


Abstract
Protein kinase inhibitors (KIs), which are mainly biotransformed by CYP3A4-catalyzed oxidation, represent a rapidly expanding class of drugs used primarily for the treatment of cancer. Ligand- and structure-based methods were applied here to investigate whether computational approaches may be used to predict the site(s) of metabolism (SOM) of KIs and to identify amino acids within the CYP3A4 active site involved in KI binding. A data set of the experimentally determined SOMs of 31 KIs known to undergo biotransformation by CYP3A4 was collated. The structure-based (molecular docking) approach employed three CYP3A4 X-ray crystal structures to account for structural plasticity of this enzyme. Docking pose and SOM predictivity were influenced by the X-ray crystal template used for docking and the scoring function used for ranking binding poses. The best prediction of SOM (77%) was achieved using the substrate (bromoergocryptine)-bound X-ray crystal template together with the potential of mean force score. Binding interactions of KIs with CYP3A4 active site residues were generally similar to those observed for other substrates of this enzyme. The ligand-based molecular superposition approach, using bromoergocryptine from the X-ray cocrystal structure as a template, poorly predicted (42%) the SOM of KIs, although predictivity improved to 71% when the docked conformation of sorafenib was used as the template. Among the web-based approaches examined, all web servers provided excellent predictivity, with one web server predicting the SOM of 87% of the data set molecules. Computational approaches may be used to predict the SOM of KIs, and presumably other classes of CYP3A4 substrates, but predictivity varies between methods.
Introduction
Protein kinases catalyze the phosphorylation of proteins, an essential post-translational signaling mechanism that influences numerous cellular processes. Abnormal regulation of kinase signaling cascades is known to cause a myriad of diseases, including cancer. For instance, mutation of kinase receptors (e.g., the epidermal growth factor receptor and vascular endothelial growth factor receptor families) and downstream signaling proteins (e.g., ras sarcoma oncoproteins, rapidly accelerated fibrosarcoma kinase, mitogen-activated extracellular signal-regulated kinase, mitogen-activated protein kinase etc.) is associated with constitutive pathway activation, uncontrolled cell proliferation, and malignancy (Rowland et al., 2017). Protein kinase inhibitors (KIs) compete with ATP at the ATP-binding site of the kinase, thereby preventing phosphorylation of the target protein (Duckett and Cameron, 2010). Consequently, strategies to target both the active and inactive conformations of protein kinases have been used to develop inhibitors. However, specific targeting to a single kinase has proved challenging. Nonetheless, KIs have led to significantly improved outcomes for several difficult-to-treat malignancies, such as renal cell and hepatocellular carcinomas (e.g., sorafenib, axitinib), metastatic melanoma (e.g., vemurafenib, dabrafenib), HER-2–positive breast cancer (e.g., lapatinib), and pancreatic cancer (e.g., sunitinib) (Druker et al., 2006; Burstein et al., 2008; Chapman et al., 2011; Zhu et al., 2011; Ferraro and Zalcberg, 2014; Faivre et al., 2017; Hochhaus et al., 2017).
As of November 2016, the Food and Drug Administration had approved 31 KIs for clinical use (Rowland et al., 2017; Zhang et al., 2017), although several others have been approved since that time (e.g., brigatinib, neratinib) and more are in clinical development (Wu et al., 2015, 2016). It is thus anticipated that the number of KIs available for the treatment of cancer and certain other diseases (e.g., rheumatoid arthritis, idiopathic pulmonary fibrosis) will increase in the future.
The metabolism of KIs occurs primarily in the liver and gastrointestinal tract. Although multiple enzymes have been implicated in KI biotransformation (Rowland et al., 2017), CYP3A4 is the main enzyme involved in the metabolism of the majority of drugs in this class (Table 1). Consequently, CYP3A4 activity may influence KI clearance and, hence, dosage and response. Moreover, clinically significant drug-drug interactions have been reported for several KIs when simultaneously dosed with CYP3A4 inhibitors or inducers (Duckett and Cameron, 2010; Keller et al., 2018). In addition, the CYP3A4-catalyzed biotransformation of several KIs (e.g., erlotinib, dasatinib, gefitinib, and lapatinib) is known to result in the formation of reactive metabolites capable of covalently binding to cellular macromolecules (Duckett and Cameron, 2010). Despite the importance of CYP3A4 in KI metabolism, the binding of these drugs within the CYP3A4 active site, and how this determines the site of metabolism (SOM), has received little attention. Numerous computational approaches have been developed for SOM prediction. The in silico methods provide a useful starting point for the characterization of drug-metabolism pathways and complement experimental data (Tarcsay and Keseru, 2011; Kirchmair et al., 2012; Ford et al., 2015).
Contribution of CYP3A4 and other enzymes to the metabolism of kinase inhibitors based on biotransformation data from in vivo and in vitro studies
CYP3A4 is a major drug-metabolizing enzyme which is responsible for the metabolism of approximately 30% of clinically used drugs (Zanger and Schwab, 2013; Rendic and Guengerich, 2015). This enzyme is abundantly expressed in the liver and, in addition to KIs, metabolizes drugs from numerous therapeutic classes, including benzodiazepines (e.g., alprazolam, midazolam), macrolide antibiotics (e.g., clarithromycin, erythromycin), calcium channel blockers (e.g., diltiazem, verapamil), carbamazepine, and human immunodeficiency virus-1 protease inhibitors (e.g., indinavir, saquinavir). A feature of CYP3A4 substrates is the diverse range of sizes (molecular mass) and shapes. CYP3A4 has a large active site, ranging in volume from 950 to 2000 Å3 in the absence and presence of bound ligand (Yano et al., 2004; Ekroos and Sjögren, 2006). Not surprisingly, X-ray crystal structures demonstrate that CYP3A4 displays significant structural plasticity to accommodate structurally diverse ligands (Sevrioukova and Poulos, 2013, 2017). Almost 40 X-ray crystal structures of human CYP3A4 have been published to date. Three of the structures were crystallized without a bound ligand (1W0E, 1TQN, 4I3Q), while five structures have been cocrystallized with a substrate [viz., progesterone, bromoergocryptine (two independent structures), midazolam, and erythromycin A] and 31 with an inhibitor, mostly ritonavir analogs. Despite advances in CYP3A4 structure function, there is limited structural information on how KIs are recognized by this enzyme and which domains within the active site are associated with substrate recognition and binding.
The objectives of the present study were to 1) collate SOM data for kinase inhibitors metabolized by human CYP3A4 from published in vivo and in vitro studies, 2) investigate whether molecular docking of KIs in selected CYP3A4 X-ray crystal structure(s) and ligand-based alignment approaches using the structure cocrystallized with bromoergocryptine may be used as templates to predict the SOM of Kis, 3) evaluate existing web-based methods for SOM prediction of KIs metabolized by CYP3A4, and 4) identify amino acids important for KI binding within the CYP3A4 active site from the docking analyses.
Materials and Methods
KI Data Set.
A data set of 31 marketed KIs was collated (Fig. 1). Information on the SOMs of the KIs metabolized by human CYP3A4 was obtained from the published literature and from product information (Glivec, 2001; Iressa, 2003; McKillop et al., 2004; Gschwind et al., 2005; Ling et al., 2006; Nexavar, 2006; Sutent, 2006; Tarceva, 2006; Sprycel, 2007; Tykerb, 2007; Christopher et al., 2008b; Minami et al., 2008; Tasigna, 2008; Shilling et al., 2010; Votrient, 2010; Castellino et al., 2012; Speed et al., 2012; Zelboraf, 2012; Bershas et al., 2013; Caprelsa, 2013; Deng et al., 2013; Jakavi, 2013; Stivarga, 2013; Tafinlar, 2013; Xalkori, 2013; Bosulif, 2014; Ding, 2014; Dowty et al., 2014; Ho et al., 2014; Iclusig, 2014; Mekinist, 2014; Smith et al., 2014; Goldinger et al., 2015; Imbruvica, 2015; Johnson et al., 2015; Lacy et al., 2015; Loi et al., 2015; Scheers et al., 2015; Xeljanz, 2015; Zydelig, 2015; Abbas and Hsyu, 2016; Cotellic, 2016; Dickinson et al., 2016; Dubbelman et al., 2016; Lenvima, 2016; Tagrisso, 2016; Takahashi et al., 2016; Zykadia, 2016; Alecensa, 2017; Ibrance, 2017; Inlyta, 2017; Morcos et al., 2017; Ye et al., 2017; Cabometyx, 2018; Gerisch et al., 2018). Additionally, data from in vitro studies with human liver microsomes, hepatocytes, and/or recombinant CYP3A4, where available, were used for confirmation of metabolite formation and the role of CYP3A4 in metabolite formation (Christopher et al., 2008a; Wang et al., 2008; Ghassabian et al., 2012; Stopfer et al., 2012; Inoue et al., 2014; Lawrence et al., 2014; Jin et al., 2015; Zientek et al., 2016; Nakagawa et al., 2018). The major SOM was identified as the predominant primary metabolite(s), determined from in vivo data (fecal/urine excretion and/or plasma concentrations). In cases where multiple major sites of metabolism were reported, all sites were considered. Precise sites of metabolism were not identified experimentally for some of the data set molecules (alectinib, ceritinib, cobimetinib, ponatinib); in these cases, the SOM(s) was taken as a substructure within the molecule (e.g., morpholine, phenyl rings, etc.) where biotransformation was reported (Fig. 1).



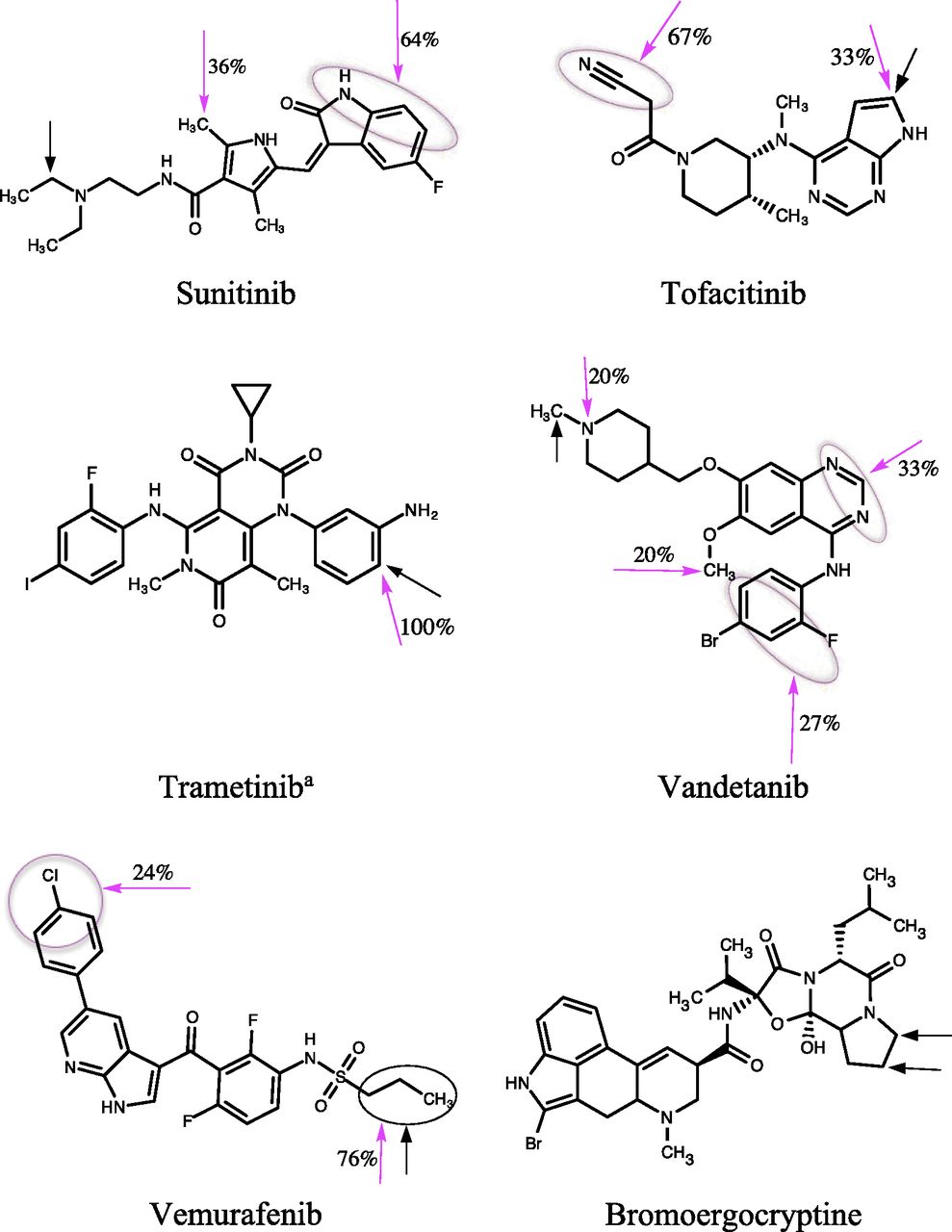
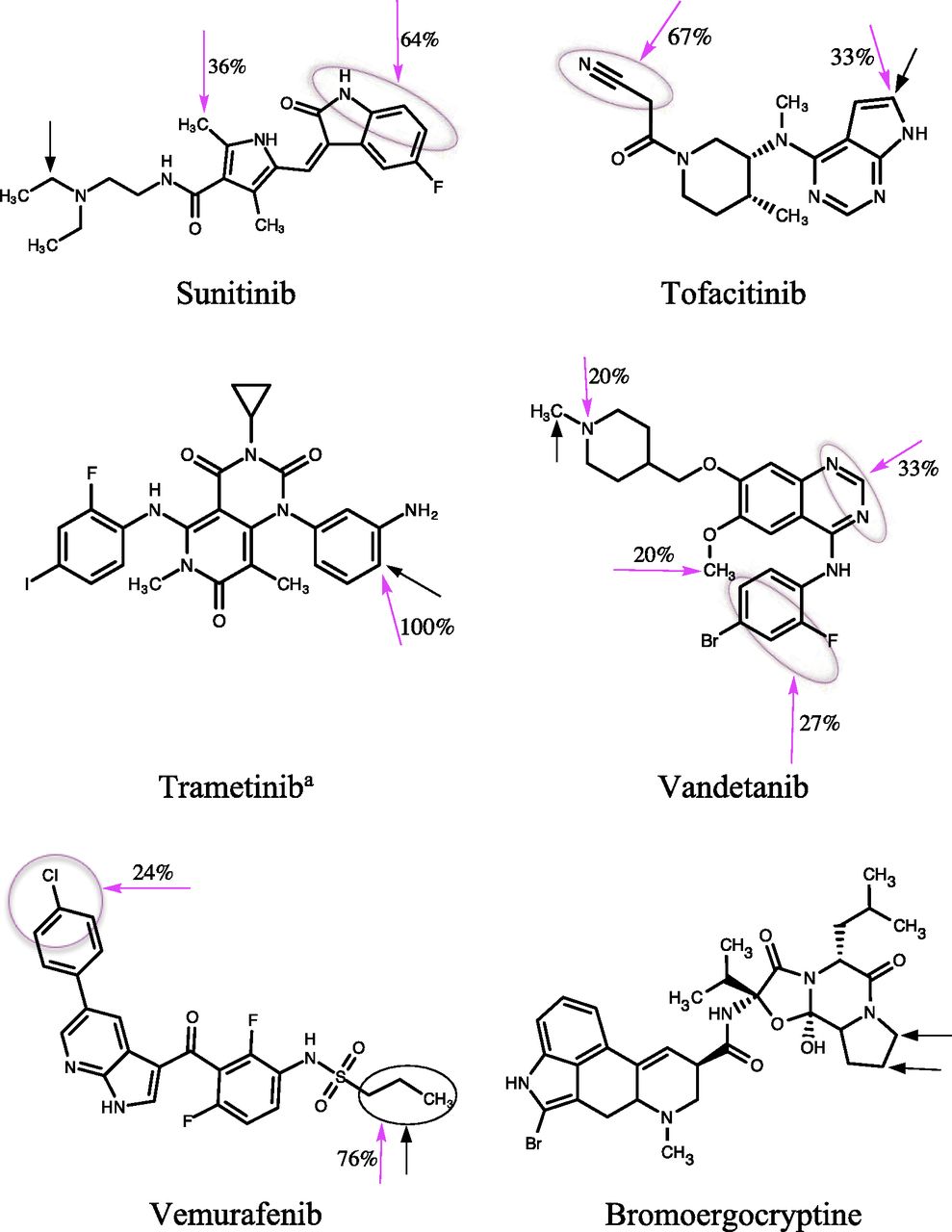
Chemical structures of KIs and bromoergocryptine. The experimentally known major SOMs of KIs are shown by black arrows, and the predicted SOM by magenta arrows. The number adjacent to the magenta arrow shows the ratio (expressed as a percentage) of the number of docked poses with the indicated SOM relative to the total number of successful docked poses (see Results: Molecular Docking). Black and magenta circles represent multiple sites identified from experimental and docking predictions, respectively. aThe deacetylated product was investigated in this study.
CYP3A4 appears not to be the main enzyme involved in afatinib and nintedanib metabolism. The major metabolites of afatinib are apparently formed nonenzymatically (by Michael addition), although in vitro studies indicate some involvement of FMO3 and CYP3A4 in the metabolism of this drug (Giotrif, 2013). Nintedanib is primarily metabolized by esterases and UGT1A1, with a lesser contribution of CYP3A4 (Ofev, 2015; Roth et al., 2015).
CYP3A4 Structure and Molecular Modeling.
Three X-ray crystal structures were selected for docking studies to take into account the plasticity of the CYP3A4 protein. These included the 5VCC structure, which was obtained from a codon-optimized cDNA and resolved to 1.7 Å (the highest resolution achieved thus far for a CYP3A4 protein); the 3UA1 structure, cocrystallized with bromoergocryptine (a substrate); and the ritonavir (an inhibitor)-bound structure, 5VCO (http://www.rcsb.org/). The 5VCC structure was cocrystallized with glycerol and ethylene glycol (from crystallization medium) but not with a drug substrate or inhibitor. Thus, it is subsequently referred as the “unliganded” structure. The CYP3A4 structures were prepared for docking studies by adding unresolved residues/atoms, H atoms, and assigning Kollman all-atom charges to the protein data bank (pdb) structures using the biopolymer module of SYBYL X-2.1 (Certara, Princeton, NJ).
The three-dimensional coordinates (structure data format) of data set molecules were obtained from the PubChem server (https://pubchem.ncbi.nlm.nih.gov/). The molecules were imported into SYBYL (version X-2.1) and assigned a protonation state at pH 7.4, calculated using the calculator plugins implemented in ChemAxon (https://www.chemaxon.com). The structures were additionally assigned Gasteiger-Huckel partial atomic charges (Gasteiger and Marsili, 1980) and energy minimized by Powell’s conjugate gradient method using the Tripos force field (Powell, 1977). A minimum energy difference of 0.001 kcal/mol was set as the convergence criterion. All molecular modeling was performed using SYBYL installed on a Linux workstation running the Red Hat Linux 6.9 operating system.
Molecular Docking Calculations.
Docking experiments were conducted using the Surflex-Dock docking suite (Jain, 2003), which combines Hammerhead’s empirical scoring function with a molecular similarity method (morphologic similarity) to generate putative poses of ligand fragments. First, an idealized binding site is generated from the protein structure by placing three different types of molecular fragments (CH4, C=O, and NH) into putative sites in multiple positions, which are then optimized for binding to the protein. High-scoring nonredundant fragments, collectively referred to as the “protomol,” serve as a target for alignment of ligands (or ligand fragments) based on molecular similarity. The ligand to be docked is fragmented, and each fragment is conformationally searched and aligned to the protomol to yield poses that maximize molecular similarity to the protomol. The aligned fragments are scored and pruned on the basis of the scoring function and the degree of protein interpenetration (Jain, 2003).
The putative binding-site protomol for ligand docking was calculated for the three CYP3A4 structures described previously. Ligands and water molecules were removed from the structures before protomol creation and docking calculations. The protomol for the 5VCC structure was defined automatically, whereas the respective cocrystallized ligands were used as a reference for generating the protomol with the 5VCO and 3UA1 structures. The active site, which includes the heme porphyrin moiety, was identified as the principal site for docking by the protomol. The docking module Surflex-Dock GeomX (SFXC) was used for all docking calculations.
The aim of the docking studies was to assess whether the first-ranked pose identified by each of the scoring functions can accurately predict the experimentally determined major SOMs for KIs biotransformed by CYP3A4 (i.e., only the top-ranked binding pose obtained from different scoring functions was evaluated for prediction of the experimentally identified SOM, which essentially tests scoring accuracy). Surflex-Dock predicts the ligand binding poses based on the total score (as the default). However, binding poses based on other scoring functions [D-score, potential of mean force (PMF)-score, G-score, ChemScore, and consensus score (CScore)] can also be evaluated using this program [Kuntz et al., 1982; Eldridge et al., 1997; Jones et al., 1997; Muegge and Martin, 1999; Clark et al., 2002; SYBYL X-2.1 (Certara)].
The D-score is derived from the molecular docking algorithm DOCK, which is based on the charge and van der Waals interactions between the protein and the ligand (Kuntz et al., 1982). Here, the van der Waals energy is calculated with a Lennard-Jones 12-6 potential, and the electrostatic energy is calculated with the Coulombic equation. The PMF-score is drawn from the work of Muegge and Martin (1999), who analyzed a large set (697) of protein-ligand complexes drawn from the Protein Data Bank and developed a set of Helmholtz free energies of interactions for protein-ligand atom pairs. The ChemScore is an empirical scoring function that includes terms for hydrogen bonding, metal-ligand interactions, lipophilic contacts, and rotational entropy, along with an intercept term (Wang et al., 2003). This scoring function was originally calibrated by reproducing the measured dissociation constants of 82 protein-ligand complexes (Eldridge et al., 1997). The G-score is based on the scoring function implemented in the molecular docking program GOLD [Cambridge Crystallographic Data Centre (CCDC) Software Limited] and is the sum of three terms: protein-ligand complexation, hydrogen bonding, and internal energy (Jones et al., 1997). In this scoring function, the complexation term is calculated with a reparameterized Lennard-Jones 8-4 potential, while the hydrogen bonding term is the sum of the individual energies from all donor-acceptor pairs. The internal energy of the ligand includes a dispersion-repulsion energy and a torsional energy calculated according to the Tripos force field. The G-score was validated by testing the scoring function of a set of 100 protein-ligand complexes. Total score is the Surflex-Dock scoring function, which is an empirically derived scoring function based on the binding affinities of protein-ligand complexes from a set of X-ray crystal structures. The scoring function primarily comprises hydrophobic, polar, entropic, and solvation terms (Jain, 1996). The CScore allows integration of different scoring functions for ranking the affinity of ligands bound to the active site of a receptor. The strengths of individual scoring functions combine to produce a consensus that is more robust and accurate than any single function for evaluating ligand-receptor interactions (Clark et al., 2002). In this study, the CScore was derived based on the consensus of D-score, PMF-score, ChemScore, and G-score.
The binding poses of the docked substrates were considered positive if the experimentally known SOM occurred within 6 Å from the Fe atom of the heme moiety, as reported by others (de Graaf et al., 2007; Vasanthanathan et al., 2010). Ligands docked away from the active site or with an SOM distance >6 Å from the Fe atom were considered to be in an unproductive pose, and the docking was classified as unsuccessful.
Molecular Superposition by Morphologic Similarity.
In addition to the molecular docking method, we also examined the molecular superposition approach to predict the SOM of the data set molecules. The structural superposition (or overlay) of molecules was undertaken using the Surflex-Sim program (Jain, 2000, 2004), which utilizes the morphologic similarity approach to generate alignments of molecules. Similarity is defined as a Gaussian function of the differences in the molecular surface distances of two molecules at weighted observation points on a uniform grid. The computed surface represents distances to the nearest atomic surface and distances to donor and acceptor surfaces. Bromoergocryptine (BEC) from the 3UA1 CYP3A4 X-ray structure was used as a template for the molecular superpositioning of the data set molecules, as other substrate-bound CYP3A4 structures were considered suboptimal for the comparison of the aligned data set molecules with respect to the CYP3A4 structure. In particular, in one structure, the substrate (progesterone) bound at a site distant from heme, and binding of the other structure (midazolam) caused substantial alteration in the active site. Neither of these substrates can be used as templates because they cannot be aligned efficiently for SOM prediction. In addition to BEC, we also used the docked conformation of three KIs with differing chemical structures, molecular masses, and shapes—namely, tofacitinib, sorafenib, and lapatinib (see Results and Discussion)—as templates for the molecular superpositioning of data set molecules.
Web-Based SOM Prediction.
In addition to molecular docking and molecular superposition, three web servers were assessed for SOM prediction: MetaPrint2D (www-metaprint2d.ch.cam.ac.uk/metaprint2d/), Regioselectivity (RS)-WebPredictor 1.0 (http://reccr.chem.rpi.edu/Software/RS-WebPredictor/), and XenoSite (http://swami.wustl.edu/xenosite/). MetaPrint2D utilizes a database of atom environments based on the biotransformation of xenobiotics from a curated database extracted from the literature (Carlsson et al., 2010). SOM prediction is achieved by calculating the atom environments within a molecule, which are then searched and compared for similar environments in the molecular database. An occurrence ratio is calculated for each atom in the molecule by measuring how often a similar environment has been found at a reaction center relative to how many times it has been observed in total. Ratios generated in this manner represent the relative likelihood of metabolism occurring at each atom, which is scaled and normalized to an occurrence ratio of 1. In MetaPrint2D, the predicted metabolic sites/atoms are represented by a color code, indicating the probability of biotransformation. The most probable SOM is shown in red and the least probable in gray, with probability values ranging from 0 to 1 [red (0.66–1), orange (0.33–0.66), green (0.33–0.15), white (0.15–0.00), and gray (little/no data)]. Thus, multiple probable major SOMs may be identified for some molecules. Where five-color codes are not noted for a molecule, the major predicted SOM is based on the probability scale of red to gray. For example, if the site(s) of metabolism is predicted by green and gray codes only (e.g., dabrafenib, ibrutinib), then the positions coded green would be considered as the major predicted SOM based on the probability scale.
RS-WebPredictor is a quantitative structure activity relationship-based method for predicting the SOM by specific cytochrome P450 enzymes that takes into account the regioselectivity of substrate metabolism (Zaretzki et al., 2012). The models used for predictions are trained using a combination of topological, quantum chemical (atom-pair based), and “SMARTCyp reactivities” applied to substrate sets (i.e., substrate databases) of individual cytochrome P450 enzymes (1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A4). Models are generated using MIRank, a support vector machine–like ranking and multiple instance learning method specifically designed to correctly rank metabolophores associated with oxidized SOM(s) over metabolophores of nonoxidized SOMs on the same substrate.
XenoSite uses a machine-learning approach based on neural networks for predicting the SOM of substrates of cytochrome P450 enzymes (Zaretzki et al., 2013). Each atom in a molecule is associated with a vector of numbers, with each number encoding a chemical property of the SOM. This approach is somewhat similar to RS-WebPredictor, which also uses a combination of several descriptors (topological, quantum chemical, SMARTCyp reactivity, refined quantum chemical, molecule-level, and fingerprint similarity), which are analyzed by machine-learning algorithms. The server predicts a score for each atom, which ranges from 0 to 1, that can be interpreted as a probability or statistical likelihood of metabolism occurring at a particular atom. The major SOM of the data set molecules is identified based on a color-coded probability scale.
Results
Kinase Inhibitors and Their Sites of Metabolism.
In general, KIs are metabolized by CYP3A4 to produce numerous products, several of which are classified as “major” metabolites. Hence, in many cases, multiple major SOMs were considered. A precise site of primary biotransformation was unavailable for some drugs, such as the morpholine ring in alectinib and the piperidine ring in crizotinib, which have multiple potential oxidation sites (Fig. 1). These regions were considered for SOM predictions. Approximately 50 primary biotransformation pathways for the 31 KI substrates were collated (Fig. 1).
Molecular Docking.
Given the structural flexibility of CYP3A4, molecular docking was performed using three CYP3A4 X-ray crystal structures as templates. The binding poses of the 31 KIs based on the distance criterion (viz., within 6 Å of the heme iron) for the prediction of SOM were evaluated using six scoring functions. In total, 558 binding poses for the data set molecules were assessed, which comprised 18 individual poses of each KI obtained from docking in the three different templates. The number of data set molecules predicted correctly by each scoring function in the X-ray crystal structures is shown in Tables 2, 3, and 4 and Fig. 2.
Prediction of the SOM of KIs by molecular docking in the 5VCC structure using different scoring functions
Prediction of the SOM of KIs by molecular docking in the 3UA1 structure using different scoring functions
Prediction of the SOM of KIs by molecular docking in the 5VCO structure using different scoring functions

Number of KIs for which the major SOM was correctly predicted by molecular docking using the six scoring functions: 5VCC as the template structure for docking (A), 3UA1 as the template structure for docking (B), and 5VCO as the template structure for docking (C).
The PMF-score and CScore predicted the experimentally known major SOM of 23 and 21 of the 31 data set molecules, respectively, docked in the unliganded X-ray crystal structure 5VCC (Fig. 2; Table 2). By contrast, the total score and G-score predicted the major SOM of 18 data set molecules. The docked poses ranked by D-score were poorer still, predicting the SOM of 16 KIs. Docking in the 5VCC (unliganded) structure failed to generate binding poses (using four or more scoring functions) of nine KIs (viz., afatinib, alectinib, crizotinib, dabrafenib, imatinib, nintedanib, palbociclib, ruxolitinib, and sunitinib) that predicted the experimentally determined SOM.
SOM prediction using the 3UA1 structure (CYP3A4 cocrystallized with bromoergocryptine) provided better outcomes compared with docking in the 5VCC structure. The best prediction was obtained with the PMF-score and total score, with each scoring function predicting the SOM of 24 (77%) data set molecules (Fig. 2; Table 3). As with docking in the 5VCC structure, the D-score performed poorly in ranking binding poses in the 3UA1 structure with the experimentally known SOM. The majority of the failed predictions from ranking with the D-score either docked the KI distant from the active site or showed a binding orientation where the major SOM was >6 Å from the heme. Molecular docking in the 3UA1 structure resulted in fewer failed predictions. The major SOM of only six KIs (afatinib, crizotinib, dabrafenib, imatinib, palbociclib, and sunitinib) was predicted inaccurately by four or more scoring functions.
Docking in the CYP3A4 structure (5VCO) cocrystallized with ritonavir provided slightly better predictivity compared with the 5VCC structure but not SOM predictions obtained from docking in the 3UA1 structure (Fig. 2; Table 4). Use of 5VCO as the template failed to predict the SOM of nine KIs (bosutinib, crizotinib, dabrafenib, imatinib, nintedanib, osimertinib, palbociclib, tofacitinib, and vandetanib). The PMF-score predicted the SOM of 21 data set molecules, whereas the G-score and CScore each correctly predicted the SOM of 20 KIs. In contrast to the 3UA1 and 5VCC structures, the D-score performed relatively better and correctly predicted the SOM of 19 data set molecules. The docked conformations of the data set molecules in the 5VCC structure were more widely distributed due to the larger active site compared with the substrate-bound structure. Of note, prediction of the SOM of four KIs (crizotinib, dabrafenib, imatinib, and palbociclib) consistently failed with all three X-ray crystal templates. Overall, the PMF-score provided best predictivity of the known SOMs of the data set KIs with all three CYP3A4 structures.
The ratio (expressed as a percentage) of the number of docked poses with a specific SOM relative to the total number of successful docked poses obtained from the six different scoring functions was calculated to identify the most probable SOM for each KI (Fig. 1). All binding orientations of KIs were taken into account for calculating the ratio, with the exception of those that were classified as unproductive (i.e., docked at a distant site or SOM >6 Å from heme). The ratios correctly predicted the experimentally known major SOM of 24 (77%) KIs (Fig. 1). Furthermore, the docking-based SOM prediction was useful for predicting the metabolic sites of those molecules whose experimental SOM was poorly defined (e.g., ceritinib, cabozantinib, and ponatinib). Several binding poses with multiple SOMs were predicted for osimertinib and vandetanib, both of which are known to undergo extensive metabolism mediated by CYP3A4 (Fig. 1).
To investigate the role of individual amino acids in KI binding, CYP3A4-ligand binding interactions were explored based on a consensus docked pose (maximum number of poses with a common SOM) from different scoring functions with the 3UA1 (substrate-bound) structure. The docked complexes were analyzed by the protein-ligand interaction profiler (PLIP) server (https://projects.biotec.tu-dresden.de/plip-web/plip/index) to classify the types of interactions between the data set molecules and individual amino acids within the CYP3A4 active site (Salentin et al., 2015). Representative docked poses (bosutinib, dasatinib, ibrutinib, and sorafenib) are shown in Fig. 3. For each of the four substrates, the SOM [represented as a sphere(s)] is located within 5 Å of the heme Fe atom. By way of example, key residues within the CYP3A4 active site in close proximity to bosutinib are Arg212 and Arg372 (H-bonding), Glu374 (salt-bridge interaction with the positively charged N atom of the piperazine ring), Phe57 (aromatic interaction), and Phe108 (aromatic pi stacking interaction).
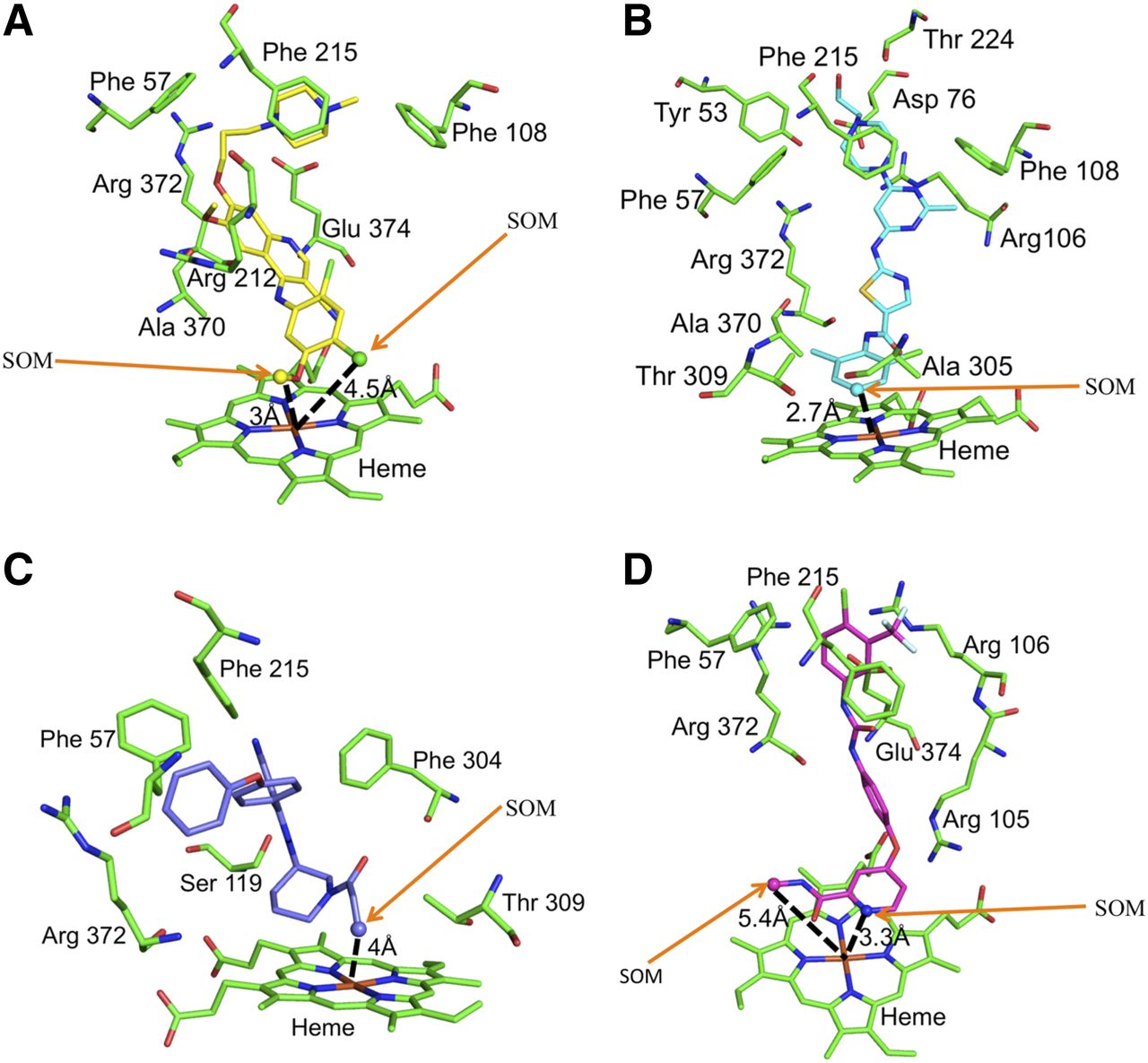
Consensus docked poses of kinase inhibitors in the CYP3A4 catalytic site: bosutinib (C atoms shown as yellow) (A), dasatinib (C atoms shown as cyan) (B), ibrutinib (C atoms shown as purple) (C), and sorafenib (C atoms shown as magenta) (D). The SOM atoms are shown as spheres (with arrows to aid identification). Protein C atoms are in green. O, N, S, and halogens (Cl/F) are shown in red, blue, yellow, and green, respectively. The SOM distances are shown in Å (dashed black lines).
Most KIs are predicted to bind primarily via a hydrophobic interaction(s) within the CYP3A4 active site. Table 5 shows CYP3A4 active site residues that are associated with KI binding. Phe57 and/or Phe215 is associated with the binding of approximately 80% of the data set molecules via hydrophobic/aromatic contacts (e.g., Fig. 3). Pi-cation interactions are observed most commonly with Arg106 (e.g., cabozantinib, ceritinib) but may also involve Arg105 (e.g., alectinib, dasatinib) and Arg212 (e.g., nilotinib and ruxolitinib). The data set molecules form H-bonds with a range of residues, most importantly Ser119, Arg212, Thr224, Arg372, and Glu374. Several kinase inhibitors contain nonaromatic rings, such as piperidine (e.g., ceritinib, cobimetinib) and piperazine (e.g., bosutinib, ponatinib), or an aliphatic tertiary amine group(s) (e.g., afatinib, osimertinib), which are predicted to exist in a protonated state at physiological pH. The catalytically favorable docked poses of several of these protonated KIs show a charge (salt-bridge) interaction(s) with Asp76 and/or Glu374 (Table 5).
CYP3A4 amino acids involved in noncovalent interactions with KIs based on docking (PMF-score) in the 3UA1 structure
Ligand Superposition by Morphological Similarity.
Molecular overlay is a useful approach for predicting the structural similarity of ligands and has been used to predict the SOM of substrates of several cytochrome P450 enzymes (Sykes et al., 2008; de Bruyn Kops et al., 2017). The CYP3A4 substrate BEC, which was cocrystallized in the 3UA1 structure, was initially used as the template for ligand superposition. Importantly, BEC is the only substrate bound within the active site of a CYP3A4 X-ray crystal structure in a catalytically favorable orientation and which has a molecular mass similar to many of the data set KIs. However, the known SOM was predicted for only 13 of the data set molecules using BEC as the template for structural overlay (Table 6).
Prediction of the SOM of KIs using different template structures for ligand superposition
As there was no other suitable cocrystal substrate that could be used for ligand superposition, we hypothesized that SOM prediction might be improved if a template from the data set was selected based on its docked conformation. First, we evaluated the docked conformation of sorafenib as the template for the structural overlay. The docked conformation of sorafenib is positioned at a catalytically favorable distance such that the major metabolic sites, N-oxide and hydroxylation (Fig. 1), are 3.3 and 5.4 Å from the Fe atom of the heme. Ghassabian et al. (2019) similarly reported that the distance of the site of N-oxidation from the heme (Fe atom) was 3.3 Å when sorafenib was docked in another unliganded CYP3A4 structure (pdb code 1TQN). Use of sorafenib as the template for ligand superposition predicted the SOM of 22 of the 31 data set molecules (prediction accuracy of ∼71%) (Table 6). Alignment of the representative substrates cabozantinib, dasatinib, lenvatinib, and regorafenib with sorafenib is shown in Fig. 4. Interestingly, the SOMs of dabrafenib and imatinib, which were not predicted by docking in the X-ray crystal structures, were similarly not predicted using the ligand superposition method with sorafenib as the template. On the other hand, imatinib overlaid well with the respective SOM positions (separated only by 1.2 Å) using lapatinib as the template. Overall predictivity with lapatinib as the template was similar to that with sorafenib (Table 6). SOM prediction was also evaluated using the structurally dissimilar and lower-molecular-mass KI tofacitinib. However, the data set molecules showed a poor overlay (Supplemental Fig. 1) with this template; the known major SOM was predicted correctly for only 10 KIs (Table 6).

Predicted SOM(s) of KIs by ligand superposition using sorafenib (C atoms in yellow) as the template: cabozantinib (A), dasatinib (B), lenvatinib (C), and regorafenib (D). The major SOMs are shown as spheres (with arrows to aid identification). C atoms for the overlayed molecules (A–D) are shown in magenta. O, N, and halogen (F/Cl) atoms are shown in red, blue, and green, respectively.
Web-Based SOM Prediction.
Three web-based methods were used to predict the SOM of data set molecules. MetaPrint2D, RS-WebPredictor, and XenoSite correctly predicted the major SOM of 24, 26, and 27 of the 31 KIs, respectively (Supplemental Table 1). MetaPrint2D failed to predict the major SOM for crizotinib, lapatinib, lenvatinib, and sunitinib, whereas both RS-WebPredictor and XenoSite correctly predicted the major SOM of these compounds. Additionally, MetaPrint2D failed to predict the known SOM for tofacitinib. On the other hand, MetaPrint2D was the only web server that correctly predicted the major SOM for dasatinib and pazopanib. The performance of RS-WebPredictor and XenoSite was similar, correctly predicting the SOM of 84% and 87% of the KIs, respectively. All the web servers failed to predict the SOM of dabrafenib, as also noted for the docking and ligand superposition methods.
Discussion
Metabolism provides a clearance mechanism for approximately 75% of clinically used drugs. Although biotransformation primarily functions as a deactivation and detoxification mechanism, metabolism may result in the formation of compounds with enhanced pharmacological, physiologic, and/or toxicological properties. Thus, metabolism is a significant consideration in drug efficacy and safety. Accordingly, metabolite characterization is of fundamental importance in drug discovery and development. SOM prediction tools provide a complementary approach to in vitro and in vivo approaches, such as metabolite profiling of structural analogs prior to synthesis (Kirchmair et al., 2015). In this study, several in silico approaches were compared to assess their ability to predict the SOM of KIs, which are mainly, or in part, metabolized by CYP3A4. The studies further aimed to use computational approaches to identify binding modes and interactions of KIs with specific amino acids in the CYP3A4 active site.
The SOM prediction of substrates using docking approaches is not straightforward due to the known plasticity of cytochrome P450 proteins that may adopt substantially different conformations upon ligand binding (Nair et al., 2016). Hence, three X-ray crystal structures were investigated as templates for molecular docking to account for the molecular flexibility. Studies of the docking of the data set molecules within the CYP3A4 active site showed that the choice of X-ray crystal template is an important consideration. Docked ligands overlapped well with the structure of bound BEC in the 3UA1 cocrystal structure. By contrast, the binding modes of ligands docked in the 5VCC and 5VCO structures only partially overlapped with bound BEC. Notably, of the three X-ray crystal structures used as templates, the 3UA1 structure has a relatively constricted active site that is narrower and lower in volume (779 Å3) compared with both the unliganded (5VCC, 823 Å3) and the inhibitor-bound (5VCO, 954 Å3) structures (http://sts.bioe.uic.edu/castp/index.html?201l). The data indicate that multiple ligand-bound X-ray crystal templates may need to be assessed to identify the most suitable structure for docking studies.
As noted earlier, the docked KIs aligned well with BEC in the active site of the 3UA1 structure, with the SOM of ∼77% of the data set molecules located within ∼3 Å of the SOM of BEC. Six scoring functions were evaluated to ascertain whether the top-ranked binding poses could correctly predict the SOM of the data set molecules. Predictions based on the scoring functions were generally dependent on the X-ray crystal template used, although the PMF-score was superior among the scoring functions evaluated in ranking the binding poses of KIs with the experimentally known SOM. By contrast, the D-Score was relatively poor for predicting the correct SOM in all X-ray crystal templates. The ranking of binding poses by different scoring functions for SOM prediction improved when the ligand-bound crystal structures (e.g., 3UA1) were used as a template for docking studies, except with the D-score. Overall, however, the docking approach provided very good SOM predictivity for most of the data set molecules.
Docking in the 3UA1 structure in combination with the various scoring functions failed to predict the SOM of only four KIs (afatinib, dabrafenib, imatinib, and sunitinib) (Table 2). No obvious molecular fingerprint could be identified for these molecules. The inability of the docking approach to predict the SOMs could be due to multiple reasons. First, the ligands might bind in a unique conformation that may involve reshaping of the CYP3A4 active site or changes in the conformation of amino acid side chains, as noted in multiple CYP3A4 X-ray crystal structures (Sevrioukova and Poulos, 2013, 2017). In these cases, the scoring functions may fail to rank the pose(s). Second, where KIs are modified nonenzymatically or metabolism by CYP3A4 is a minor metabolic pathway, binding may be weak. For instance, afatinib, a poor CYP3A4 substrate, was incorrectly ranked in the 3UA1 and 5VCC structures by more than four scoring functions in each structure. Third, as KIs are a new drug class, metabolic pathways for some drugs may not be completely characterized. Thus, currently unidentified SOM(s) may exist. For example, although methyl hydroxylation of dabrafenib (Fig. 1) is a known metabolic pathway, this SOM was not predicted by any of the in silico approaches investigated here, which suggests the possibility of an alternate site of metabolism.
An additional benefit of the docking approach is that it provides insights into the structure of the ligand-enzyme complex. Docking in the active site of the CYP3A4 structures identified a number of residues important for KI binding, particularly Phe57, Asp76, Arg105, Arg106, Ser119, Arg212, Phe215, Thr224, Arg372, and Glu374 (Table 5). The binding interactions of the docked data set molecules were consistent with the BEC (substrate)-bound CYP3A4 X-ray structure and, more generally, those reported to be important for CYP3A4 ligand recognition. For example, based on the 3UA1 X-ray crystal structure, the lysergic acid moiety of BEC is sandwiched between the side chains of Arg106 and Phe215 due to hydrophobic contacts. Similarly, the Thr224 and Arg212 side chains are known to form H-bonds with the lysergic acid group of BEC, while Ser119 has been shown to be associated with H-bonding/polar interactions in multiple CYP3A4 X-ray crystal structures (Sevrioukova and Poulos, 2013, 2017; Kaur et al., 2016).
The ligand superposition approach evaluated in this study performed poorly (<50% prediction of SOM) using BEC as the template. This contrasts with the success of the alignment-based approach reported previously from this laboratory for CYP2C9 substrates and UGT2B10 inhibitors (Sykes et al., 2008; Pattanawongsa et al., 2016). It should be noted, however, that the CYP2C9 substrate flurbiprofen, used for molecular overlay, exhibited marked structural similarity with the CYP2C9 data set molecules (Sykes et al., 2008), most of which contained an acidic functional group (e.g., the carboxylate of nonsteroidal anti-inflammatory drugs) that forms a salt bridge with Arg108 (Wester et al., 2004). However, substrates metabolized by CYP3A4 are more diverse, both chemically and structurally (Rendic and Di Carlo, 1997), and KIs are no exception in this regard. Thus, the selection of a suitable template for ligand superposition and SOM prediction using this approach is less straightforward. The structure of BEC is comparatively more rigid, with significant chemical differences compared with KIs (e.g., bosutinib, dasatinib, gefitinib), which influences alignment and overlay between the template and data set molecules. Therefore, we additionally explored the novel approach of using the docked conformation of selected KIs to predict the SOM of other compounds in the data set. Use of sorafenib as the template correctly predicted the SOM of ∼71% of the data set molecules. It is acknowledged, however, that this approach may be data set–sensitive given the structural diversity of CYP3A4 ligands. Nevertheless, the data strongly suggest that SOM prediction can be improved by using an appropriate template molecule from the data set under investigation when there are limited experimental data relating to substrate binding mode.
The web-based approaches evaluated in this study used three online servers. The servers predicted the major SOM for 24–27 KIs. RS-WebPredictor, which is a predecessor of XenoSite, predicted the SOM of ∼84% of the data set molecules. The performance of XenoSite was marginally superior, predicting ∼87% of the data set molecules. These data suggest that the web-based methods are useful for SOM prediction, even for relatively large, complex molecules such as KIs.
In summary, data presented here demonstrate that the docking pose and SOM predictivity are influenced by both the CYP3A4 X-ray crystal template used for docking and the scoring function used for ranking the poses, although the PMF-score consistently proved superior for ranking the binding poses of KIs against the experimentally determined SOM. The results underscore the importance of accounting for cytochrome P450 protein plasticity in ligand docking studies and indicate that the performance of scoring functions may vary for data sets of structurally diverse ligands. Nevertheless, very good predictivity of the SOM of the KIs was achieved for several protein template–scoring function combinations (e.g., 3UA1/PMF-score). The study highlighted the challenges with the ligand superposition approach, where the lack of availability of a suitable substrate cocrystal template limits the effectiveness of this method. However, the use of a representative data set substrate as the template for ligand superposition may provide an alternative approach. While the use of web-based methods predicted the SOM of the data set KIs extremely well, this approach, unlike ligand docking, does not provide insights into substrate binding within the CYP3A4 active site and the contribution of individual amino acids to substrate binding. The data indicate that computational approaches may be used to predict the SOM of KIs, but predictivity is dependent on the approach adopted. Similar considerations would be expected to be important for other classes of CYP3A4 substrates.
Authorship Contributions
Participated in research design: Nair, Miners, McKinnon.
Conducted experiments: Nair.
Performed data analysis: Nair, Miners, McKinnon.
Wrote or contributed to the writing of the manuscript: Nair, Miners, McKinnon.
Footnotes
- Received November 2, 2018.
- Accepted March 18, 2019.
This study was supported by a project grant [1120137] from the National Health and Medical Research Council of Australia. P.C.N. acknowledges Flinders Foundation and Flinders Centre for Innovation in Cancer for Early Career Research funding. R.A.M. is a recipient of a Beat Cancer Professorial Fellowship from Cancer Council SA.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- BEC
- bromoergocryptine
- CScore
- consensus score
- KI
- kinase inhibitor
- PMF
- potential of mean force
- SOM
- site(s) of metabolism
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}