


Abstract
Limited understanding of species differences in kidney transporters is a critical knowledge gap for prediction of drug-induced acute kidney injury, drug interaction, and pharmacokinetics in humans. Here, we report protein abundance data of 19 transporters in the kidney cortex across five species (human, monkey, dog, rat, and mouse). In general, the abundance of all of the 19 membrane transporters was higher in preclinical species compared with human except for multidrug resistance protein 1 (MDR1), organic cation transporter (OCT) 3, and OCTN1. In nonhuman primate, the total abundance of 12 transporters for which absolute data were available was 2.1-fold higher (P = 0.025) relative to human but the percentage of distribution of these transporters was identical in both species. Multidrug resistance-associated protein (MRP) 4, OCTN2, organic anion transporter (OAT) 2, sodium/potassium-transporting ATPase, MRP3, SGLT2, OAT1, MRP1, MDR1, and OCT2 were expressed differently with cross-species variabilities of 8.2-, 7.4-, 6.1-, 5.9-, 5.4-, 5.2-, 4.1-, 3.3-, and 2.8-fold, respectively. Sex differences were only significant in rodents and dog. High protein-protein correlation was observed in OAT1 versus MRP2/MRP4 as well as OCT2 versus MATE1 in human and monkey. The cross-species and sex-dependent protein abundance data are important for animal to human scaling of drug clearance as well as for mechanistic understanding of kidney physiology and derisking of kidney toxicity for new therapeutic candidates in drug development.
Introduction
In the early toxicity assessment of candidate therapeutics in drug development, the data obtained from animal models need to be interpreted with caution for predicting human pharmacokinetics (PK) (Tang and Mayersohn, 2011) and risk of organ injury as well as drug-drug interactions (Fisel et al., 2014). Clinical and preclinical species differences in the mRNA expression, protein abundance, and activity of transporters in organs relevant to drug disposition (i.e., intestine, liver, and kidney) remain a major reason for the poor allometric scaling (Wang et al., 2015). Although significant progress has been made toward the understanding of species and sex differences of drug transporters in the liver (Wang et al., 2015), limited data exist in the kidney and intestine. Particularly, kidney transporters can affect systemic drug clearance by regulating drug secretion and/or reabsorption and contribute to kidney toxicity by affecting intracellular drug concentration (Filipski et al., 2009). For example, kidney toxicity of tenofovir, methotrexate, cisplatin, ifosfamide, ciprofloxacin, and sitagliptin is associated with solute carrier transporters such as organic anion transporters (OATs) and organic cation transporters (OCTs) (Fisel et al., 2014) (Fig. 1). Allometric scaling methods are used for extrapolation of preclinical renal disposition data to human (Zou et al., 2012); however, these methods are not always successful, particularly when a drug undergoes kidney transport and metabolism. For example, tenofovir is taken up into proximal tubules mainly by OAT1 and effluxed into the urine by multidrug resistance-associated protein (MRP) 4, and both of these transporters are shown to be associated with tenofovir kidney toxicity (Kohler et al., 2011). Furthermore, examples exist where kidney toxicity is different in males and females, e.g., cisplatin, an OCT2 substrate, is more toxic to male rats (Nematbakhsh et al., 2013). In addition, rise in serum creatinine (sCr) is used as a surrogate of kidney function. However, since a fraction of sCr is secreted by active transporters, cross-species and sex-dependent variability in sCr transport could lead to false positive or negative conclusions regarding kidney function. In particular, a rise in sCr could be a result of inhibition of renal transporters without direct kidney injury (Chu et al., 2016).
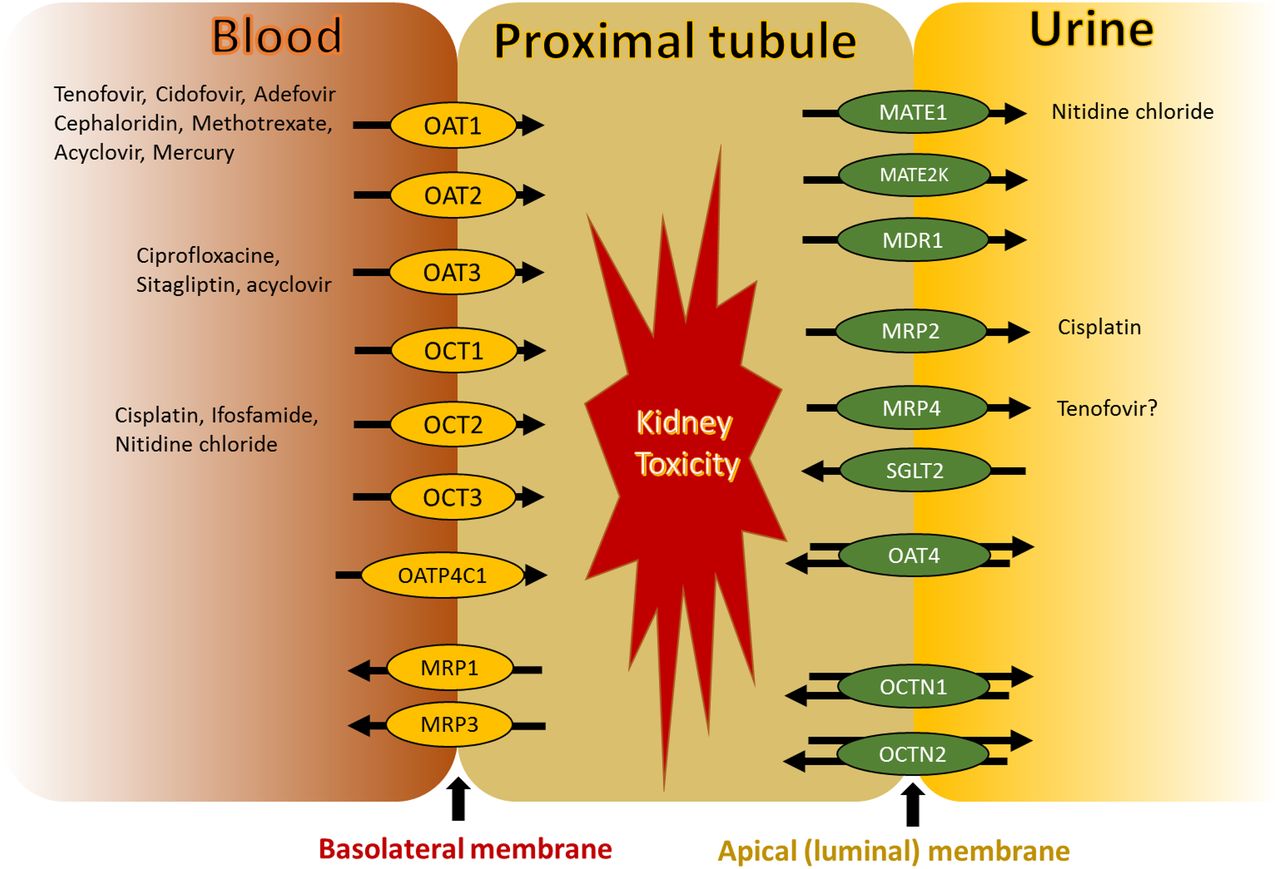
Localization of drug transporters in proximal tubule. Kidney proximal tubule contains OAT1-3, OATP4C1, OCT1-3, and MRP1/3 at the basolateral membrane and MATE1, MATE2k, MDR1, MRP2/4, SGLT2, OAT4, and OCTN1/2 at the apical membrane. Both uptake and efflux drug transporters are involved in nephrotoxicity of certain drugs shown in the figure.
Therefore, characterization of the cross-species and sex-dependent differences in protein abundances of kidney transporters is important for scaling and better prediction of renal secretion, reabsorption, and toxicity in humans. Accordingly, we hypothesized that mapping interspecies and sex differences in the abundance of kidney cortical transporters would enable development of physiologically based PK (PBPK) models that will improve the prediction of PK, kidney toxicity, and the potential risk of drug-drug interactions.
Materials and Methods
Chemicals and Reagents.
Liquid chromatography mass spectrometry (MS) grade acetonitrile, methanol, chloroform, and formic acid were purchased from Fisher Scientific (Fair Lawn, NJ) and formic acid was purchased from Sigma-Aldrich (St. Louis, MO). The ProteoExtract native membrane protein extraction kit was procured from Pierce Biotechnology (Rockford, IL). The protein quantification bicinchoninic acid kit and in-solution trypsin digestion kit were purchased from Pierce Biotechnology. Iodoacetamide, dithiothreitol, and pierce trypsin protease (MS grade) were purchased from Thermo Fisher Scientific (Rockford, IL). Ammonium bicarbonate buffer (98% purity) was purchased from Acros Organics (Geel, Belgium). Human serum albumin and bovine serum albumin were obtained from Calbiochem (Billerica, MA) and Thermo Fisher Scientific, respectively. Surrogate light and heavy peptides were obtained from New England Peptides (Boston, MA) and Thermo Fisher Scientific, respectively. OAT1 antibody was procured from Abcam (Cambridge, MA), anti-mouse IgG horseradish peroxidase–linked secondary antibody was purchased from (Cell Signaling Technologies) and SDS gel (Mini-PROTEAN TGX) was obtained from Bio-Rad (Hercules, CA).
Procurement of Normal Kidney Cortices from Experimental Animals.
Normal kidney cortical tissue (approximately 100–150 mg) was collected consistently at autopsy from the same kidney region from the preclinical species, i.e., cynomolgus monkey (n = 11; five males and six females), beagle dog (n = 12; six males and six females), Wistar Han rat (n = 20; 10 males and 10 females), and CD-1 mouse (n = 18; eight males and 10 females). The autopsies were conducted at the Pfizer Worldwide R&D facility (Cambridge, MA), which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. After collection, kidney tissue was flash frozen and stored at −80°C before shipping to the University of Washington. The study was conducted in accordance with the National Research Council Guide for the Care and Use of Laboratory Animals (https://grants.nih.gov/grants/olaw/Guide-for-the-Care-and-use-of-laboratory-animals.pdf) and in compliance with the Animal Welfare Act and its implementing regulations, under Institutional Animal Care and Use Committee approved protocols.
Membrane Protein Isolation from Kidney Tissue.
Total membrane protein was extracted from the kidney cortex (∼30–60 mg) of preclinical species using a previously described protocol (Xu et al., 2018). In brief, 30–60 mg of kidney cortical tissue was homogenized using a handheld rotary homogenizer with plastic probes. The homogenate was centrifuged at 16,000g for 30 minutes at 4°C. The supernatant (cytosolic fraction) was transferred to a new tube and the remaining pellet (membrane fraction) was resuspended with 600 µl of solubilization buffer (Pierce Biotechnology) and incubated at 4°C for 30 minutes with continuous mixing. The membrane fraction was used for transporter quantification.
Peptide Selection and Liquid Chromatography–Tandem Mass Spectrometry Protein Quantification of Kidney Drug Transporters.
We applied an optimized liquid chromatography–tandem mass spectrometry (LC-MS/MS) methodology, which relies on selective quantification of surrogate peptides of drug transporters. Whenever applicable, conserved peptides across species were selected for precise comparison. When conserved peptides were not available, a novel matrix approach (Supplemental Fig. 1) was used.
Peptide selection for individual drug transporters (Supplemental Table 1) in kidney across species was performed using a previously discussed in silico approach (Bhatt and Prasad, 2018). Total protein in kidney samples was quantified using a bicinchoninic acid assay kit (Pierce Biotechnology) following the vendor protocol. Samples were digested as described previously (Bhatt et al., 2019). Briefly, 80 µl of the membrane sample (2 mg/ml total protein) was mixed with 30 µl ammonium bicarbonate buffer (100 mM), 10 μl of human serum albumin (10 mg/ml), and 20 μl of bovine serum albumin (0.02 mg/ml) in a 1.5 ml microcentrifuge tube. Proteins were denatured and reduced with 10 μl of 250 mM dithiothreitol at 95°C for 10 minutes with gentle shaking at 300 rpm. The sample was cooled at room temperature for 10 minutes, and the denatured protein was alkylated with 20 μl of 500 mM iodoacetamide; the reaction was carried out in the dark for 30 minutes. Ice-cold methanol-chloroform (600 µl, 5:1 v/v) and water (400 µl) were subsequently added to each sample. After vortex mixing and centrifugation at 16,000g (4°C) for 5 minutes, the upper aqueous and lower organic layers were carefully removed without disturbing the protein pellet by using vacuum suction. The protein pellet was dried at room temperature for 10 minutes and then washed with 500 µl ice-cold methanol, followed by centrifugation at 8000g (4°C) for 5 minutes. The supernatant was removed and the pellet was dried at room temperature for 30 minutes and resuspended in 60 µl of ammonium bicarbonate buffer (50 mM, pH 7.8). Finally, the reconstituted protein sample was digested by adding 20 μl of trypsin (the protein:trypsin ratio was approximately 50:1) and incubated at 37°C for 16 hours. The reaction was quenched by the addition of 20 μl of peptide internal standard cocktail (prepared in 80% acetonitrile in water containing 0.5% formic acid) and 10 μl 80% acetonitrile in water containing 0.5% formic acid. The sample was mixed by vortex mixing and centrifuged at 4000g for 5 minutes. The supernatant was collected in a liquid chromatography MS vial for analysis. LC-MS/MS data acquisition was performed on a Waters Acquity UPLC system coupled with a SCIEX API-6500 triple quadrupole mass spectrometer using the optimized parameters outlined in Supplemental Tables 2 and 3 as per the validated method described previously (Prasad et al., 2016). LC-MS/MS data analysis is discussed in the Supplemental Material. Previously published human transporter levels were used as the reference data (Prasad et al., 2016), whereas an archived pooled (n = 21) human kidney tissue sample from our previous study was analyzed along with the animal samples as a quality control.
To control for cross-species variability in the membrane isolation recovery, we used total membrane protein per gram of kidney, which served as a scaling factor to express data in picomoles per gram unit. This scaling was particularly important since the total membrane protein per gram of kidney value for human (Supplemental Fig. 2) was significantly lower among species. To validate the LC-MS/MS results, western blotting analysis of a representative protein, OAT1, was performed using an established protocol (Neradugomma et al., 2017) (Supplemental Material).
Results
Cross-Species Kidney Cortical Transporter Abundances.
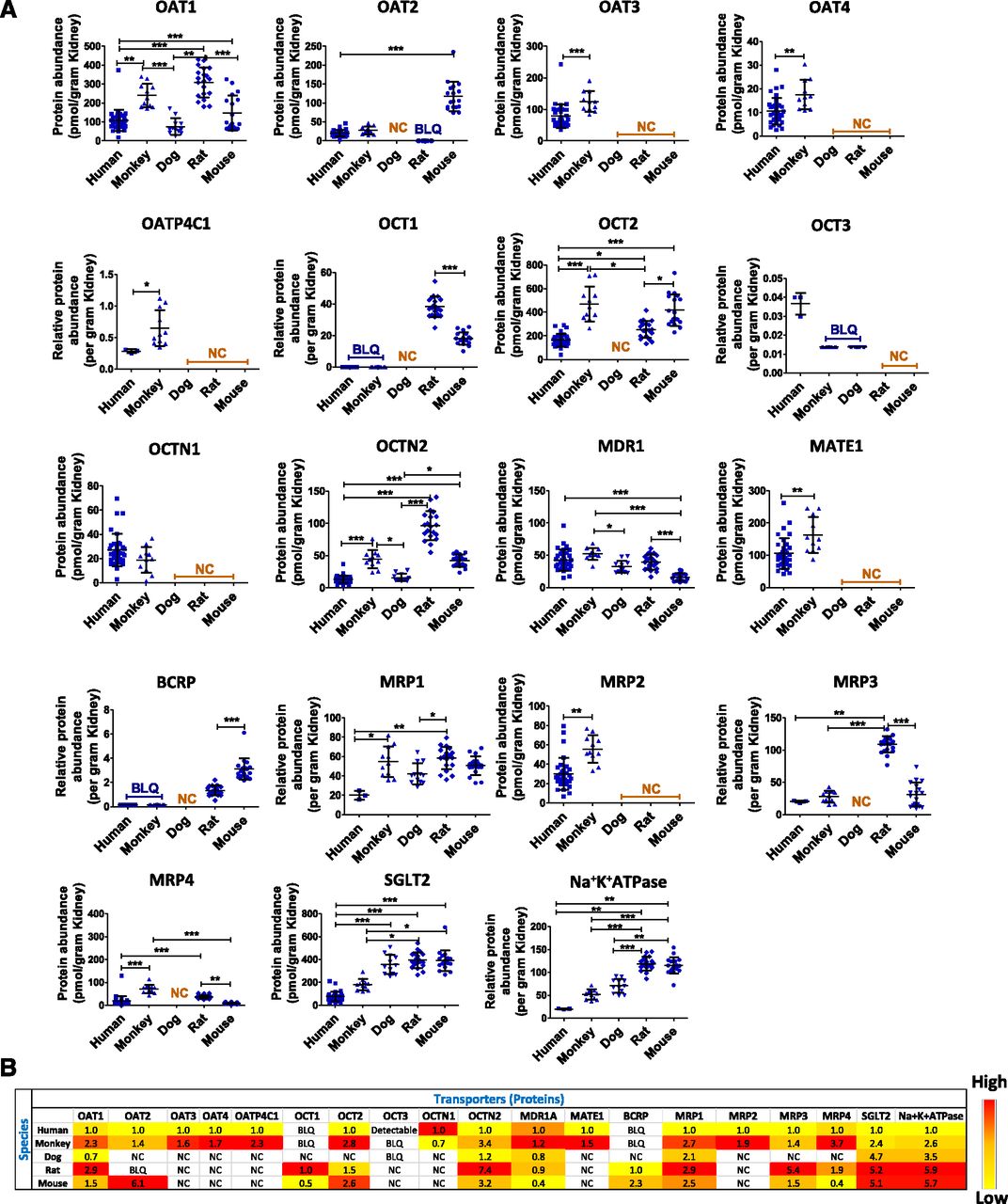
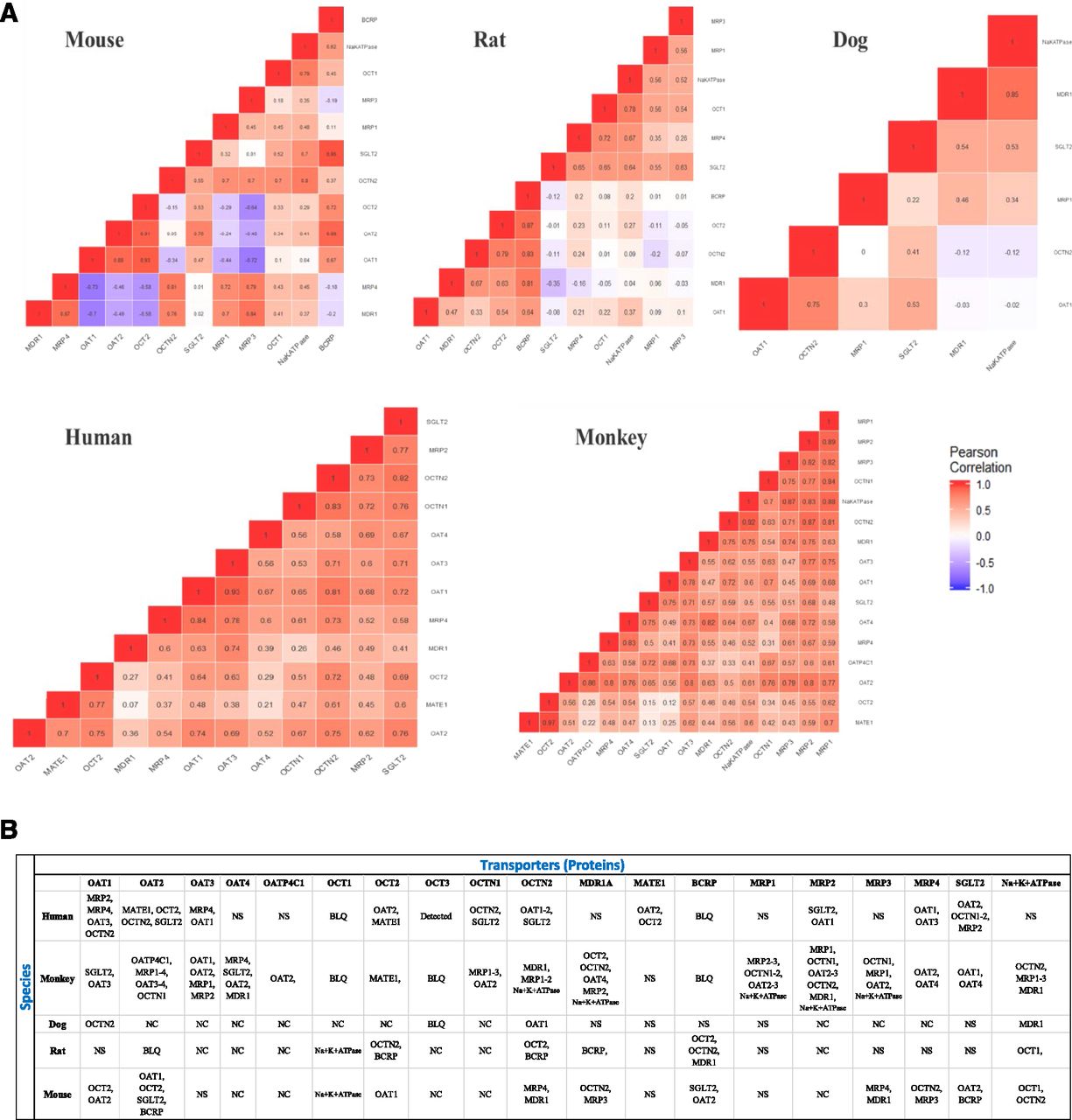
We quantified and investigated 19 basolateral and apical membrane transporters in normal kidney cortices obtained from four commonly used preclinical species in toxicity studies and drug safety evaluation and development, i.e., nonhuman primates (cynomolgus monkeys), beagle dogs, Wistar Han rats, and CD-1 mice. These data were compared with our previously generated human kidney data (Prasad et al., 2016). We report absolute data for 12 transporters (picomoles per gram of tissue) and relative abundance (normalized to total protein) data for seven transporters (Fig. 2; Supplemental Table 4). In general, the abundance of all of the 19 membrane transporters was higher in preclinical species compared with human except for multidrug resistance protein 1 (MDR1), OCT3, and OCTN1 (Fig. 2B). Human MDR1 abundance was similar to monkey, dog, and rat but it was 2.5-fold higher than mouse MDR1 (P < 0.05). OCT3 was only detectable in human and OCTN1 was similar in human and monkey. In nonhuman primate, the total abundance of 12 transporters for which absolute data were available was 2.1-fold higher (P = 0.025) relative to human but the percentage of distribution of these transporters was identical in both species (Supplemental Fig. 3). The abundance of OAT1, OCTN2, SGLT2, MRP1, sodium/potassium-transporting ATPase, and MRP3 was highest in rat and lowest in human with a fold range of 2.9–7.4 (P < 0.05). MRP4 was highest in nonhuman primate and lowest in mouse. The abundance of OAT2 was ∼5-fold (P < 0.0001) higher in mouse compared with human and nonhuman primate and was undetectable in rat. MATE1, MRP2, OAT3, OAT4, and OATP4C1 were 1.5- to 2.3-fold (P < 0.05) higher in nonhuman primate than human. OCT1 and breast cancer resistance protein were only detected in rodents. SGLT2 and sodium/potassium-transporting ATPase were 2- to 6-fold higher in all four preclinical species than human (P < 0.05). A significant correlation was observed between transporters (Fig. 3).

(A) Kidney cortical transporter cross-species comparison of protein abundance (picomoles per gram tissue) expression. Each dot represents the individual sample and data are represented as mean ± S.D. MATE2K was not detectable in any species because of poor sensitivity of the surrogate peptide. Only six transporters were detected in dog kidney because of the lack of conserved peptides. (B) Heat map of relative quantitative abundance of kidney transporters across species. Value in the cell represents the relative abundance of transporters across different species compared with transporter abundance in human kidney. Yellow to red color indicates increasing abundance of a particular transporter across species.

(A) Correlation plot for kidney transporter proteins across all five species. Values in the cell represent the coefficient of correlation (r2) between two transporter proteins. (B) Positive correlation between the transporter proteins with r2 > 0.70. The number of samples from individual species used in this study (with few exceptions) was human (n = 34), monkey (n = 11), dog (n = 12), rat (n = 20), and mouse (n = 18). Not conserved (NC) indicates that a conserved peptide was not found for that particular species. BLQ denotes below limit of quantification but the peptide was conserved. The BLQ values were derived by extrapolation of the signal-to-noise ratio to 3. No significant (NS) correlation (r2 < 0.70) was found between transporter proteins.
Sex Differences in Kidney Cortical Transporter Abundances.
Transporter abundance in human and nonhuman primate kidney did not show a sex-dependent pattern. Significant sex differences in transporter abundance were observed in mouse followed by rat > dog (Table 1). OAT1 was higher by 3.2- and 1.3-fold (P < 0.05) in male mouse and rat, respectively. OAT2 was 1.6-fold higher in male mouse than female. OCT2 was higher in male rat and mouse. MDR1 was 2- and 1.4-fold higher (P < 0.05) in female in mouse and dog, respectively, whereas MDR1 abundance was higher in male rat by 1.6-fold. MRP4 is predominantly expressed in male mouse (2.4-fold, P < 0.05) kidney. OCTN2 abundance was 1.3- and 1.4-fold higher in female mouse and male rats, respectively. SGLT2 was only 1.2-fold higher (P < 0.05) in female rats.
Sex differences in protein abundance of transporters across species
Comparison of LC-MS/MS Proteomics and Western Blot Data of Representative Transporter OAT1.
Antibodies are not available for all the studied transporters and when available, these do not work for all species. Nevertheless, we performed OAT1 quantification by western blotting using an anti-mouse OAT1 antibody to validate the LC-MS/MS data (the method is discussed in the Supplemental Material). The sex difference in OAT1 abundance in mouse was qualitatively confirmed by the western blot data (Supplemental Fig. 4). The higher signal of OAT1 in the rat sample was consistent with the proteomics data; however, the antibody did not work for human OAT1 (data not shown).
Discussion
Here, we report a comparison of 19 clinically relevant kidney drug transporters between human and four common preclinical animal models used in drug development. The presented data are the total membrane expression because it is technically difficult to reproducibly extract purified plasma membrane from frozen tissues (Kumar et al., 2015). While plasma membrane isolation involves multiple steps, we have previously demonstrated that total membrane extraction from different parts of the kidney is highly reproducible using the total membrane extraction kit (Prasad et al., 2016).
The quantitative information on cross-species transporter abundance is useful in interpreting kidney safety data in humans. In particular, drug-induced modest rises (20%–30%) in sCr in preclinical models could be due to direct tubular damage (i.e., drug-induced kidney injury) or indirect inhibition of kidney transporters. For example, an investigational Janus kinase inhibitor, INCB039110 (itacitinib), resulted in rises in sCr in a healthy volunteer’s study (Zhang et al., 2015). A follow-up dedicated kidney investigative clinical study revealed no changes in Food and Drug Administration/European Medicines Agency qualified nonclinical kidney toxicity biomarkers (e.g., Cystatin C) or glomerular filtration rate, but rather the rises in sCr were attributed to interference with kidney uptake transporters such as OCT2, OCT3, and OAT2 (Zhang et al., 2015) and efflux transporters such as MATE1 and MATE2K (Lepist et al., 2014; Zhang et al., 2015). Thus, understanding which transporters are potentially involved in uptake or efflux of the therapeutic candidate and knowing the relative abundance of the transporter across nonclinical species and humans will allow better interpretation of clinical data.
This data set for species and sex-specific abundances for kidney transporters is also useful to the biomedical community and drug developers in predicting the drug clearance and safety of new molecule entities. In agreement with our data, OCT1 and breast cancer resistance protein were not detected in human and nonhuman primate but were abundant in rodents (Bleasby et al., 2006; Prasad et al., 2016). The relatively low abundance of MDR1 in mouse kidney compared with human and rat is consistent with the mRNA data (Bleasby et al., 2006). Similarly, both nonhuman primate and rat have been shown to be associated with kidney toxicity with repeated dosing of tenofovir; however, such toxicity was not observed in the mouse at a similar dose (Ng et al., 2015; Ustianowski and Arends, 2015). The high correlation between OAT1 versus MRP2 and MRP4 indicates that the latter are important for the apical efflux of organic anions. Similarly, the high correlation of cation transporters OCT2 and MATE1 in human and nonhuman primate perhaps suggests that these transporters are coregulated. We also observed that OAT2 (a sCr secreting transporter) is a highly abundant anion transporter in mouse, whereas OAT1 is predominantly expressed in other species. Therefore, results of individual OAT knockout mouse versus rat are expected to be significantly different.
Differential sex abundance in mouse and rat (Table 1) was supported by negative correlations between transporters (Fig. 3). In particular, in the mouse the basolateral uptake transporters such as OATs and OCT2 were higher in male but the apical transporters (primarily efflux) were higher in females. In the rat, sex-dependent regulation of the basolateral uptake transporters was consistent with mouse; however, OCTN2, breast cancer resistance protein, and P-glycoprotein were also higher in males. Although the mechanisms of sex-dependent expression of these proteins are unknown, these transporters are important in secretion of conjugated sex hormones (Bush et al., 2017), and therefore can be regulated by the latter. Consistent with our data, the abundance and activity of some OATs and OCTs have been shown to be sex dependent (Cerrutti et al., 2002; Groves et al., 2006; Breljak et al., 2010; da Silva Faria et al., 2015).
Good correlation of OAT1 and OAT3 abundance is supported by the fact that these are transcriptionally coregulated (Prasad et al., 2016). Because most of the coregulated proteins work in tandem (OCT vs. multidrug and toxin extrusion protein and OAT vs. MRP) in the vectorial transport process, substrate-mediated regulation could be tested as a potential mechanism of correlations.
Regarding the limitations of this study, it does not allow comparison of all transporters because of the lack of MS-quantifiable or conserved peptides across species (e.g., dog). Nevertheless, our novel LC-MS/MS proteomics method allowed quantification of a majority of kidney transporters across species, which is not currently feasible using conventional immunoquantification. We also demonstrated that the quality of the LC-MS/MS proteomics data is superior to western blot data using OAT1 as an example (Supplemental Fig. 4). In addition, the protein abundance data (in the absence of in vitro functional data) are limited for the prediction of species differences in drug-induced kidney injury or PK. However, the transporter abundance data are a critical piece of information (i.e., a prerequisite), which constitute key physiologic parameters for PBPK modeling (Harwood et al., 2013). In particular, these data can be integrated with transport kinetics data (e.g., Km in eq. 1) of a drug or new chemical entity to predict in vivo transporter activity in animal versus human models (eq. 2). Furthermore, sex-dependent quantitative differences in the transporter abundance can be directly integrated into a PBPK model to extrapolate drug PK or drug toxicity (eqs. 1 and 3); where Km can be assumed similar between male versus female in a single species: (1)Thus,
(1)Thus, (2)
(2) (3)where CL is the intrinsic clearance (picomoles per minute per milligram protein or picomoles per minute per gram of kidney); Vmax is the maximum transport activity; Km is substrate affinity to a transporter; Kcat is the turnover number; and [S] is the substrate concentration. Here, Vmax depends on the transporter abundance, whereas Km and Kcat are independent of protein levels (Bhatt and Prasad, 2018). It is also noteworthy that the data obtained from the absolute peptide approach should not be considered as absolute molar protein abundance data because complete trypsin digestion may not be confirmed. However, these data (absolute peptide levels) can be used in allometric scaling of animal-to-human transporter-mediated clearances using eqs. 1–3. Because the scaling factor is derived by dividing the peptide abundance values, it does not matter whether absolute protein or absolute peptide values are used in the scaling as long as the trypsin digestion and the sample extraction are reproducible and consistent across species/genders. We have previously described this scaling approach (Bhatt and Prasad, 2018).
(3)where CL is the intrinsic clearance (picomoles per minute per milligram protein or picomoles per minute per gram of kidney); Vmax is the maximum transport activity; Km is substrate affinity to a transporter; Kcat is the turnover number; and [S] is the substrate concentration. Here, Vmax depends on the transporter abundance, whereas Km and Kcat are independent of protein levels (Bhatt and Prasad, 2018). It is also noteworthy that the data obtained from the absolute peptide approach should not be considered as absolute molar protein abundance data because complete trypsin digestion may not be confirmed. However, these data (absolute peptide levels) can be used in allometric scaling of animal-to-human transporter-mediated clearances using eqs. 1–3. Because the scaling factor is derived by dividing the peptide abundance values, it does not matter whether absolute protein or absolute peptide values are used in the scaling as long as the trypsin digestion and the sample extraction are reproducible and consistent across species/genders. We have previously described this scaling approach (Bhatt and Prasad, 2018).
In summary, our data on cross-species kidney transporter abundances, particularly in human, monkey, and rodents, provide useful quantitative information, which can be leveraged by: 1) allowing for preclinical-to-clinical translation of transporter-mediated secretary clearance using PBPK modeling, 2) distinguishing direct drug-induced kidney toxicity from indirect impact on kidney transporters (e.g., transporter-mediated creatinine clearance), and 3) utilizing cross-species transporter levels in conjunction with kidney injury biomarkers to better understand kidney safety signals and human safety risk assessment.
Acknowledgments
The authors thank Dr. Naveen Neradugomma for help with the western blotting analysis.
Authorship Contributions
Participated in research design: Basit, Radi, Vaidya, Prasad.
Conducted experiments: Basit, Karasu.
Performed data analysis: Basit, Prasad.
Wrote or contributed to the writing of the manuscript: Basit, Radi, Vaidya, Prasad.
Footnotes
- Received January 25, 2019.
- Accepted May 20, 2019.
This project was supported and funded by Pfizer Inc.
Z.R. and V.S.V. are employed by Pfizer Inc.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- CL
- clearance
- LC-MS/MS
- liquid chromatography–tandem mass spectrometry
- MDR1
- multidrug resistance protein 1
- MRP
- multidrug resistance-associated protein
- MS
- mass spectrometry
- OAT
- organic anion transporter
- OCT
- organic cation transporter
- PBPK
- physiologically based pharmacokinetics
- PK
- pharmacokinetics
- sCr
- serum creatinine
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}