


Abstract
Carfilzomib (CFZ) is a proteasome inhibitor used for oncology indications including treating multiple myeloma. CFZ is a potent cytotoxic agent with an IC50 value in the nanomolar range in various cancer cell lines and was considered as a potential payload for antibody drug conjugates (ADCs); however, the conjugated CFZ to anti-CD22 or anti-HER2 antibody totally abolishes the in vitro potency. This was a surprise since with other payloads such as monomethyl auristatin E (MMAE), where potent antiproliferation efficacy was retained as MMAE alone or as a payload in an ADC. Further investigations were conducted using CFZ alone, CFZ with a linker, and CFZ-ADC with tissue matrices including lysosomal enzymes. With CFZ linked to the ADC, cathepsin B (a lysosomal enzyme) was efficient in liberating CFZ from the ADC by cleavage of the valine-citrulline linker. At the same time, the liberated CFZ in the lysosome was inactivated due to further metabolism by lysosomal enzymes. The products from epoxide and amide hydrolysis were identified from these incubations. These results suggested that the CFZ-ADC upon uptake and internalization specifically delivers CFZ payload to the lysosomes, where CFZ was inactivated. On the other hand, CFZ by itself is not as vulnerable and could reach its target. Therefore, lysosomal stability is an important criterion in the selection of a payload for making the next generation of potent ADC therapeutics.
Introduction
Carfilzomib (CFZ) is an anticancer drug that can selectively inhibit the proteasome function in human cancer cells. Derived from a peptide epoxyketone nature product called expomicin, CFZ possesses a labile epoxy ketone pharmacophore (Kim and Crews, 2013). The epoxy ketone warhead in CFZ was demonstrated to have high selectivity to form a covalent adduct with the 20S proteasome, and thus disrupt the cellular protein homeostasis to induce cell death. Despite the essential role of proteasomes in all eukaryotic cells, a reasonable therapeutic window was still demonstrated for the proteasome inhibitors to selectively kill cancer cells (Adams, 2004a,b). In 2012, CFZ was approved by the Food and Drug Administration to treat multiple myeloma in patients who have received at least two prior therapies (the first-generation proteasome inhibitor bortezomib and an immunomodulatory agent such as thalidomide) and demonstrated disease progress within 60 days after the last therapy was finished (Herndon et al., 2013).
In multiple clinical trials, the treatment by CFZ alone or combined with dexamethasone has been demonstrated as tolerable and effective to induce responses in patients. However, the risk of severe adverse events was also very high in patients receiving CFZ, and the treatment was even discontinued in some patients (Lendvai et al., 2014; Dimopoulos et al., 2016; Schlafer et al., 2017). The common adverse events induced by CFZ include anemia, hypertension, thrombocytopenia, and pneumonia. Like other small molecular cytotoxic agents, CFZ may largely decrease the health-related quality of life for patients in chemotherapy. Therefore, CFZ was tested as a payload of antibody drug conjugates (ADCs) to investigate the potential of improving its efficacy and safety. ADC technology is a demonstrated strategy to effectively enlarge the therapeutic windows of cytotoxic agents and minimize their side effects (Flygare et al., 2013; Polakis, 2016; Zhang et al., 2018). Also, because of the broad cytotoxicity to all cancer cells containing proteasomes, conjugation to antibodies can help explore the usage of CFZ to treat cancers other than multiple myeloma. Recently, Staben et al. (2016) reported a new linker design strategy for payloads containing tertiary and heteroaryl amines, which was applied in the conjugation of CFZ to antibodies. The current article describes the challenges faced in investigating the feasibility of using CFZ as an ADC payload.
Materials and Methods
Experimental Procedure
Ammonium acetate, sodium citrate, Tris-HCl, formic acid, and cathepsin B were purchased from Sigma-Aldrich (St. Louis, MO). Pooled human liver S9 fractions were purchased from BD Biosciences (Bedford, MA). Human cellular lysosomal fractions prepared from human liver (mixed genders) were purchased from Xenotech (Lenexa, KS). Cancer cell lines including MDA-MB-231, MES-SA, MES-SA/Dx5, KPL-4, HCT116, A2058, BJAB, SK-BR-3, and WSU-DLCL2 were obtained from ATCC (Manassas, VA). These selected cell lines that include human breast cancer cell lines MDA-MB-231 and KPL-4, human sarcoma cell lines MES-SA and MES-SA/Dx5, human colon cancer cell line HCT116, human melanoma cell line A2058, and human lymphoma cell line BJAB comprise a panel of human cancer cell lines representing various human cancers. The cell culture medium used was Dulbecco’s modified Eagle’s medium/F-12 supplemented with 10% FBS. The medium and drug solution was not changed and was replenished daily because the incubation time was short. The final DMSO concentration was 0.1%. Vehicle controls were included in the study and cell viability in the vehicle controls served as the baseline (0% changes) in order to determine cell viability at different drug concentrations. The maleimidocaproyl (MC)/valine-citrulline (ValCit)/p-aminobenzyl quaternary ammonium (PABQ) linker drug of CFZ (Sigma-Aldrich) was synthesized as described previously (Staben et al., 2016). Human CD22 and HER2 antibodies with two engineered cysteine residues were generated as described previously (Polson et al., 2010; Bhakta et al., 2013). The anti-HER2 and anti-CD22 conjugates for CFZ were prepared and characterized as described previously (Zhang et al., 2016). The drug-to-antibody ratio (DAR) and aggregation percentage for the anti-HER2/CFZ conjugate were characterized as 1.9% and 2.2%, respectively. The DAR and aggregation percentage for the anti-CD22/CFZ conjugate were characterized as 1.9% and 4%, respectively. The DAR and aggregation percentage for the anti-HER2/monomethyl auristatin E (MMAE) conjugate were characterized as 1.9% and <5%, respectively. The DAR and aggregation percentage for the anti-CD22/MMAE conjugate were characterized as 1.9% and <5%, respectively.
Cell Viability Assay.
Cells were seeded in 96-well plates and allowed to adhere overnight at 37°C in a humidified atmosphere of 5% CO2. The cell culture medium was then removed and replaced by 100 µl fresh medium containing various concentrations of small molecule drugs (stock solutions in DMSO) or antibody drug conjugates [stock solutions in 20 mM histidine-acetate (pH 5.5), 240 mM sucrose, and 0.02% Tween-20]. After 4 days of drug incubation, the cell viability was determined using Promega CellTiter-Glo luminescent reagent, which measures the ATP level (an indirect measure of cell number). The luminescent intensity was measured on a PerkinElmer Envision reader. The relative cell viability was calculated by normalizing to nondrug treatment control and was graphed using GraphPad Prism software. The IC50 value was determined as the concentration needed to obtain 50% of maximum cell killing.
In Vitro Immolation of Linker Drug by Cathepsin B.
The linker drug was incubated at 15 µM in 100 mM citrate buffer (pH 5.5) containing 25 µg/ml cathepsin B and 5 mM cysteine at 37°C. Aliquots were taken at 0, 1, 4, and 24 hours during the incubation. The samples were mixed with an equal volume of acetonitrile, vortexed, and then centrifuged at 13,500g for 15 minutes. The supernatant was then analyzed by liquid chromatography/high-resolution mass spectrometry (LC-HRMS).
Stability of CFZ in Cancer Cell Homogenates/Human Liver S9 Fractions.
Cancer cells (5 × 106) were suspended and homogenized by beads in 0.5 ml ice-cold 100 mM Tris-HCl buffer (pH 7.4). The protein concentrations of the cancer cell homogenates were determined by Bradford assay using bovine serum albumin as the standard. CFZ (5 µM) was incubated with 1 mg/ml cell homogenates at 37°C, and then aliquots were taken, processed, and analyzed in the same way as previously described. The stability of CFZ was also tested in the same buffer containing 1 mg/ml human liver S9 fractions.
Metabolite Identification of CFZ by LC-HRMS.
Identification of CFZ metabolites was conducted by LC-HRMS on a 1290 Infinity LC System coupled with a Thermo Orbitrap Elite mass spectrometer. The liquid chromatography conditions were as follows: column, Waters Atlantis dC18, 100 Å, 3 μm, and 75 μm × 100 mm; mobile phase A, 0.1% formic acid in 10 mM ammonium acetate in H2O; mobile phase B, 0.1% formic acid and 10 mM ammonium acetate in 90% acetonitrile; gradient, 0–1.0 minutes 5% B, 1.0–16.0 minutes 5%–34% B, 16.0–29.0 minutes 34%–56% B, 29.0–30.0 minutes 56%–95% B, 30.0–31.0 minutes 95% B, and 31.0–31.5 minutes 95%–5% B; flow rate, 0.4 ml/min; and column temperature, 25°C. The cysteine adduct of MC/ValCit/PABQ/CFZ was identified by the molecular ion [M+H]2+ at the mass-to-charge ratio (m/z) found (698.3723) and calculated (698.3727), C72H103N12O15S2+, and by major fragments at m/z: 720.4323, 659.2857, 633.3065, 571.2540, 554.2278, and 549.3073 (retention time: 24.0 minutes). CFZ was identified by the molecular ion [M+H]+ at the m/z found (720.4312) and calculated (720.4331), C40H58N5O7+, and by major fragments at m/z 702.4203, 566.3317, 549.3047, 521.3104, 432.2843, 402.2372, 289.1537, and 261.1589 (retention time: 27.9 minutes). Metabolite (M) 0 was identified by the molecular ion [M+H]+ at the m/z found (589.2653) and calculated (589.2650), C24H41N6O9S+, and by major fragments at m/z 572.2375, 546.2584, 528.2480, 414.1688, and 386.1738 (retention time: 6.4 minutes). M1 was identified by the molecular ion [M+H]+ at the m/z found (420.2492) and calculated (420.2493), C22H34N3O5+, and by major fragments at m/z 289.1537 and 261.1589 (retention time: 13.7 minutes). M2 was identified by the molecular ion [M+H]+ at the m/z found (307.1653) and calculated (307.1652), C16H23N2O4+, and by major fragments at m/z 261.1592 and 100.0754 (retention time: 7.9 minutes). M3 was identified by the molecular ion [M+H]+ at the m/z found (738.4417) and calculated (738.4436), C40H60N5O8+, and by major fragments at m/z 549.3052, 521.3109, 450.2951, 402.2375, 337.2112, 289.1540, and 261.1591 (retention time: 22.4 minutes).
Results
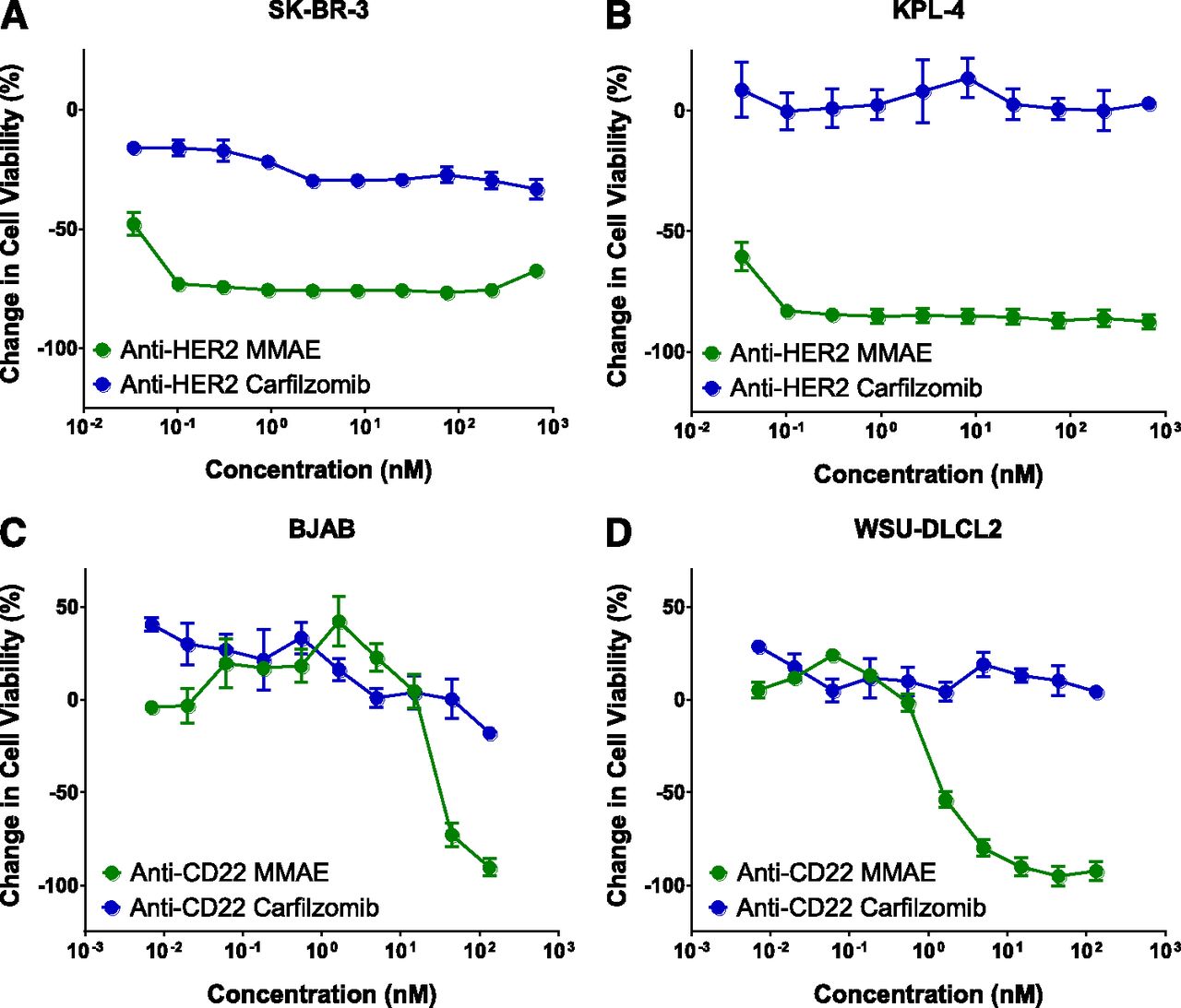
As a free drug, CFZ is an excellent cytotoxic agent that can effectively kill cancer cells at nanomolar concentrations. Consistent with previous studies, free CFZ was tested and found to be very cytotoxic to multiple cancer cell lines in our study (Fig. 1) (Kuhn et al., 2007; Baker et al., 2014). However, when CFZ was conjugated to antibodies, the conjugates were ineffective in killing cancer cells. An ADC comprising anti-HER2 antibody and CFZ is unable to inhibit the proliferation of the HER2-positive human breast cancer cell lines SKBR-3 and KPL-4 at a concentration equivalent to as high as 1 µM CFZ (Fig. 2, A and B). Also, CFZ conjugated to anti-CD22 antibody shows no efficacy in the CD22-positive cancer cell lines BJAB and WSU-DLCL2 (Fig. 2, C and D). As the positive control, MMAE, a peptide-like cytotoxic agent employed as the payload in the marketed ADC brentuximab vedotin (Adcetris), shows great cell-killing activity in these cell lines after being conjugated to the same anti-HER2 or -CD22 antibodies (Fig. 2).

Cytotoxicity of CFZ in various cancer cell lines. The IC50 values were as follows: MDA-MB-231, 9.3 nM; MES-SA, 4.1 nM; MES-SA/Dx5, 11.8 nM; KPL-4, 19.7 nM; HCT116, 4.8 nM; A2058, 4.6 nM; and BJAB, 1.9 nM.

Cytotoxicity of anti-HER2 and CD22 antibody conjugates of CFZ and MMAE in various cancer cell lines: (A) SK-BR-3, (B) KPL-4, (C) BJAB, and (D) WSU-DLCL2.
To determine whether CFZ can be successfully released from ADC in lysosomes, an in vitro assay was conducted using purified cathepsin B and the linker drug as a surrogate to determine whether proteolytic cleavage of the linker can occur (Fig. 3A). LC-HRMS analysis indicated the rapid disappearance of the cysteine adduct of MC/ValCit/PABQ/CFZ in the incubation (Fig. 3A). In the first hour, more than 95% of the linker drug disappeared. The cysteine adduct of the MC/ValCit linker residue (M0) was detected as a product from the cleavage, and the peak areas of M0 at 1, 4, and 24 hours were comparable to each other. This is consistent with the previous study in which the MC/ValCit/PABQ linker has been demonstrated to be efficient in releasing payloads conjugated via a quaternary ammonium (Staben et al., 2016). Unfortunately, no free CFZ was detected in the incubation, nor was the possible PABQ/drug intermediate detected (Fig. 3A). Given the fact that CFZ has a peptide backbone, the proteolytic cleavage by the lysosomal cathepsin(s) might result in the liability of unconjugated CFZ in the lysosomes.

Selected ion chromatograms of lysosomal metabolic products of CFZ or its linker drug. (A) MC/ValCit/PABQ/CFZ in incubation with cathepsin B at pH 5.5; (B) CFZ in incubation with lysosomal fractions prepared from human liver at pH 5.5; (C) CFZ in incubation with human liver S9 at pH 7.4.
In vitro incubations of CFZ with lysosomal fractions provide some insights. The incubation of CFZ with lysosomal fractions prepared from human livers at pH 5.5 showed that almost 100% of CFZ disappeared rapidly in 1 hour (Fig. 3B). Two metabolites were detected in the incubation, which were more hydrophilic based on retention times and had lower molecular weights than the parent CFZ. Based on the exact mass and mass spectrometry fragmentation patterns, the two metabolites were identified as hydrolytic metabolites (Fig. 4, B and C). The same metabolites (M1 and M2) were also observed in the incubation of MC/ValCit/PABQ/CFZ with cathepsin B (Fig. 3A), indicating that cathepsin B hydrolyzed not only the amide bond in the valine-citrulline dipeptide and the p-aminobenzyl spacer, but also the amide bonds within CFZ. Besides cathepsin B, many other members in the cathepsin family have been identified in the lysosomes of human cancer cells (Nomura and Katunuma, 2005; Caculitan et al., 2017). It is unknown whether cathepsins other than cathepsin B can also catalyze the hydrolysis of CFZ.

Product ion scans of CFZ (A) and metabolites M1 (B), M2 (C), and M3 (D) formed in lysosomal and S9 incubations.
Further investigation was conducted to determine whether the nonlysosomal hydrolases inside the cells can also contribute to the hydrolysis of CFZ. At 37°C and pH 7.4, moderate hydrolysis of CFZ was observed in the homogenates of all three cancer lines (KPL4, SKBR3, and BJAB) tested. After 1-hour incubation in the homogenates of KPL4, SKBR3, and BJAB cells, approximately 40% of the CFZ (starting concentration 5 µM) disappeared (Fig. 5). The hydrolytic metabolites with cancer cell fractions were similar to those observed in the lysosomal fraction hydrolysis of CFZ. For comparison, we also conducted the incubation of CFZ in human liver S9 fractions at pH 7.4 because it provides an abundant resource of hydrolysis enzymes (Fiddler et al., 1979). Interestingly, the peptide backbone of CFZ was found to be very stable in human liver S9 fractions incubated at physiologic pH, but the epoxy group was hydrolyzed to give dihydrodihydroxy metabolite M3 (Fig. 3C; Fig. 4D). This reaction is likely catalyzed by an epoxide hydrolase under a neutral pH condition (Arand et al., 2005). We proposed that hydrolysis of CFZ in the cytosol of cancer cells can impair its efficacy. However, the low IC50 values of free CFZ in various cancer cell lines indicated that the impact of cytosolic hydrolysis on its efficacy is very limited, perhaps because the hydrolysis rates are relatively slow and enough free CFZ can bind to the proteasomes before extensive hydrolysis occurs.

Disappearance of CFZ in incubations with various cellular fractions of cancer cells or buffers.
Discussion
In the current study, the feasibility of utilizing CFZ, a drug used to treat multiple myeloma, was investigated as an ADC payload. An ADC underwent antigen binding, tissue uptake, cell internalization, and biochemical transformation (proteolytic degradation of antibody, linker cleavage, and immolation) to release payload in lysosomes. The biotransformation pathways associated with CFZ and its linker drug in lysosomes are summarized in Fig. 6. CFZ was hydrolyzed to multiple metabolites in lysosomes. CFZ is highly cytotoxic to cancer cell lines by diffusing into the cells and covalently binding to the 20S subunit of proteasomes to illicit its action (Fig. 7A). However, the mechanism of action of the ADC is much more complicated and involves a series of events including antibody-antigen binding, endosome incorporation, lysosomal catalysis, linker cleavage (and immolation), payload relocation, and finally the cell-killing event (Schrama et al., 2006). The ADC may fail to show efficacy if any one of these steps fails. In the current study, in vitro assays using cathespin B and lysosomal fractions have demonstrated that CFZ is susceptible to hydrolysis in the lysosomes. As part of the incorporated ADC, CFZ may be rapidly hydrolyzed by cathepsin(s) during lysosomal catalysis before or after any CFZ molecule is released from the ADC. It should be noted that due to lysosomal preparations, the protein concentration in the in vitro incubation was diluted to 0.3 mg/ml, while the actual protein concentration inside the lysosomes of living cells can be as high as 200 mg/ml (Dean and Barrett, 1976). It can be expected that there is no or very limited escape of CFZ off an ADC from the lysosomes to reach the target and thus no proteasome adduction and dysfunction will occur (Fig. 7B). In contrast, MMAE exemplifies that good lysosomal stability can be critical in making an excellent payload for the ADC. MMAE embodies four amide bonds in its structure, but it is 100% resistant to lysosomal hydrolysis under similar in vitro incubation conditions (Doronina et al., 2003). After ADC incorporation and linker cleavage and immolation, the release of free MMAE from lysosomes to cytosol is a necessary step before MMAE can block the polymerization of tubulin and exert antineoplastic effects (Francisco et al., 2003). Retrospectively, a conclusion can be drawn that the stability of MMAE in lysosomes is a prerequisite for the efficacy and successful development of brentuximab vedotin. The experiences and lessons learned from MMAE and CFZ have indicated that lysosomal stability should be considered as one of the criteria in evaluating the potential of a compound to serve as the cytotoxic warhead in an ADC. At the early stage in the discovery of new payloads, in vitro incubation with cathepsin(s) or lysosomal fractions is a very helpful assay in evaluating the lysosomal stability of the lead compounds, especially those with functional groups vulnerable to hydrolases (e.g., amides and esters). In the future, a more quantitative relationship between the rate of hydrolysis in vitro and anticancer efficacy may be established for new payloads with designing experiments to learn the kinetics of hydrolysis reactions.
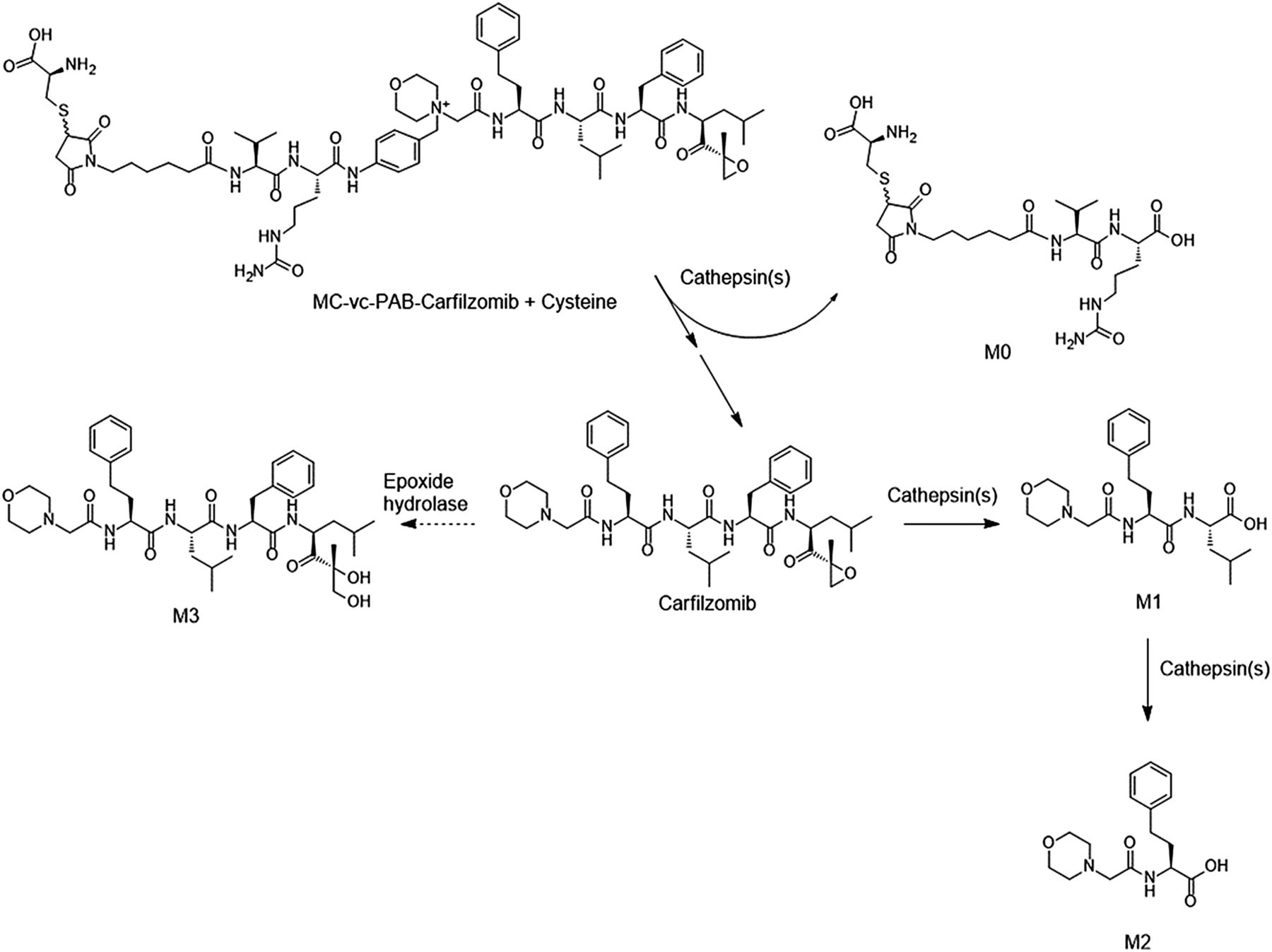
Lysosomal degradation pathways of CFZ linker drug.
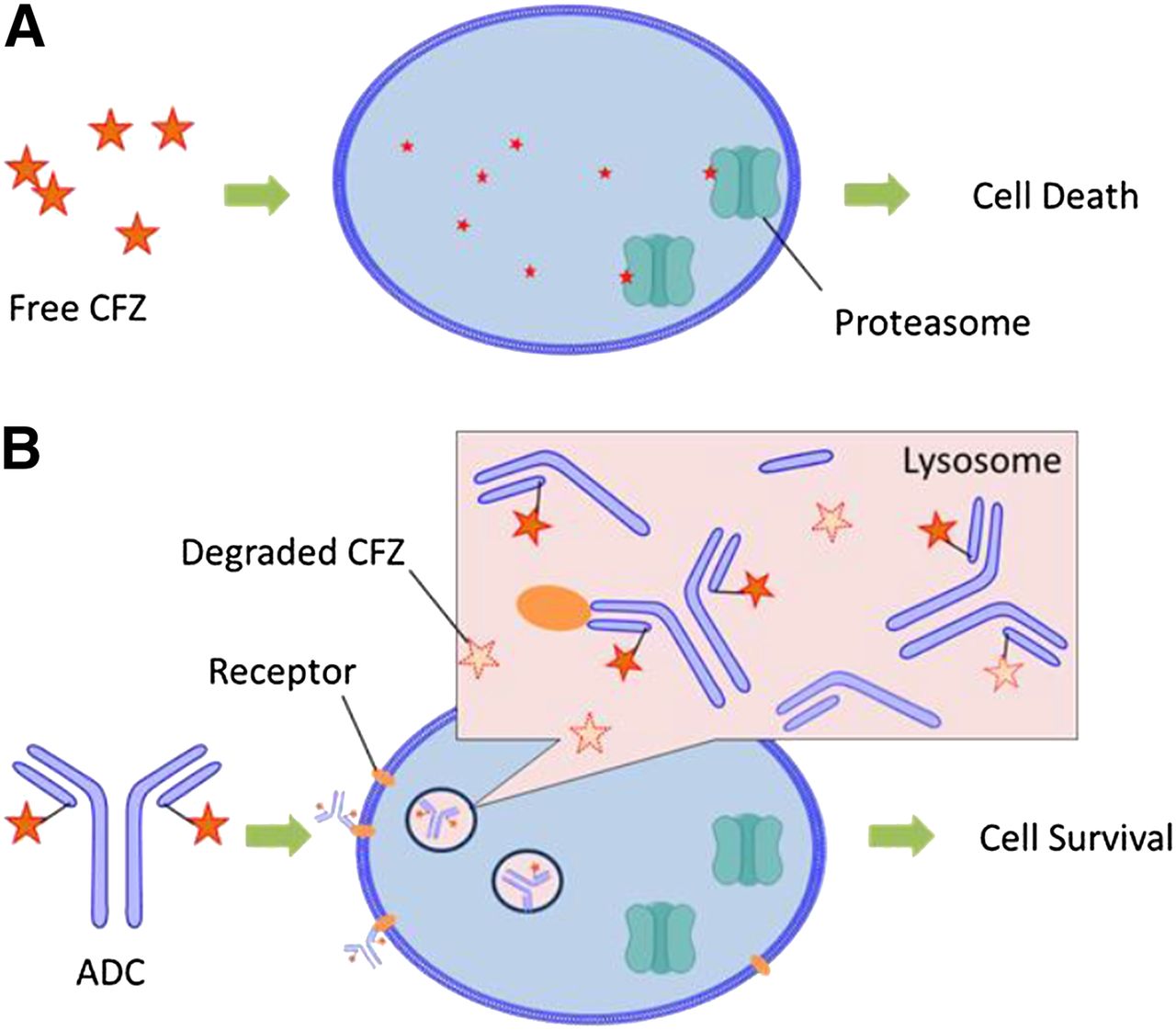
Proposed cellular distribution and trafficking of CFZ (A) and antibody-CFZ conjugate (B).
ADC technology makes it possible to deliver cytotoxins to specific antigen-bound cancer cells, and possibly minimize the toxicity-induced side effects in normal tissues and organs, enabling the application of these cytotoxins in cancer chemotherapy (Flygare et al., 2013; Polakis, 2016). In addition, ADC could enable the delivery of payloads with poor cell membrane permeability into cancer cells, and consequently enhance their efficacy. Several categories of substances are usually being investigated as ADC payloads, which are mainly antimitotic and DNA-damaging agents (McCombs and Owen, 2015). Meanwhile, researchers are also looking for novel ADC payloads that can act via new targets or pathways, or trying to conjugate existing small molecule drugs to antibodies for improving their efficacy or safety.
In summary, CFZ is a potent cytotoxic agent in various cancer cell lines; however, CFZ is an inactive payload in ADCs. Our studies demonstrated that CFZ is highly vulnerable to hydrolysis in lysosomal fractions. CFZ conjugated to antibodies loses its cytotoxic potency because of the extensive hydrolysis of the drug occurring in lysosomes, which is necessary for ADC catabolism. The results indicate that when selecting an appropriate payload in the development of an ADC, lysosomal stability is a property of payloads thath should be taken into consideration.
Authorship Contributions
Participated in research design: Ma, Dela Cruz-Chuh, Khojasteh, Dragovich, Pillow, Zhang.
Conducted experiments: Ma, Dela Cruz-Chuh.
Contributed new reagents or analytic tools: Pillow, Ma, Khojasteh, Dragovich, Zhang.
Performed data analysis: Ma, Dela Cruz-Chuh, Khojasteh, Zhang.
Note Added in Proof—The title of this article was changed from that used for the Fast Forward version published May 9, 2019.
Footnotes
- Received January 26, 2019.
- Accepted April 29, 2019.
↵1 Current affiliation: Calithera Biosciences, South San Francisco, California.
At the time of this study all authors were employees of Genentech, Inc., and no financial support was received.
Abbreviations
- ADC
- antibody drug conjugate
- CFZ
- carfilzomib
- DAR
- drug-to-antibody ratio
- LC-HRMS
- liquid chromatography/high-resolution mass spectrometry
- M
- metabolite
- MC
- maleimidocaproyl
- MMAE
- monomethyl auristatin E
- m/z
- mass-to-charge ratio
- PABQ
- p-aminobenzyl quaternary ammonium
- ValCit
- valine-citrulline
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}