetrapib in plasma and epididymal white adipose tissue (eWAT) of male C57Bl/6J mice (n = 3 mice per time point). Anacetrapib concentrations were determined in plasma and epididymal white adipose tissue (eWAT) (A) 30 minutes to 24 hours after a single oral dose (100 mg/kg) and (B) for 28 days of once-daily dosing (100 mg/kg, oral gavage), with tissue/plasma collection occurring 24 hours after the previous dose (trough). Data points represent the mean ± S.E.M.
Because anacetrapib entered adipose tissue readily within 24 hours, acute dosing could be used to examine the localization of anacetrapib in adipose tissue using a radiolabeled compound. A separate group of mice were treated with [14C]-anacetrapib to track the location of anacetrapib-associated radioactivity in adipose tissue.
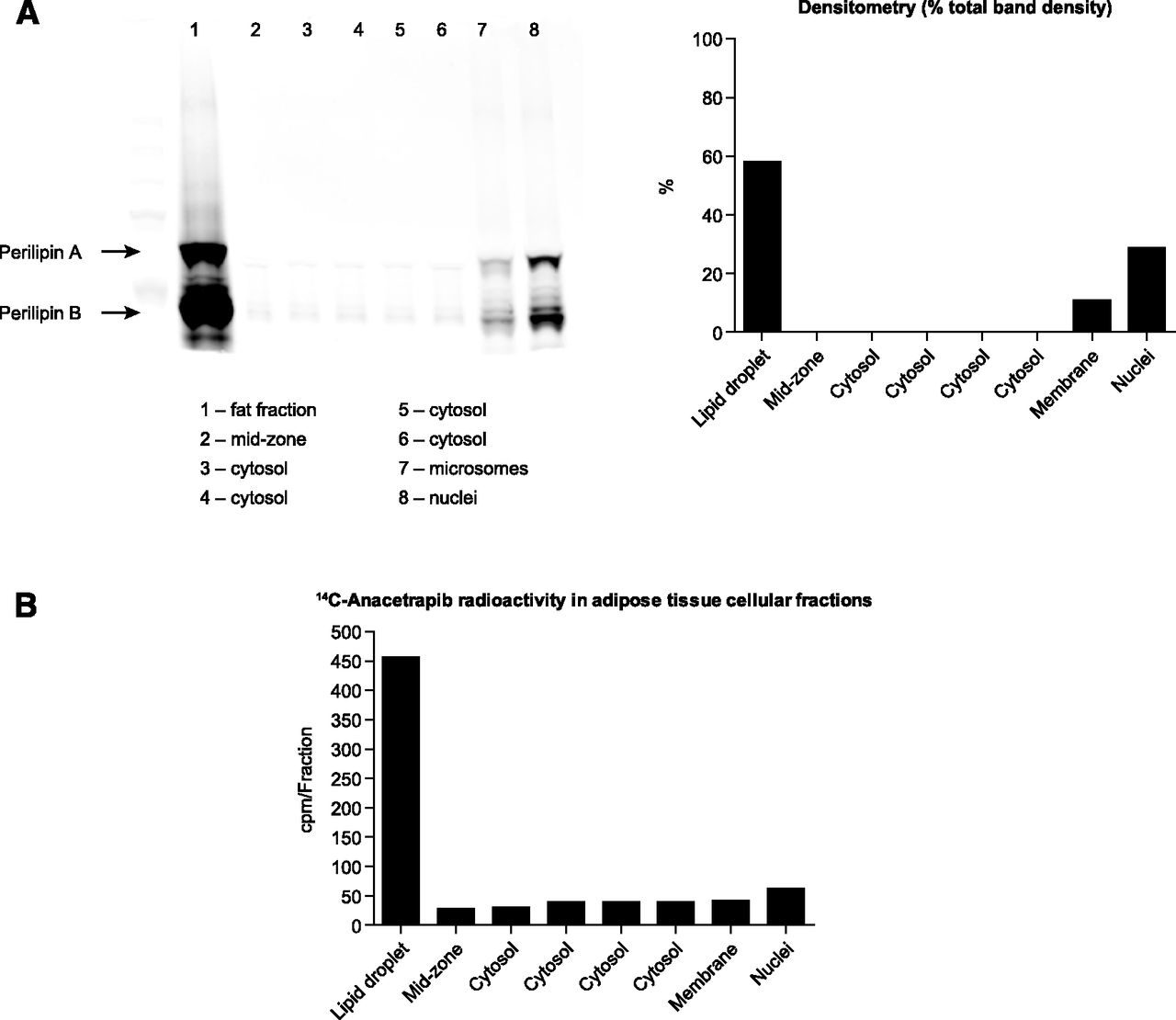
After density-gradient ultracentrifugation of adipose tissue, we used the lipid droplet-associated protein perilipin (Brasaemle, 2007) to confirm the identity/purity of the fraction containing the lipid droplet (Fig. 2A). The majority of [14C]-anacetrapib-associated radioactivity colocalized in the same fraction as perilipin, which suggested that the site of anacetrapib deposition in adipose tissue is the lipid droplet (Fig. 2B).

Localization of radiolabeled anacetrapib to the lipid droplet of adipocytes from mice treated with [14C]-anacetrapib (pooled adipose samples from n = 8 mice). (A) Left panel: Immunoblot of perilipin A and B, showing the greatest signal in the fat fractions, with small amounts located in the microsome and nuclear fraction. Right panel: Densitometry of perilipin signal. (B) Graph depicting radioactivity associated with subcellular fractions of adipose tissue from WT mice treated for 24 hours with 10 μCi of [14C]-anacetrapib (in a dosing solution of cold anacetrapib to deliver 10 mg/kg total), indicating the greatest amount of anacetrapib-associated radioactivity occurring in the lipid droplet fraction.
Entry of Anacetrapib into Cultured Human Adipocytes Is Facilitated by Presence of Lipid Droplet, but Does Not Require Active Transport.
To further our understanding of the mechanisms of entry of anacetrapib into adipocytes, cultured human adipocytes were differentiated from preadipocytes into mature adipocytes as described in Materials and Methods, and the uptake of [14C]-anacetrapib was examined before and after differentiation. The maturation of the adipocytes was confirmed by the presence of a lipid droplet. After a 48-hour incubation with radiolabeled anacetrapib, the intracellular levels of [14C]-anacetrapib-associated radioactivity were greater in the mature/differentiated adipocytes compared with preadipocytes (Fig. 3), suggesting the lipid droplet increases anacetrapib partitioning and further supporting the lipid droplet as the primary site of anacetrapib deposition. Uptake of radiolabeled anacetrapib was also compared between mature adipocytes and Chinese hamster ovary (CHO) cells, which served as a nonadipocyte cell type. The intracellular levels of radiolabeled anacetrapib were significantly greater in mature adipocytes compared with CHO cells (Fig. 3).

Entry of [14C]-anacetrapib in human cultured adipocytes, compared with preadipocytes and nonadipocyte cells (CHO-K1 cells). Cells were treated with 1.44 μCi/ml of [14C]-anacetrapib mixed with 10 μM of nonlabeled anacetrapib and cell lysates were counted in a scintillation counter. Anacetrapib-associated radioactivity increased in a time-dependent manner to a greater extent in mature adipocytes (○) compared with preadipocytes (●) and CHO cells (▴). aP < 0.05 comparing adipocytes to preadipocytes. bP < 0.05 comparing adipocytes to CHO cells. Data points represent the mean cpm ± S.E.M. from triplicate replicates from one of three experiments with similar results.
To determine whether the uptake of anacetrapib requires active transport, we treated cells with a cocktail of compounds to inhibit synthesis of ATP (NaN3, rotenone, antimycin A, and 2-deoxyglucose). Blockade of active transport was confirmed by the inhibition of adenosine uptake (Fig. 4A). Under the same conditions that blocked adenosine uptake, anacetrapib uptake was unaffected (Fig. 4B), indicating that the entry of anacetrapib into adipocytes does not require active transport.

Blockade of active transport does not impair entry of [14C]-anacetrapib into adipocytes. (A) Blockade of radiolabeled adenosine transport into adipocytes by 1 μM rotenone, 1 μM antimycin A, and 10 mM 2-deoxy-d-glucose (2-DG) confirms active transport blockade under these conditions. S-(4-nitrobenzyl-6-thioinosine), an adenosine transporter antagonist, was used as a positive control for blockade of adenosine uptake. ***P < 0.001 versus vehicle for rotenone/antimycin/2-DG treatment. (B) Treatment of adipocytes with rotenone/antimycin/2-DG had no effect on time-dependent [14C]-anacetrapib uptake (left panel) or concentration-dependent [14C]-anacetrapib uptake (right panel). *P < 0.05 versus control. Data points represent the mean cpm ± S.E.M. from one of three independent experiments with similar results.
Mechanism of Anacetrapib Entry into Adipose Is Independent of Lipase Activity.
Given that anacetrapib-associated radioactivity was found to primarily localize to the lipid droplet of adipocytes, we performed experiments to determine whether mobilization of fatty acids by lipase-activity plays a role in the initial uptake of anacetrapib into adipose tissue. WT mice were treated with a single dose of anacetrapib in the presence or absence of 1000 mg/kg of P-407 to inhibit hepatic, endothelial, and lipoprotein-associated lipases (13–15). The P-407 treatment was associated with a large increase in plasma lipids (Fig. 5A), consistent with inhibition of lipase and subsequent prevention of fatty acid mobilization and transfer from blood to tissues. The plasma levels of anacetrapib were significantly increased in the P-407-treated mice compared with the PBS-treated controls (Fig. 5B), and the plasma anacetrapib levels followed a similar time course as plasma triglyceride. Despite this large difference in plasma anacetrapib levels between mice treated with P-407 and PBS, the levels of anacetrapib in adipose tissue were not different between each treatment group, except at the 0.5- and 1-hour time points where the adipose levels of anacetrapib were actually higher in the animals treated with P-407 (Fig. 5B).

Lipase inhibition does not impair entry of anacetrapib into adipose tissue in mice. Male WT mice were treated with 100 mg/kg anacetrapib 1 hour after pretreatment with P-407 (1000 mg/kg), and plasma and epididymal white adipose tissue (eWAT) were collected at various time points over 24 hours. (A) Plasma triglyceride (TG, left panel) and plasma total cholesterol (TC, right panel) were significantly increased after P-407 treatment of mice, compared with PBS control. (B) Plasma (left panel) and eWAT (right panel) concentrations of anacetrapib after P-407 treatment. *P < 0.05; **P < 0.01; ***P < 0.001 versus PBS control. Each data point represents the mean ± S.E.M. from n = 3 animals.
Discussion
The long terminal half-life of anacetrapib is thought to be due prolonged retention in adipose tissue. We previously reported sustained levels of anacetrapib in white adipose tissue for at least 35 weeks after cessation of dosing in mice (Hartmann et al., 2016); in humans, low levels of anacetrapib persist in plasma for years after dosing (Gotto et al., 2014). A recent report by Krishna et al. (2017) described the accumulation of anacetrapib during dosing and persistence of anacetrapib in adipose tissue after dosing in human subjects, similar to what was observed in mice. Taken together, these observations support the notion that retention and very slow elimination from adipose tissue explains both the long terminal plasma half-life and the persistence of anacetrapib levels in plasma years after dosing is stopped.
In the current study, anacetrapib entered adipose tissue quickly in mice, reaching a maximum concentration (∼10 μM) approximately 2 hours after a single 100 mg/kg dose, falling to a trough level at 24 hours which remained stable for the 28-day dosing period. Harvesting and fractionating white adipose tissue from [14C]-labeled anacetrapib-treated mice suggested that anacetrapib resided predominantly in the lipid droplet, evidenced by colocalization of anacetrapib-associated radioactivity with perilipin, a protein marker that is present in the lipid droplet fraction. Furthermore, in cultured cells, the accumulation of anacetrapib-associated radioactivity was greatest in cells containing a mature lipid droplet (i.e., human differentiated adipocytes) compared with undifferentiated/preadipocytes or CHO cells, which do not contain a lipid storage vacuole.
The preference of anacetrapib for neutral lipid is not unexpected, given that anacetrapib is a neutral molecule and its lipid partition coefficient (log D) is approximately 7.1, a value associated with high lipophilicity (Krishna et al., 2017). However, the mechanism by which anacetrapib enters the adipocyte is unclear. It is important to note that due to the location of the radiolabel on [14C]-anacetrapib, the radioactivity measured in the subcellular localization experiments may not represent 100% anacetrapib as a parent molecule and could include radiolabeled metabolites of anacetrapib. Given that the unlabeled anacetrapib that accumulates in adipose tissue to micromolar concentrations, as reported in Fig. 1, and is measured as “parent” anacetrapib, the majority of radioactivity measured in the radiolabeled anacetrapib experiments is assumed to be parent. However, it cannot be ruled out that a proportion of the radioactivity reported in the subcellular lipid droplet fraction, and in the cell lysate of cultured cells, could be metabolites of anacetrapib.
Krishna et al. (2017) reported that the majority of plasma anacetrapib in humans is carried in lipoproteins. Although some anacetrapib is expected to be bound to CETP in the lipoprotein fraction (specifically HDL), as described by Ranalletta et al. (2010), the concentrations of plasma/lipoprotein-associated anacetrapib, as described by Krishna et al. (2017), are higher than the reported plasma concentrations of CETP in humans (∼25 nM) (Marcel et al., 1990). Therefore it is likely that lipoprotein-associated anacetrapib (not bound to CETP) is carried in the neutral lipid core. The WT mice used in this study lack CETP; therefore, presumably, the anacetrapib carried in the lipoprotein fraction is located in the neutral lipid core of the particle. The use of CETP-deficient/WT mice to study anacetrapib distribution into adipose tissue is justified by our previous report that the lack of CETP has no effect on the ability of anacetrapib to accumulate into adipose tissue (Hartmann et al., 2016).
If lipoproteins “deliver” anacetrapib to adipose tissue, then mechanisms responsible for mobilization and transport of fatty acids into adipose for storage (i.e., active transport, lipase-mediated lipolysis) could also be responsible for transport of anacetrapib into adipocytes. The cocktail of rotenone, antimycin A, and 2-deoxyglucose was used to inhibit ATP production from mitochondrial electron transport/oxidative phosphorylation, and glycolysis (Wick et al., 1957; Palmer et al., 1968; Giudicelli et al., 1977; Bashan et al., 1993; Kim et al., 1999), at the minimum concentrations found to maximally block adenosine uptake into adipocytes, a process dependent upon ATP-mediated active transport (Gray et al., 2004; Molina-Arcas et al., 2009). Active transport blockade had no effect on anacetrapib entry into cells, indicating that anacetrapib must enter these cells via a mechanism independent of ATP.
Poloaxamer 407 (P-407) inhibits systemic lipase activity in vivo (Wasan et al., 2003; Millar et al., 2005; Johnston, 2010), and pretreatment of mice with P-407 resulted in a large increase in plasma triglyceride, evidence that lipase-mediated lipolysis (and therefore uptake of lipid) was inhibited. This increase in plasma lipid was accompanied by large increases in plasma anacetrapib, which is consistent with the notion that since lipoproteins are responsible for carrying neutral lipid (i.e., triglyceride, cholesterol ester) in plasma, the capacity of lipoproteins to carry anacetrapib is increased with increased lipoprotein-associated triglyceride, given the high logD of anacetrapib. This observation is also consistent with our previous report in mice, where plasma anacetrapib levels were higher in obese/hyperlipidemic WT mice (diet-induced obesity) compared with lean WT controls (Hartmann et al., 2016).
If lipase-mediated lipolysis was involved in the mobilization and entry of anacetrapib into adipose tissue, a reduction in adipose anacetrapib would have been observed. However, no reduction in adipose anacetrapib levels was observed. Taken together, these data suggest that anacetrapib does not follow a path into adipose tissue similar to the mechanisms known to mediate fatty acid storage. Candidate mechanisms for anacetrapib entry into adipose tissue include passive diffusion or holoparticle uptake of lipoproteins via scavenger receptors; however, these mechanisms require additional study.
In conclusion, anacetrapib rapidly partitions into adipose tissue and localizes into the lipid droplet component of the adipocyte via a process independent of active transport or lipase activity. Because lipolysis does not play a role in the mobilization of anacetrapib from lipoprotein, it is interesting to speculate that the reverse process, lipolysis of triglyceride to mobilize fatty acids from adipose to the plasma compartment, is also not involved in mobilization of anacetrapib from these deep tissue stores. Although additional study is required to identify the precise mechanisms of anacetrapib entry and egress from adipose tissue, the findings from the studies described herein could partially explain the extremely slow elimination of anacetrapib from adipose and the persistence of anacetrapib in the plasma of patients years after cessation of dosing.
Authorship Contributions
Participated in research design: Johns, LeVoci, Lu, Wang.
Conducted experiments: LeVoci, Krsmanovic, Lu, Xu, Wang, Chen.
Contributed new reagents or analytic tools: Xu.
Performed data analysis: Johns, LeVoci, Krsmanovic, Lu, Xu, Wang, Chen.
Wrote or contributed to the writing of the manuscript: Johns, Lu, Hartmann, Bateman, Blaustein.
Footnotes
- Received September 14, 2018.
- Accepted November 29, 2018.
This research was supported by Merck & Co., Inc., Kenilworth, NJ.
Abbreviations
- BSA
- bovine serum albumin
- CETP
- cholesteryl ester transfer protein
- CHO
- Chinese hamster ovary cells
- DMEM
- Dulbecco’s modified Eagle’s medium
- HDL
- high-density lipoprotein
- LDL
- low-density lipoprotein
- PBS
- phosphate-buffered saline
- P-407
- poloxamer 407
- VLDL
- very-low-density lipoprotein
- WT
- wild type
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}