


Abstract
2-(N-acetyl-D-tyrosyl-trans-4-hydroxy-L-prolyl-L-asparaginyl-L-threonyl-L-phenylalanyl) hydrazinocarbonyl-L-leucyl-Nω-methyl-L-arginyl-L-tryptophanamide monoacetate (TAK-448, RVT-602), a kisspeptin analog, has been developed as a therapeutic agent for prostate cancer. The purpose of the present study is to clarify the mechanism of the less than dose-proportional nonlinear pharmacokinetics of TAK-448 after subcutaneous administration to rats. The plasma pharmacokinetics of TAK-448 and radiolabeled TAK-448 ([14C]TAK-448) were examined after subcutaneous and intravenous administrations to rats. [14C]TAK-448 was also subcutaneously injected together with protease inhibitors. The effects of the protease inhibitors on the in vitro metabolism of [14C]TAK-448 were investigated using rat skin homogenates. In a dose-ascending study, less than dose-proportional nonlinear pharmacokinetics were observed after subcutaneous administration with limited absorption of TAK-448 at the highest dose level contrary to the linear pharmacokinetics following intravenous dosing, indicating enhancement of subcutaneous metabolism with dose escalation. The systemic absorption of unchanged TAK-448 recovered when protease inhibitors were subcutaneously coadministered, suggested the involvement of subcutaneous proteases in the first-pass metabolism. An in vitro metabolism study suggests that serine protease could be responsible for the subcutaneous metabolism of TAK-448. Dose-dependent enhancement of first-pass metabolism appears to contribute to the less than dose-proportional nonlinear pharmacokinetics of TAK-448 after subcutaneous administrations to rats.
Introduction
For many years, prostate cancer has been treated by bilateral orchiectomy to eliminate testosterone production and by the administration of gonadotropin-releasing hormone agonists. Continuous administration of gonadotropin-releasing hormone agonists suppresses testosterone secretion by the downregulation of gonadotropin secretion from the pituitary gland (Steinberg, 2009).
Kisspeptin was isolated from human placenta as a ligand of the human G-protein–coupled receptor OT7T175/GPR54 (Kotani et al., 2001; Muir et al., 2001; Ohtaki et al., 2001). Although kisspeptin was originally considered to be a metastasis-suppressing peptide (Hori et al., 2001; Ohtaki et al., 2001; Stafford et al., 2002), it has been shown in mammals that kisspeptin has a pivotal role in the regulation of hypothalamic/pituitary/gonadal function. Deletions and mutations in GPR54 were also identified in patients with hypogonadotropic hypogonadism in the clinic (de Roux et al., 2003; Funes et al., 2003; Seminara et al., 2003). Administration of exogenous kisspeptin (45–54), the C-terminal 10 amino acid residue peptide of kisspeptin, induces gonadotropin secretion in the mouse, rat, monkey, and sheep, as well as in humans (Matsui et al., 2004).
2-(N-acetyl-D-tyrosyl-trans-4-hydroxy-L-prolyl-L-asparaginyl-L-threonyl-L-phenylalanyl) hydrazinocarbonyl-L-leucyl-Nω-methyl-L-arginyl-L-tryptophanamide monoacetate (TAK-448, RVT-602) is a peptide analog of kisspeptin chemically synthesized by Takeda Pharmaceutical Company Limited. In vitro and in vivo pharmacological studies have indicated that TAK-448 is a potent agonist of the kisspeptin receptor and continuous administration of TAK-448 rapidly reduced testosterone levels in rats (Matsui et al., 2012). The compound is being developed as a treatment for prostate cancer.
With the rapid advances in recombinant gene technology, biologically active peptide drugs have become widely prevalent such as the glucagon-like peptide-1 analog liraglutide, erythropoietin, interferon analogs, and gonadotropin-releasing hormone analogs such as leuprolide (Brogden and Heel, 1987; Okada et al., 1988, 1994; Ogawa et al., 1989; Periti et al., 2002; Hadziyannis and Papatheodoridis, 2003; Elliott et al., 2008; Croom and McCormack, 2009). Since these peptide drugs would be orally inactive, they are generally administered via the parenteral route. We previously reported the disposition of TAK-448 after single subcutaneous and intravenous administrations to rats and dogs with radiolabeled TAK-448 ([14C]TAK-448) (Moriya et al., 2019). That study revealed that the dosed radioactivity was rapidly and almost completely absorbed after subcutaneous administration at pharmacologically effective doses and an appreciable amount of TAK-448 together with a hydrolyzed metabolite, N -Acetyl-D-tyrosyl- trans -4-hydroxy-L-prolyl-L-asparaginyl-L-threonine (M-I), were detected in the plasma of rats and dogs. In dogs, the bioavailability (BA) of TAK-448 was 92.4% after subcutaneous administration. This value is comparable to the 96.8% ratio (subcutaneous/intravenous) for total radioactivity at a dose of 1 mg/kg, suggesting that the subcutaneously dosed TAK-448 was almost completely absorbed in the unchanged form in dogs and the effect of first-pass metabolism at subcutaneous administration was minimal. On the other hand, although the radioactivity ratio of 94.1% for subcutaneous/intravenous administration was also comparable in rats, the BA of TAK-448 was much lower with a value of 66.3% in rats. This result suggests that TAK-448 underwent subcutaneous first-pass metabolism in rats. As described previously, since TAK-448 is a synthetic peptide analog, some peptidases or proteases could be involved in the subcutaneous metabolism of TAK-448. Metabolic enzymes have been reported to be expressed in rat skin (Oesch et al., 2007); however, drug metabolism in skin is poorly understood compared with that in the primary drug metabolizing tissues.
The present study revealed that TAK-448 exhibited marked nonlinear pharmacokinetics with less than dose-proportional increases in the area under the plasma concentration-time curve (AUC) at doses of 0.1, 1, and 10 mg/kg after subcutaneous administration to rats, although the linear pharmacokinetics was observed after subcutaneous administration to dogs and monkeys (Supplemental Tables 1 and 2). Particularly in the dose range from 1 to 10 mg/kg, the AUC was decreased even though the dose increased. The results of a pharmacokinetic study after intravenous administration to rats showed clear linearity over the same dose range, suggesting that subcutaneous metabolism may be a critical factor in the nonlinearity of TAK-448 in rats. It is essential to examine the mechanism of the nonlinearity in preclinical studies; otherwise, it might be difficult to estimate the effective concentration level from the results of early clinical trials. In the present study, we investigated the mechanism for the less than dose-proportional nonlinear pharmacokinetics involved in the first-pass metabolism of TAK-448 after subcutaneous administration to rats. The effects of protease inhibitors on the absorption of TAK-448 and the in vitro metabolism of TAK-448 in rat skin homogenates were also examined to clarify the properties of the subcutaneous metabolism in rats.
Materials and Methods
Materials.
TAK-448, M-I, and a structurally related internal standard were synthesized by Takeda Pharmaceutical Company Limited (Osaka, Japan). [14C]TAK-448 was synthesized by GE Healthcare UK, Ltd. (Little Chalfont, Buckinghamshire, UK) and purified by Nemoto Science Co., Ltd. (Tokyo, Japan) with a specific activity of 5.72–6.59 MBq/mg. The chemical structures of [14C]TAK-448, its M-I metabolite, and the internal standard 2-(NAcetyl-D-tyrosyl-D-tryptophyl-L-asparaginyl-L-threonyl-Lphenylalanyl) hydrazinocarbonyl-L-leucyl-Nω-methyl-L-arginyl-L-tryptophanamidemonoacetate (T-1532583), with only one amino acid substitution with TAK-448 from Hyp to D-Trp, are shown in Fig. 1. The radiochemical purity of [14C]TAK-448, as verified by high-performance liquid chromatography (HPLC) equipped with an online liquid scintillation counter detector, was more than 96.7%. A protease inhibitor cocktail for general use was purchased from Sigma-Aldrich Japan (Tokyo, Japan). 4-(2-Aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF), bestatin, leupeptin hemisulfate monohydrate, and aprotinin from bovine lung were purchased from Wako Pure Chemical Industries (Osaka, Japan). EDTA-Na and [(2S,3S)-3-carboxyoxirane-2-carbonyl]-L-leucine (4-guanidinobutyl) amide hemihydrate (E-64) were purchased from Dojindo Laboratories (Kumamoto, Japan) and the Peptide Institute, Inc. (Osaka, Japan), respectively. All other chemicals and reagents were purchased from commercial sources.

Chemical structures of TAK-448, its M-I metabolite, and the internal standard, T-1532583.
Animals.
Male Sprague-Dawley rats were purchased from Charles River Laboratories Japan, Inc. (Kanagawa, Japan). Animals were fed laboratory chow (CR-LPF; Oriental Yeast Co., Ltd., Tokyo, Japan), had free access to water, and were housed in temperature- and humidity-controlled rooms with 12-hour light and dark cycles. Animal studies were conducted in accordance with the “Guideline for the Care and Use of Laboratory Animals” from the Pharmaceutical Research Division, Takeda Pharmaceutical Company Limited, and were approved by the Ethical Committee for animal experiments for our division.
In Vivo Study.
TAK-448 and [14C]TAK-448 were dissolved in N,N-dimethylacetamide/5% glucose solution (1:9, v/v) for single subcutaneous and intravenous administrations to rats. For single subcutaneous administration, the dosing solution and/or protease inhibitor cocktail were injected dorsally to nonfasted male rats. For single intravenous administration, the dosing solution was injected into the femoral veins of nonfasted male rats. After subcutaneous and intravenous administrations of TAK-448 or [14C]TAK-448 to rats, blood was collected from the tail veins at the designated time points and centrifuged at approximately 8000g at 4°C for 3 minutes to obtain plasma; the total radioactivity or the concentration of TAK-448 was measured as described subsequently.
For the preparation of the injection formulation for the protease inhibitor, a bottle of protease inhibitor cocktail (Sigma-Aldrich Japan) was added with 2 ml of 10 mM PBS. To examine the effect of the subcutaneous coadministration of the protease inhibitor cocktail on the pharmacokinetics of [14C]TAK-448 after subcutaneous dosing, the dosing solution of [14C]TAK-448 and the protease inhibitor cocktail solution were mixed in a volume ratio of 3:2 to achieve final concentrations of [14C]TAK-448 from 1 to 10 mg/ml per kilogram, and the concentrations of the protease inhibitors were as follows: 40 mM AEBSF, 20 mM EDTA, 2.6 mM bestatin, 0.28 mM E-64, 0.02 mM leupeptin, and 0.006 mM aprotinin. The effect of the subcutaneous dosing of the protease inhibitor cocktail on the pharmacokinetics of [14C]TAK-448 after intravenous administration to rats was also examined. Protease inhibitor cocktail was dorsally injected and [14C]TAK-448 was subsequently injected into the femoral vein of rats.
Preparation of Skin Homogenates.
Male Sprague-Dawley rat was sacrificed by exsanguination from the abdominal aorta under anesthesia with ether, and beagle dog and cynomolgus monkey were sacrificed by overdosing with sodium pentobarbital at an intravenous dose of about 50 mg/kg, followed by excision of about 25 cm2 of the dorsal skin. Human skin (female, Caucasian, 22 cm2) was obtained from a commercial source (KAC, Kyoto, Japan). Rat skin was collected just before preparation of the homogenates, and the others were stored in a freezer with a controlled range of –90 to –70°C until used. The following procedure was conducted under ice-chilled conditions. After trimming off excessive muscle and connective tissue the skin was finely minced and weighed. The minced skin was transferred into a flask and homogenized in 10 mM PBS to prepare a homogenate at a concentration of 10% (w/v). The homogenate was centrifuged at approximately 1500g at 4°C for 10 minutes, and the middle layer was obtained as a skin homogenate. The skin homogenate was diluted with 10 mM PBS for measurement of protein concentrations, and the remaining homogenate was stored in a freezer with a controlled temperature range of –90 to –70°C and shielded from light until used. Protein concentrations were determined using an assay kit (Coomassie Plus Reagent) with bovine serum albumin as a standard.
In Vitro Metabolism of [14C]TAK-448 in the Skin Homogenate.
One milliliter of PBS containing 900 μg protein of the skin homogenate with or without protease inhibitors and the homogenate containing 100 μg/ml of unlabeled TAK-448 were preincubated at 37°C for 5 minutes except for the samples for time 0 and incubation at 4°C. The metabolism study was initiated by addition of 10 μl of [14C]TAK-448 PBS solution (1 μg/ml at the final concentration) followed by incubation at 4 or 37°C for the designated times. After the incubation for the designated times, 1 ml of the stop solution (methanol/0.5% formic acid, 1000/1, v/v) was added to terminate the reaction. For the incubation time of 0 minutes, a 1 ml aliquot of the stop solution was added to 1 ml of the skin homogenate mixture, followed by the addition of 10 μl of [14C]TAK-448 under ice-chilled conditions. For inactivation of the metabolic enzymes, the rat skin homogenates were incubated in a hot water bath at 80°C for 15 minutes followed by storage at 4°C until used in the metabolism study. The protease inhibitor cocktail used in this study contained 6 mM AEBSF, 3 mM EDTA-Na, 390 μM bestatin, 43 μM E-64, 3 μM leupeptin, and 0.9 μM aprotinin, and the effects of each component were examined.
Liquid Chromatography–Tandem Mass Spectrometry Analysis.
Concentrations of TAK-448 in rat plasma were determined by liquid chromatography–tandem mass spectrometry using the internal standard, 2-(N-Acetyl-D-tyrosyl-D-tryptophyl-L-asparaginyl-L-threonyl-Lphenylalanyl) hydrazinocarbonyl-L-leucyl-Nω-methyl-L-arginyl-L-tryptophanamide monoacetate (T-1532583). The liquid chromatography system was an LC-20A system (Shimadzu Co., Kyoto, Japan), while the mass spectrometer was an API4000 system (Applied Biosystems/MDS Sciex, Foster City, CA) equipped with an electrospray ionization source. The column used was an Inertsil ODS-3 (150 × 2.1 mm i.d., particle size 5 μm; GL Science, Tokyo, Japan). Column temperature was maintained at 40°C. Purified water/acetic acid (500:1, v/v) as the mobile phase A and methanol/acetic acid (500:1, v/v) as the mobile phase B were used with varying compositions at a flow rate of 0.2 ml/min. The HPLC gradient program was the time (minutes)/% of B (v/v): 0/15, 1/15, 4/70, 8.5/70, 8.6/15, and 15/15. The turbo ion spray interface was operated in the positive ion mode at an ionization voltage of 5500 V with a turbo gas heater at 550°C. Curtain gas, ion source gas 1, ion source gas 2, and collision gas were at 40 psi (276 kPa), 50 psi (345 kPa), 80 psi (552 kPa), and 10, respectively. Quantitation was performed by selected reaction monitoring. The mass transition was from m/z 613.5 to 511.0 for TAK-448 and from m/z 649.8 to 754.2 for the internal standard. The collision energies for TAK-448 and the internal standard were 23 V and 17 V, respectively, and the dwell time for each compound was 0.15 seconds. The analytical data were processed with Analyst software, version 1.4.1 (Applied Biosystems/MDS Sciex).
Analytical Methods for the 14C Samples.
The radioactivity in the biologic samples, organic solvent extract, and the samples for HPLC was determined by the liquid scintillation counter as reported previously (Tanayama et al., 1978; Kondo et al., 1995). The radioactivity in the plasma and organic solvent was directly counted in a toluene-phosphor mixture containing nonionic detergent. [14C]TAK-448 and its metabolite in the plasma and the reaction mixture after metabolism in the skin homogenates were analyzed by an HPLC equipped with an online liquid scintillation counter detector as reported previously (Moriya et al., 2019). The HPLC consisted of a Waters 2795 HPLC system (Waters Corp., Milford, MA) with a Capcellpak C18 MG II column (250 mm × 4.6 mm i.d., particle size 5 μm; Shiseido Co. Ltd., Tokyo, Japan) for separation of TAK-448 and its related compounds. The column temperature was maintained at 40°C. Then, 0.05 mol/l ammonium formate/distilled water/acetonitrile/formic acid (10:80:10:0.05, by vol.) as the mobile phase A and 0.05 mol/l ammonium formate/acetonitrile/formic acid (10:90:0.05, by volume) as the mobile phase B were used with varying compositions at the flow rate of 1.0 ml/min. The HPLC gradient program was time (minutes)/% of B (v/v): 0/0, 10/0, 35/40, 40/90, 40.1/0, and 55/0. The radioactive materials in plasma were extracted with five volumes of the extracting solution (methanol/formic acid, 1000:1, v/v). After evaporation of the organic solvent under a nitrogen stream, the residue was dissolved in a small volume of mobile phase A. For samples obtained in the in vitro study, after the termination of the metabolic reaction, the samples were centrifuged at approximately 1500g at 10°C for 10 minutes to obtain the supernatants. The supernatants were evaporated to dryness under a nitrogen stream and the residues were dissolved in mobile phase A, followed by composition analysis. Radioactivity was monitored online by Radiomatic 625TR (PerkinElmer Inc., Waltham, MA). Under these conditions, TAK-448 and M-I were eluted at about 30 and 7 minutes, respectively.
Data Analysis.
Concentrations of total radioactivity in the biologic samples and those of the metabolite in the plasma were expressed as TAK-448 equivalent values. The data are reported as mean or mean ± S.D. Metabolic profiles of the radioactivity in the rat plasma were determined using pooled samples from three animals. The Cmax, time to reach Cmax, and concentration at 5 minutes after administration were established directly from the plasma concentration data. The half-life (t1/2), AUC, area under the moment curve (AUMC), volume of distribution at steady state, and total clearance were calculated by noncompartmental methods in WinNonlin (version 6.3; Pharsight Corp., Mountain View, CA). Following intravenous bolus dosing, AUMC/AUC is the mean residence time in the systemic circulation. Frequently, the mean absorption time (MAT) is calculated as the difference between AUMC/AUC following nonintravenous bolus administration (frequently oral, but here subcutaneous) and mean residence time. However, this difference is, in fact, the mean loss time from the absorption site (MLTab site), since both absorption from and degradation at the absorption site affect this parameter. The values for t1/2 were calculated by the least-squares method and AUC and AUMC were calculated by the linear trapezoidal method.
The ratio of the concentration of TAK-448 in the skin homogenate after the reaction (the remaining percentage of initial concentration) was calculated by the following equation: where CT is the concentration of TAK-448 in the skin homogenate after incubation at 4 or 37°C for designated time, and C0 is the concentration at time 0.
where CT is the concentration of TAK-448 in the skin homogenate after incubation at 4 or 37°C for designated time, and C0 is the concentration at time 0.
Results
Nonlinear Pharmacokinetics of TAK-448 after a Single Subcutaneous Administration to Rats.
The linearity in the plasma pharmacokinetics of TAK-448 was investigated after single subcutaneous and intravenous administrations to rats (Figs. 2 and 3; Table 1). After subcutaneous administration of TAK-448 at a dose of 0.1 mg/kg, the concentration of TAK-448 reached a maximum of 90.0 ng/ml (Cmax) at 0.5 hours (time to reach Cmax), and then the compound was rapidly eliminated with a t1/2 value of 0.5 hours. The AUC was calculated to be 123 ng∙h/ml at that dose. At the subcutaneous dose of 1 mg/kg, the Cmax and AUC values were increased at a less than dose-proportional manner to 381.3 ng/ml and 804 ng∙h/ml, respectively. However, when the dose was increased to 10 mg/kg, the Cmax and AUC values decreased to 284.6 ng/ml and 309 ng∙h/ml, respectively. Although the t1/2 values were scarcely affected by the dose escalation, the time to reach Cmax, AUMC/AUC, and MLTab site values were decreased at the highest subcutaneous dose. On the other hand, almost linear pharmacokinetics results were observed after intravenous administration of TAK-448 over the dose range of 0.01–10 mg/kg (Table 1). The concentration at 5 minutes after administration, the first sampling time, and the AUC showed almost dose-proportional increases, as shown in Table 1. The t1/2 and mean residence time values were almost constant and the t1/2 values were comparable with those observed after subcutaneous administrations. The values of the volume of distribution at steady state and total clearance were constant over the intravenous dose range of 0.01–10 mg/kg.

Time profiles for the plasma concentrations of TAK-448 after subcutaneous administration of TAK-448 to rats. TAK-448 was dorsally injected into male Sprague-Dawley rats at doses of 0.1 mg/kg (open circle), 1 mg/kg (open square), and 10 mg/kg (open triangle). Each point and vertical bar represents the mean ± S.D. of five animals.

Time profiles for the plasma concentrations of TAK-448 after intravenous administration of TAK-448 to rats. TAK-448 was injected into the femoral vein of male Sprague-Dawley rats at doses of 0.01 mg/kg (open circle), 0.1 mg/kg (open square), 1 (open triangle) and 10 mg/kg (open diamond). Each point and vertical bar represents the mean ± S.D. for five animals.
Pharmacokinetic parameters of TAK-448 after subcutaneous and intravenous administrations of TAK-448 to rats
The MLTab site after subcutaneous administration was calculated by subtracting the AUMC/AUC value after intravenous administration (i.e., mean residence time) from the AUMC/AUC value after subcutaneous administration at the same dose. Mean ± S.D. (n = 5).
Nonlinearity of the First-Pass Metabolism after a Single Subcutaneous Administration of [14C]TAK-448 to Rats.
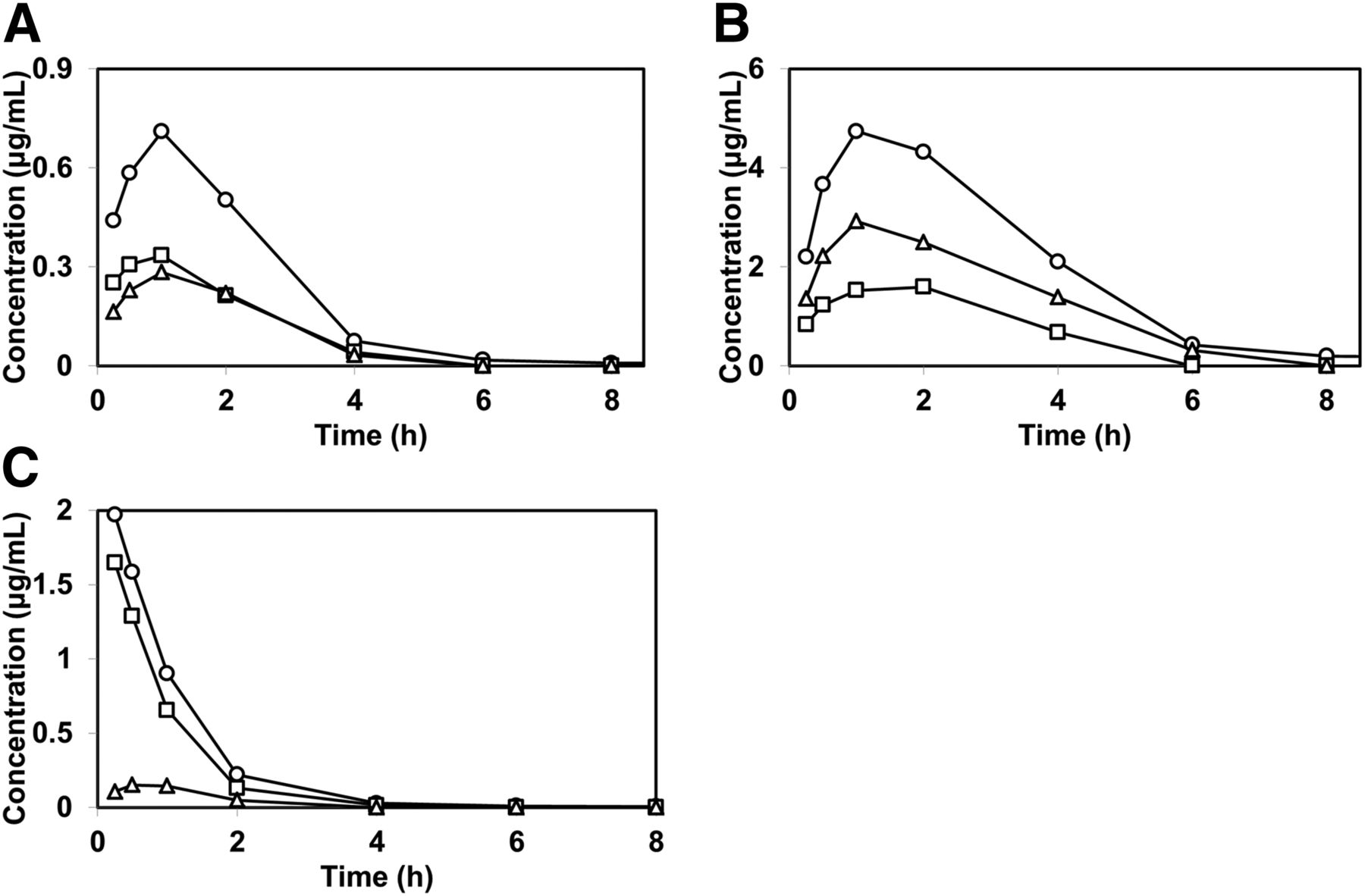
The concentrations of total radioactivity, TAK-448, and M-I (a hydrolyzed metabolite) in the plasma of rats were investigated after subcutaneous and intravenous administrations of [14C]TAK-448 (Fig. 4; Table 2). After subcutaneous administration, the increases in the Cmax and AUC values for total radioactivity were slightly less than dose proportional; the Cmax values were 1.124 and 6.361 μg/ml and the AUC values were 2.326 and 16.020 μg∙h/ml at 1 and 10 mg/kg, respectively. On the other hand, ascending doses of [14C]TAK-448 from 1 to 10 mg/kg showed noticeable decreases in the Cmax and AUC values for TAK-448; the Cmax values were 0.624 and 0.410 μg/ml and the AUC values were 1.184 and 0.386 μg·h/ml at the subcutaneous doses of 1 and 10 mg/kg, respectively. Based on the AUCs, 50.9% and 2.4% of the total radioactivity were accounted for by TAK-448 after subcutaneous administration of [14C]TAK-448 to rats at doses of 1 and 10 mg/kg, respectively. In contrast, the Cmax and AUC values of M-I showed more than dose-proportional increases within the subcutaneous dose range of 1 and 10 mg/kg. Composition analysis of the rat plasma after intravenous administration of [14C]TAK-448 at a dose of 1 mg/kg indicated that TAK-448 was the major component accounting for 76.4% of the total radioactivity based on the AUCs.

Plasma concentrations of total radioactivity, TAK-448, and M-I after subcutaneous and intravenous administrations of [14C]TAK-448 to rats. [14C]TAK-448 was subcutaneously injected at doses of 1 mg/kg (A) and 10 mg/kg (B), and intravenously administered at a dose of 1 mg/kg (C) to male Sprague-Dawley rats. Data for the total radioactivity (open circle) represent the mean plasma concentrations for three animals and those for TAK-448 (open square) and M-I (open triangle) represent the concentrations for the pooled plasma from three animals.
Pharmacokinetic parameters of total radioactivity, TAK-448, and M-I in the plasma of rats after subcutaneous and intravenous administrations of [14C]TAK-448
Parameters for total radioactivity are calculated from the mean plasma concentrations for three animals. Parameters for TAK-448 and M-I are calculated from the concentrations for the pooled samples from three animals. Numbers in parentheses denote percentage of total radioactivity. Data are taken from Fig. 3 and Moriya et al. (2019).
Effect of the Subcutaneous Coadministration of the Protease Inhibitor Cocktail on the Pharmacokinetics of TAK-448.
The influence of a subcutaneously coadministered protease inhibitor cocktail on the pharmacokinetics of the total radioactivity, TAK-448, and M-I in rats was examined after subcutaneous and intravenous administrations of [14C]TAK-448 (Fig. 5; Table 3). The concentrations of the protease inhibitors were 40 mM AEBSF, 20 mM EDTA, 2.6 mM bestatin, 0.28 mM E-64, 0.02 mM leupeptin, and 0.006 mM aprotinin in the dosing formulation. After subcutaneous coadministration of [14C]TAK-448 and the protease inhibitors to rats, in contrast to the results depicted in Fig. 4, the Cmax and AUC values of TAK-448 were not decreased but increased less than dose proportionally, while those of M-I increased dose proportionally with the dose increase from 1 to 10 mg/kg. In addition, subcutaneous coadministration of the protease inhibitors increased the Cmax and AUC values of TAK-448 at the dose of 10 mg/kg compared with those without inhibitors, while those of M-I were less than those without protease inhibitors (Tables 2 and 3). On the other hand, the comparison of the pharmacokinetic parameters indicated that subcutaneous coadministration of the protease inhibitors had little effect on the plasma concentration profiles for all of the components after intravenous administration of [14C]TAK-448 (Tables 2 and 3).

Effect of the subcutaneously coadministered protease inhibitor cocktail on the time profiles for the plasma concentrations of TAK-448 after administration of [14C]TAK-448. [14C]TAK-448 was injected subcutaneously at doses of 1 mg/kg (A) and 10 mg/kg (B), and administered intravenously at a dose of 1 mg/kg (C) to male Sprague-Dawley rats. Data for the total radioactivity (open circle) represent the mean plasma concentrations for three animals and those for TAK-448 (open square) and M-I (open triangle) represent the concentrations for the pooled plasma from three animals. The subcutaneously administered protease inhibitor cocktail contains 40 mM AEBSF, 20 mM EDTA, 2.6 mM bestatin, 0.28 mM E-64, 0.02 mM leupeptin, and 0.006 mM aprotinin.
Pharmacokinetic parameters of total radioactivity, TAK-448, and M-I in the plasma of rats after coadministration of [14C]TAK-448 and protease inhibitor cocktail
Parameters for total radioactivity are calculated from the mean plasma concentrations for three animals. Parameters for TAK-448 and M-I are calculated from the concentrations for the pooled samples from three animals. Numbers in parentheses denote percentage of total radioactivity. Data are taken from Fig. 4.
In Vitro Metabolism of [14C]TAK-448 in Rat Skin Homogenates.
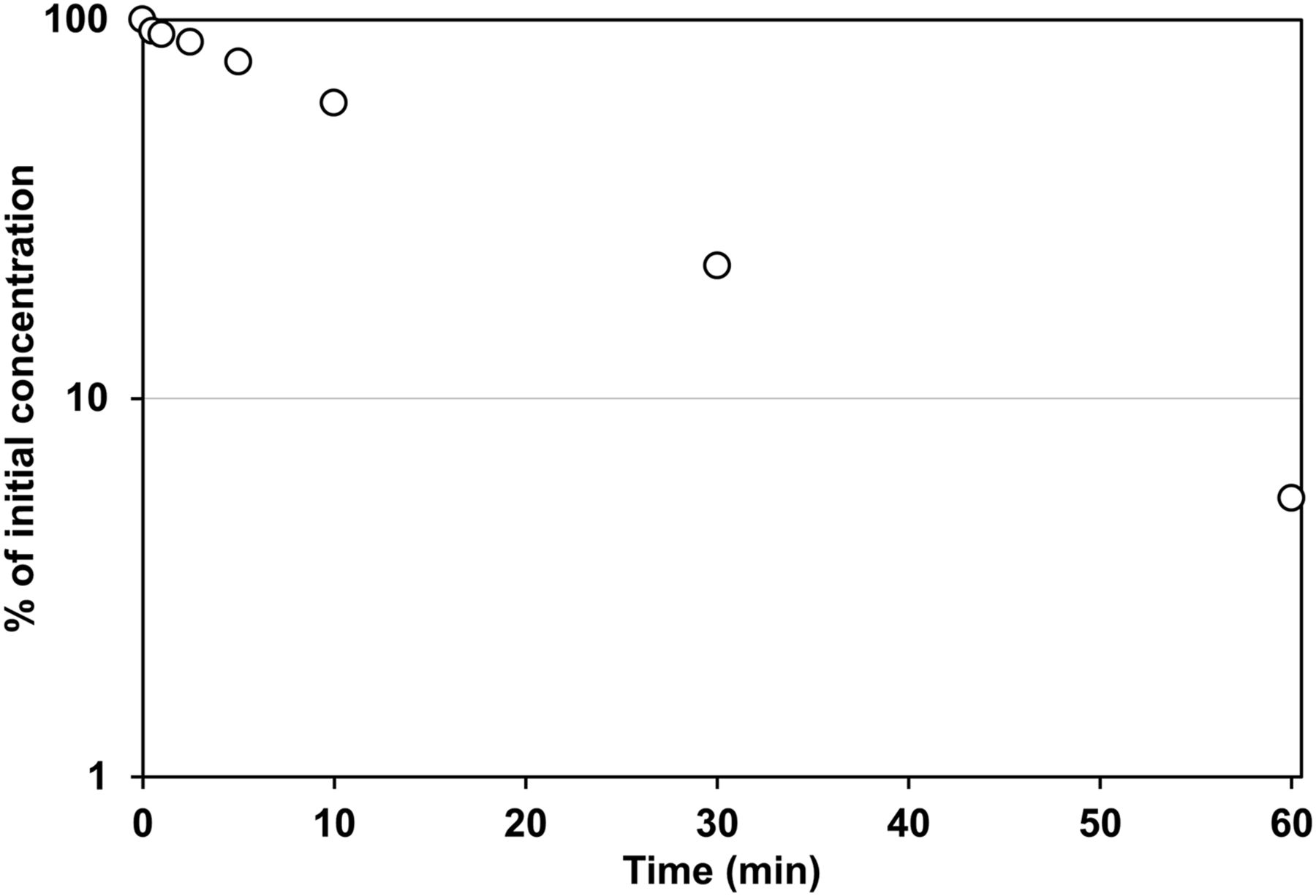
The time profiles for the degradation of [14C]TAK-448 (1 μg/ml) in the skin homogenates prepared from rats were examined (Fig. 6). The spiked [14C]TAK-448 was rapidly degraded in a time-dependent manner with a t1/2 value of 14.4 minutes at 37°C. To examine whether the degradation of TAK-448 was mediated by metabolic enzymes, the reaction was performed under ice-chilled conditions or using a preheated homogenate at 80°C for 15 minutes (Fig. 7), under which condition the metabolic enzymes were reported to be inactivated (Sato et al., 2008). After incubation for 60 minutes with rat skin homogenate, the residual ratios of TAK-448 were 91.2% and 98.2% for the ice-chilled and preheated samples, respectively. The concentration dependency of the metabolism was examined at the concentration of 100 μg/ml of TAK-448, and the residual ratio at the termination of the reaction was 3.6% (Fig. 7). In addition, the inhibitory effects of the protease inhibitor cocktail, also used in the in vivo study, and its ingredients on the metabolism of TAK-448 were investigated to determine the enzymes responsible for the subcutaneous metabolism of TAK-448 (Fig. 8). In the presence of the protease inhibitor cocktail, the metabolism of TAK-448 was significantly inhibited; the residual ratio at 60 minutes after the initiation of the reaction was 76.5%. AEBSF and EDTA also had inhibitory effects on the metabolism with the residual ratios of 71.7% and 16.7% at the termination of the reaction, respectively. The other components (bestatin, E-64, leupeptin, and aprotinin) had little effect.

Time profile of the metabolism of [14C]TAK-448 in the rat skin homogenate. Skin homogenates (900 μg) prepared from Sprague-Dawley rats were incubated at 37°C for 0, 0.5, 1, 2.5, 5, 10, 30, and 60 minutes in the presence of [14C]TAK-448 (1 μg/ml). Each point represents the mean value for two experiments.

Involvement of the metabolic enzymes on the degradation of [14C]TAK-448 in the rat skin homogenate. [14C]TAK-448 (1 μg/ml) was incubated with the rat skin homogenate (900 μg) at 37°C (control). The concentration dependency was evaluated in the presence of 100 μg/ml of unlabeled TAK-448. The incubation was carried out at 4°C to estimate the temperature dependency (ice-chilled conditions). The skin homogenate was preheated at 80°C for 15 minutes to inactivate the metabolic activity and metabolism was evaluated as for the control (heat-treated skin). Incubation time was 60 minutes for this experiment. Each column represents the mean ± S.D. of three experiments.

Effects of the protease inhibitors on the metabolism of [14C]TAK-448 in the rat skin homogenate. [14C]TAK-448 (1 μg/ml) was incubated with the rat skin homogenate (900 μg) at 37°C for 60 minutes with a protease inhibitor cocktail (6 mM AEBSF, 3 mM EDTA-Na, 390 μM bestatin, 43 μM E-64, 3 μM leupeptin, and 0.9 μM aprotinin) and its individual components.
Species Differences in the Metabolism of [14C]TAK-448 with Skin Homogenates.
The metabolism of [14C]TAK-448 (1 μg/ml) in the skin homogenates was compared among rat, dog, monkey, and human. After incubation for 60 minutes with the skin homogenates the residual ratios of TAK-448 were 5.5%, 14.8%, 14.4%, and 45.6% in rat, dog, monkey and human, respectively.
Discussion
In the present study, we attempted to clarify the mechanism for the less than dose-proportional nonlinear pharmacokinetics after single dose subcutaneous administration of TAK-448 to rats and ascertained that first-pass metabolism via proteases in the rat skin could be involved in the nonlinearity.
Our previous pharmacokinetic study with [14C]TAK-448 revealed that the BA of TAK-448 was lower than the apparent fraction of total radioactivity after subcutaneous administration to rats, suggesting that TAK-448 underwent first-pass skin metabolism (Moriya et al., 2019). In the present study the markedly less than dose-proportional nonlinear pharmacokinetics results were observed after a single subcutaneous administration of TAK-448 to rats in the dose range from 0.1 to 10 mg/kg (Figs. 2 and 3; Table 1). The AUC increased from 123 to 804 ng·h/ml after subcutaneous doses from 0.1 to 1 mg/kg. However, with the increase in subcutaneous dose from 1 to 10 mg/kg, the AUC decreased to 309 ng·h/ml. In contrast to subcutaneous administration, the pharmacokinetics was almost linear after intravenous administration in the dose range from 0.01 to 10 mg/kg, suggesting that the nonlinearity was specific to subcutaneous administration with dose-dependent enhancement of the presystemic clearance, and thus would not be caused by saturation of the plasma protein binding or induction of metabolizing enzymes. We previously reported the moderate and almost constant plasma protein binding of TAK-448 in rats (Moriya et al., 2019). It would be also quite unlikely that the enzymes were induced after a single dose. We attempted to clarify these nonlinear pharmacokinetic properties following subcutaneous administration by using [14C]TAK-448 and a protease inhibitor cocktail.
The dose-ascending study of [14C]TAK-448 after single subcutaneous administration to rats suggested that the subcutaneous metabolism of TAK-448 could be markedly facilitated by a dose increase from 1 to 10 mg/kg (Fig. 4; Table 2). Although the AUC of TAK-448 decreased from 804 to 309 ng·h/ml after subcutaneous administration of unlabeled TAK-448 at the doses of 1 and 10 mg/kg, the AUC of total radioactivity was increased from 2.326 to 16.020 μg·h/ml after subcutaneous administration of [14C]TAK-448 in the same dose range, suggesting the influence of radiolabeled metabolites. Based on AUC ratios, the composition analysis showed that 50.9% and 2.4% of total radioactivity were accounted for by TAK-448 after subcutaneous administration of [14C]TAK-448 at the doses of 1 and 10 mg/kg, respectively. On the other hand, the composition ratios of M-I, a hydrolyzed metabolite, increased from 30.6% to 84.5% in the same dose range. These results suggest that the subcutaneous metabolism of TAK-448 to M-I showed dose-dependent enhancement from 1 to 10 mg/kg.
The extent and rate of metabolism in skin have been reported to affect the absorption of subcutaneously injected drugs and much effort has been made to increase the BA. The BA of subcutaneously injected insulin was increased by coadministration of collagen or pretreatment with an ointment containing protease inhibitors in rats and humans, suggesting that the collagen or protease inhibitors prevented insulin from degradation by the proteolytic enzymes in subcutaneous tissue (Hori et al., 1989; Takeyama et al., 1991). To further characterize the subcutaneous degradation of TAK-448 in rats, we examined the effects of subcutaneous coadministration of a protease inhibitor cocktail (Fig. 5; Table 3). The subcutaneous coadministration had little effect on the pharmacokinetic parameters of total radioactivity after single subcutaneous and intravenous doses of 1 mg/kg; however, it significantly increased the AUC of TAK-448, along with a reduction of M-I, at a subcutaneous dose of 10 mg/kg compared with that observed without the protease inhibitors (Tables 2 and 3). These results suggest that subcutaneous metabolism by proteases might contribute to the reduced absorption of TAK-448 at the high dose. It has been reported that most proteases are synthesized as inactive precursors (zymogens) and are usually converted to the active enzyme by proteolytic processing mediated either by other proteases or autocatalysis (Neurath, 1999). It is possible that TAK-448 at high concentration could trigger the proteolytic cascade and activate the proteases responsible for the subcutaneous metabolism of TAK-448. A more detailed characterization of the mechanism for the dose-dependent enhancement of the subcutaneous metabolism is required to understand the nonlinearity observed for TAK-448. However, the present study highlights a previously underemphasized aspect of calculating MATs. In Table 1 we calculate the MLTab site as the difference between AUMC/AUC following subcutaneous versus intravenous administration. This mean loss time parameter is the sum of the MAT and mean degradation time at the absorption site. By apparently activating the enzymes at the absorption site we have markedly increased the rate of metabolism at the absorption site, thereby markedly decreasing the calculated MLTab site for the 10 mg/kg dose, probably with little effect on the actual MAT. That is, since so little of the dose is absorbed, the value for MLTab site is predominantly a measure of the rapid increase in degradation rate.
In vitro studies were performed to characterize the degradation of TAK-448 in rat skin homogenate (Figs. 6 and 7), and the results suggest that TAK-448 was metabolized by proteases sensitive to serine protease inhibitors. When incubated with rat skin homogenate, [14C]TAK-448 was degraded in a time-dependent manner with a t1/2 value of 14.4 minutes, and was also temperature dependent. Moreover, there was no degradation of [14C]TAK-448 when the metabolic enzymes were inactivated by preheat treatment. The time and temperature dependencies suggest that the degradation of TAK-448 was mediated by metabolic enzymes. However, concentration dependency was not observed, at least up to 100 μg/ml; indicating that the responsible enzyme has a low affinity for TAK-448. The metabolic enhancement was observed in vivo associated with enzyme activation, and there would be a limit to quantitative consideration because it would be difficult to assess the enzyme activation from the in vitro study. A sensitivity profile to specific protease inhibitors was examined to identify the class of protease responsible for the metabolism of TAK-448 in rat skin homogenate (Fig. 8). In the presence of a protease inhibitor cocktail the metabolism of TAK-448 was significantly inhibited with the remaining percentage of 76.5%. The protease inhibitor cocktail used in this metabolic study consisted of 6 mM AEBSF, 3 mM EDTA-Na, 390 μM bestatin, 43 μM E-64, 3 μM leupeptin, and 0.9 μM aprotinin, and the inhibitory effects of each component were also investigated. Among the constituents of the protease inhibitor cocktail, AEBSF and EDTA inhibited the metabolism of TAK-448 in rat skin homogenate, and the remaining ratios of TAK-448 were 71.7% and 16.7% of the initial concentration, respectively. AEBSF and EDTA are known to be an irreversible inhibitor of serine proteases and a reversible metalloprotease inhibitor, respectively. These results suggest that [14C]TAK-448 would be metabolized by enzymes sensitive to the serine protease inhibitor. Some serine proteases, such as kallikrein, plasmin, plasminogen activator, and matrix metalloproteases, are reported to be expressed in skin (Lundwall and Brattsand, 2008; Toriseva and Kähäri, 2009), and further studies will be required to identify which serine protease was involved in subcutaneous metabolism of TAK-448.
The nonclinical pharmacokinetic studies suggest that a high subcutaneous dose of TAK-448 could possibly lead to limited exposure of this compound in the clinic. The nonlinear pharmacokinetics in rats with less than dose proportionality was sensitive to protease inhibitors (Tables 2 and 3). Although the mechanism is still unclear, the enhancement of protease activity would definitely be related to the nonlinearity at high doses in rats. Even though similar enhancement could occur, dogs and monkeys showed almost linear pharmacokinetics after subcutaneous administration of TAK-448 at up to 10 mg/kg (Supplemental Tables 1 and 2), probably because the metabolic activities are lower in dogs and monkeys compared with rats, as shown in our in vitro data. If this is true, we do not think the severe nonlinear pharmacokinetics would be observed in human with the lowest metabolic activity in vitro. In addition, the low subcutaneous dose of TAK-448 showed moderate absorption with BA of more than 65% in rats (Moriya et al., 2019), suggesting that a slow-release formulation could be a reasonable approach in order to avoid unfavorable pharmacokinetics in the clinical development of TAK-448.
In conclusion, it was demonstrated that TAK-448 showed less than dose-proportional nonlinear pharmacokinetics with a reduction in the AUC after subcutaneous administration to rats in a dose range of 1–10 mg/kg. Our results suggested that subcutaneous first-pass metabolism would be enhanced for the peptide predominantly metabolized by serine proteases. This is the first report of dose-dependent enhancement of the subcutaneous metabolism of a drug. Our study showed the importance of characterization of enzymes involved in subcutaneous metabolism of peptide drugs, which leads to mitigation of the risk of low exposure of subcutaneously administered peptide drugs in the clinic.
Authorship Contributions
Participated in research design: Moriya, Kogame, Tagawa, Benet.
Conducted experiments: Moriya.
Performed data analysis: Moriya, Kogame, Tagawa.
Wrote or contributed to the writing of the manuscript: Moriya, Kogame, Tagawa, Morohashi, Kondo, Asahi, Benet.
Footnotes
- Received March 11, 2019.
- Accepted June 3, 2019.
↵1 Current affiliation: Axcelead Drug Discovery Partners, Inc.
↵2 Current affiliation: Nemoto Science. Co., Ltd., Tokyo, Japan.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AEBSF
- 4-(2-aminoethyl) benzenesulfonyl fluoride
- AUC
- area under the plasma concentration-time curve
- AUMC
- area under the moment curve
- BA
- bioavailability
- E-64
- [(2S,3S)-3-carboxyoxirane-2-carbonyl]-L-leucine (4-guanidinobutyl) amide
- HPLC
- high-performance liquid chromatography
- MAT
- mean absorption time
- MLTab site
- mean loss time from the absorption site
- t1/2
- half-life
- M-I
- N-Acetyl-D-tyrosyl-trans-4-hydroxy-Lprolyl-L-asparaginyl-L-threonine
- TAK-448
- 2-(N-acetyl-D-tyrosyl-trans-4-hydroxy-L-prolyl-L-asparaginyl-L-threonyl-Lphenylalanyl) hydrazinocarbonyl-L-leucyl-Nω-methyl-L-arginyl-L-tryptophanamide monoacetate
- Copyright © 2019 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}