


Visual Overview

Abstract
Abiraterone (Abi) acetate (AA) is a prodrug of Abi, a CYP17A1 inhibitor used to treat patients with advanced prostate cancer. Abi is a selective steroidal inhibitor that blocks the biosynthesis of androgens. It undergoes extensive biotransformation by steroid pathways, leading to the formation of pharmacologically active Δ4-Abi (D4A) and 5α-Abi. This study aimed to characterize the glucuronidation pathway of Abi and its two active metabolites. We show that Abi, its metabolites, and another steroidal inhibitor galeterone (Gal) undergo secondary metabolism to form glucuronides (G) in human liver microsomes with minor formation by intestine and kidney microsomal preparations. The potential clinical relevance of this pathway is supported by the detection by liquid chromatography–tandem mass spectrometry of Abi-G, D4A-G, and 5α-Abi-G in patients under AA therapy. A screening of UGT enzymes reveals that UGT1A4 is the main enzyme involved. This is supported by inhibition experiments using a selective UGT1A4 inhibitor hecogenin. A number of common and rare nonsynonymous variants significantly abrogate the UGT1A4-mediated formation of Abi-G, D4A-G, and 5α-Abi-G in vitro. We also identify Gal, Abi, and its metabolites as highly potent inhibitors of steroid inactivation by the UGT pathway with submicromolar inhibitor constant values. They reduce the glucuronidation of both the adrenal precursors and potent androgens in human liver, prostate cancer cells, and by recombinant UGTs involved in their inactivation. In conclusion, tested CYP17A1 inhibitors are metabolized through UGT1A4, and germline variations affecting this metabolic pathway may also influence drug metabolism.
SIGNIFICANCE STATEMENT The antiandrogen abiraterone (Abi) is a selective steroidal inhibitor of the cytochrome P450 17α-hydroxy/17,20-lyase, an enzyme involved in the biosynthesis of androgens. Abi is metabolized to pharmacologically active metabolites by steroidogenic enzymes. We demonstrate that Abi and its metabolites are glucuronidated in the liver and that their glucuronide derivatives are detected at variable levels in circulation of treated prostate cancer patients. UDP-glucuronosyltransferase (UGT)1A4 is the primary enzyme involved, and nonsynonymous germline variations affect this metabolic pathway in vitro, suggesting a potential influence of drug metabolism and action in patients. Their inhibitory effect on drug and steroid glucuronidation raises the possibility that these pharmacological compounds might affect the UGT-associated drug-metabolizing system and pre-receptor control of androgen metabolism in patients.
Introduction
Prostate cancer (PCa) is the leading cause of cancer in men and the second cause of death in Western countries (Miller et al., 2016). Much progress has been made in the last few years with new generation hormonal therapies. Abiraterone (Abi) and enzalutamide have significantly changed clinical management, increasing survival and quality of life of patients with metastatic PCa (Fizazi et al., 2012; Scher et al., 2012; Beer et al., 2014; Ryan et al., 2015; Tucci et al., 2015). In advanced disease, PCa cells remain dependent on androgens for proliferation (Montgomery et al., 2008). Therefore, Abi, orally administered as the prodrug Abi acetate (AA), is commonly used as part of the androgen deprivation therapy in advanced disease clinical settings.
Abi is a selective steroidal inhibitor of the cytochrome P450 17α-hydroxy/17,20-lyase (CYP17A1), an enzyme involved in the biosynthesis of androgens. Abi is an androgen receptor axis–targeted agent that blocks the formation of androgens in testes, adrenal, peripheral tissues, and prostate tumor cells. Recently, new metabolites of Abi formed by steroidogenic enzymes have been described, with at least two exhibiting significant pharmacological activities, Δ4-Abi (D4A) and 5α-Abi (Li et al., 2016). Abi is first converted to D4A by 3β-hydroxysteroid dehydrogenase and blocks CYP17A1, 3β-hydroxysteroid dehydrogenase, and steroid-5α-reductase, whereas D4A also antagonizes the androgen receptor (Li et al., 2015). D4A is irreversibly converted to 3-keto-5α-Abi or 3-keto-5β-Abi. Both metabolites may then be converted to their 3α-OH and 3β-OH derivatives for a total of six downstream metabolites of D4A. The 5β-Abi metabolites were not reported to be active. In contrast, the direct product of D4A, 5α-Abi, acts as an agonist of the androgen receptor (Li et al., 2016). Likewise, the structurally related CYP17A1 inhibitor galeterone (Gal) yields analogous metabolites (Alyamani et al., 2017).
We hypothesized that, similar to endogenous steroids, Abi, its metabolites, and the structurally related Gal (Supplemental Fig. 1) undergo direct metabolism by the glucuronidation pathway. A large family of 19 UDP-glucuronosyltransferase (UGT) enzymes catalyzes this phase II drug metabolic pathway (Guillemette et al., 2014). UGTs are involved in the addition of a glucuronic acid (GlcA) moiety to a large diversity of acceptor molecules, including therapeutic drugs from all classes containing hydroxyl, carboxylic acid, thiol, or amine groups (Guillemette et al., 2014). This leads to the formation of glucuronide (G) derivatives that most often lack biologic activity and that are readily excreted through bile and urine. This pathway also catalyzes the clearance of steroids such as the potent androgen dihydrotestosterone (DHT) and thereby regulates their bioavailability and the hormonal environment to which PCa cells are exposed (Beaulieu et al., 1996). The UGT pathway undergoes complex regulation. This involves regulatory loops characterized by the ability of UGT substrates to regulate the expression of genes encoding UGT enzymes involved in their metabolism (Hu et al., 2014). We further postulated that CYP17A1 inhibitors potentially influence endogenous steroid metabolism by affecting their conjugation by UGTs.
In this study, we report that Abi and its metabolites as well as Gal undergo metabolism by the UGT pathway and that they inhibit inactivation of steroids. Results of these in vitro investigations are reinforced by the detection of G derivatives of Abi, D4A, and 5α-Abi in PCa patients treated with AA.
Materials and Methods
Chemicals and Reagents.
All chemicals and reagents were of the highest grade commercially available. UDP-GlcA and bilirubin were obtained from Sigma-Aldrich (Oakville, ON, Canada). All chemicals and solvents used for the mass spectrometry were high-pressure liquid chromatography (LC) grade. Milli-Q water was produced using the Millipore system. Abi and Gal were purchased from Toronto Research Chemical (Toronto, ON, Canada). Tacrolimus (FK-506) was purchased from Cell Signaling Technology (Danvers, MA). β-glucuronidase type VII from Escherichia coli was purchased from Sigma-Aldrich (St. Louis, MO). Hecogenin was obtained from Santa Cruz Biotechnology (Dallas, TX). The Organic Synthesis Service of the CHU de Québec Research Center (Quebec, QC, Canada) synthesized D4A and 5α-Abi based on a published method (Li et al., 2016). To identify D4A and 5α-Abi synthesized by the Medical Chemistry platform, NMR spectra were recorded on a Bruker Advance 400 digital spectrometer (Billerica, MA) at 400 MHz for 1H NMR. Spectra were referenced relative to the central residual proton solvent resonance in 1H NMR (CDCl3 = 7.26 ppm, MeOH-d4 = 3.31 ppm). High-pressure LC analyses for chemical purities (D4A 96% purity and 5α-Abi 98% purity) were performed on a Shimadzu Prominence instrument (Kyoto, Japan) using a diode array detector (wavelength detection = 190). Abi-G, D4A-G, 5α-Abi-G, and Gal-G analytical standards were produced from in vitro enzymatic assays, as described (Caron et al., 2019). Briefly, the G formed was treated with β-glucuronidase type VII from E. coli (50 U) incubated at 37°C for 20 hours in phosphate buffer (pH 6.5) to cleave the GlcA group and allow quantification with a calibration curve of the corresponding aglycone. Deuterated Gs were also produced and purified according to the same procedure. Microsomal preparations from human tissues [liver mixed gender pool of 50 individuals: cat. no. H0620, lot 1410013 (24 females, 26 males); 1210267 (19 females, 31 males) and 1610016 (20 females, 30 males); kidney mixed gender pool of eight individuals, cat. no. H0610.R, lot 0810236 (four females, four males); and colon mixed gender pool of seven, cat. no 452210, lot 05886 (four females, three males)] and commercial Supersomes [UGT1A1 (cat. no. 456411, lot 6320009), 1A3 (cat. no. 456413, lot 7065003), 1A4 (cat. no. 456414, lot 6334004, 7179004, and 8073001), 1A6 (cat. no. 456416, lot 6216001), 1A7 (cat. no. 456407, lot 4308001), 1A8 (cat. no. 456418, lot 7177002), 1A9 (cat. no. 456419, lot 7065002), 2B4 (cat. no. 456424, lot 6244003), 2B7 (cat. no. 456427, lot 7101003), 2B15 (cat. no. 456435, lot 5348003), and 2B17 (cat. no. 456437, lot 6312001, 8038005, and 8177003)] were purchased from Xenotech (Kansas City, KS), BD Biosciences (Woburn, MA), and Corning (Corning, NY), respectively. Microsomal protein extracts from human embryonic kidney (HEK)293-UGT1A4 cells expressing variant enzymes as well as LNCaP microsomes were prepared by differential centrifugation and quantified by Western blot, as described (Villeneuve et al., 2003; Laverdière et al., 2011).
Glucuronidation Assays.
Enzymatic assays were conducted with 10 µg UGT membrane protein, 50 mM Tris-HCl (pH 7.5), 10 mM MgCl2, 2 mM UDP-GlcA, pepstatin, leupeptin, and 20 µg/ml alamethicin in a final volume of 100 µl. The reactions were terminated by adding 100 μl methanol and were centrifuged at 14,000g for 10 minutes before analysis. Kinetics were conducted with microsomal fractions of a pool of 50 human livers, commercial UGT1A4 supersomes, and HEK293 recombinant human UGT1A4*1 (R11P24L48), UGT1A4*2 (T24), UGT1A4*3 (V48), and UGT1A4*4 (W11). Kinetics was performed using substrate concentrations ranging from 0.1 to 400 µM for 30 minutes. Additional assays were done with 2 μM AA and human liver microsomes (HLM) with an incubation time of 30 minutes and an overnight incubation to compare with Abi. Inhibition assays were performed using concentrations (0.5, 1, 2.5, 5, 10 µM) of a selective UGT1A4 inhibitor hecogenin and a constant concentration near Michaelis constant (Km) of substrates for 30 minutes. The IC50 was then calculated using GraphPad (7.0; GraphPad Software, La Jolla, CA). The inhibitor constant (Ki) values were evaluated using concentrations of hecogenin (0.5, 1, 2.5, 5, 10 µM) and three different concentrations of each substrate (Abi: 2, 100, 200 μM; D4A: 6, 100, 200 μM; 5α-Abi: 5, 100, 200 μM; Gal: 0.5, 2, 5 μM). The inhibition of steroid glucuronidation was performed using Abi, D4A, 5α-Abi, and Gal as inhibitors and by monitoring glucuronidation of two potent androgens [testosterone (Testo), DHT] and their adrenal precursors [dehydroepiandrosterone (DHEA) and androstanediol (A5-diol)]. Those assays were performed for 30 minutes using three concentrations of inhibitors (5, 25, 200 μM) and three concentrations of substrates (5, 25, 200 μM), as indicated in the legends of figures.
Kinetics parameters were determined by nonlinear regression using Sigma Plot 11.0 assisted by Enzyme Kinetics 1.3 (SSI, San Jose, CA) using the Marquardt–Levenberg algorithm, which solves nonlinear least square equations. The best-fitting enzyme kinetics models were subsequently determined using goodness-of-fit criteria, including the coefficient of determination (r2 values), Akaike information criteria, S.E., and 95% confidence intervals, followed by a visual inspection of fitted functions. The enzyme kinetics was subsequently represented by Eadie–Hofstee plots for glucuronidation profiles or Lineweaver–Burk plots for inhibition models. Values are expressed as mean ± S.D. of triplicate determinations of at least two independent experiments. Enzymatic activities were considered statistically significant for P values <0.05, according to Student’s t test variance analysis.
Prostate Cancer Patients Treated with AA.
A study ongoing at our center recruits PCa patients treated with AA at 1000 mg daily dose (2018-4125; CHU de Québec). The study is conducted in accordance with the declaration of Helsinki, and all patients provided consent for this project. Plasma samples were collected at least 1 month after treatment initiation to assess steady state levels of Abi and its metabolites using a protocol described previously (Caron et al., 2019).
Mass Spectrometry Analyses.
Detection of analytes was performed by high-performance LC coupled to a Q-TRAP mass spectrometer (API6500 mass spectrometer; AB Sciex, Concord, ON, Canada) using a validated method, as described (Caron et al., 2019). Briefly, the LC system consisted of a Nexera 30-AD (Shimadzu Scientific Instruments, Columbia, MD) controlled through Analyst Software, version 1.6.2. The mass spectrometer was operated in multiple reactions monitoring mode and equipped with a turbo spray source set at 500°C. The resolution used for Q1 and Q3 was Unit/Unit, and the voltage was held at 5500 V in positive mode. Mass transitions, ionization mode, declustering potential, collision energy, and lower limit of quantification were reported in Caron et al. (2019). For Gal, the chromatographic separation was identical as for Abi, whereas the multiple reactions monitoring mode transition for Gal-G1 and Gal-G2 was 565.2→389.2. The detection of tacrolimus-G and bilirubin-G was performed, as described (Lévesque et al., 2007; Laverdière et al., 2011). For the detection of DHT-G, Testo-G, DHEA-G, A5-diol-3G, and A5-diol-17G, the chromatographic separation was achieved with an ACE 3 C18 HL 3.0 μm packing material, 100 × 4.6 mm (Canadian Life Science, Peterborough, ON, Canada). The mobile phases were water, 1 mM ammonium formate (solvent A) and methanol, 1 mM ammonium formate (solvent B) at a flow rate of 0.9 ml/min. DHT-G (484.4→273.2) and DHEA-G (482.3→271.3) were detected in positive mode and eluted with 75% solvent B. Testo-G (463.1→75) was detected in negative mode and eluted with 75% solvent B. A5-diol-3G and A5-diol-17G (484.4→273.2) were detected in positive mode and eluted with the following gradient: 0 to 4.3 minutes, isocratic 50% B; 4.3 to 4.4 minutes, linear gradient 50% to 90% B; 4.4 to 5.4 minutes, isocratic 90% B; 5.4 to 5.5 minutes, 90% to 50% B; 5.5 to 8.0 minutes, isocratic 50% B.
Results
Glucuronidation of Abi, D4A, 5α-Abi, and Gal in Human Liver Microsomes and Their Detection in PCa Patients Treated with Abi.
Enzymatic assays using pooled HLM revealed the formation of secondary polar metabolites of Abi, D4A, and 5α-Abi. One G derivative was observed upon D4A and 5α-Abi incubations, and two Gs were observed from Abi, named G1 and G2 according to the order of elution (Fig. 1A). The absence of polar metabolites in assays conducted without UDP-GlcA and their hydrolysis by β-glucuronidase treatment further supported that these metabolites correspond to G derivatives (Fig. 1, B and C). In vitro incubations in the same conditions, with the prodrug AA, generated similar results compared with Abi with no difference in the nature of the peaks formed and their abundance in HLM, likely due to its rapid conversion to Abi (Bouhajib and Tayab, 2019). Based on the presence of possible reactive groups, one possibly corresponds to a N-linked G of Abi (G1), whereas the other to an O-linked G (G2). This is supported by our previous analysis (Caron et al., 2019), in which we observed a loss of signal for Abi-G1 peak when analyzed in negative mode, whereas the signal for Abi-G2 remained. The loss of Abi-G1 peak is coherent with the quaternary amine being positively charged. Similarly, two Gs, G1 and G2, were observed for the structurally related CYP17A1 inhibitor Gal. A NMR-based analysis is required to clarify the identity of all G derivatives observed for Abi, D4A, 5α-Abi, and Gal.

Identification of two G metabolites of Abi. (A) Left: Chromatogram of the two polar metabolites observed after a 30-minute incubation of Abi with HLM at the transition 526.7 Da→350.2 Da. Metabolites were named G1 and G2 based on the order of elution. Right: The tandem mass spectrometry (MS/MS) fragmentation profile of the Gs. Masses of the protonated parent ion Abi-G [M+H]+ or [M]+ and of the fragment ion Abi [M-G+H]+ are consistent with the loss of the G moiety (represented by the dashed line in each Abi-G structure). (B) These metabolites are observed in assays with HLM in the presence of the UGT cosubstrate, UDP-GlcA. (C) Polar metabolites disappear when incubated for 20 hours in presence of β-glucuronidase type VII from Escherichia coli.
A subset of PCa patients under AA therapy was analyzed to establish the presence of G derivatives of Abi and its metabolites in vivo, using a validated LC–tandem mass spectrometry method recently published by our group (Caron et al., 2019). Abi-G, D4A-G, and 5α-Abi-G were detected in circulation of patients at variable concentrations. Abi-G and 5α-Abi-G were measured in all patients, whereas D4A-G was below the lower limit of quantification (>5 ng/ml) for two patients (Table 1). A larger study is required to help clarify the clinical significance of this pathway.
Detection of Abi, D4A, and 5α-Abi and their glucuronide derivatives in five prostate cancer patients under AA treatment
UGT1A4-Mediated Glucuronidation of Abi and Its Metabolites: Kinetics Characterization and UGT1A4 Variants with Impaired Activity.
Using recombinant human UGT enzymes, the hepatic UGT1A4 exhibited the highest glucuronidation activity for all four substrates, whereas UGT1A3 displayed much lower activity (Fig. 2). Screening experiments using 2 or 200 µM substrate for 2- or 16-hour incubations with any UGT did not result in the formation of Gs besides with UGT1A4 and at a much lower extent with UGT1A3 (data not shown). This is coherent with the expression profile of these two enzymes (Ohno and Nakajin, 2009; Margaillan et al., 2015), and the minor formation of G derivatives in the intestine and none detected in the kidney. Consistent with a major contribution of the UGT1A4 enzyme, kinetic parameters (apparent Km, Vmax, and clearance values) of all four substrates were almost identical for HLM and UGT1A4 (Fig. 3; Table 2). Eadie–Hofstee plots of HLM and recombinant UGT1A4 for all substrates are depicted in Supplemental Figs. 2–4 and suggest atypical and complex kinetics. Inhibition experiments with the selective UGT1A4 inhibitor hecogenin also led to highly comparable inhibition profiles and apparent Ki values (0.29–1.05 µM) between HLM and UGT1A4 for all four substrates, further supporting its major involvement (Fig. 4; Supplemental Fig. 7). Based on these results, we conclude that UGT1A4 is likely the only enzyme involved in the hepatic glucuronidation of Abi, D4A, 5α-Abi, and Gal. We also observed that these compounds were effective at inhibiting their glucuronidation, leading to similar levels of inhibition by ∼30% to 50% in assays performed at Km values in the liver and with the UGT1A4 enzyme (Supplemental Fig. 5). As an example, 2 µM Abi proficiently inhibited the formation of D4A-G by 29% and 41% in HLM and UGT1A4 recombinant system, respectively.

Abi, D4A, 5α-Abi, and Gal glucuronidation by human tissues and recombinant human UGT enzymes. Glucuronidation activity of human tissues and recombinant UGTs (commercial supersomes and microsomes isolated from HEK293 cells for UGT1A10, UGT2B10, and UGT2B11) was measured at 200 μM substrate concentration for 2 hours. Results are expressed in picomoles per minute per milligram protein and as mean ± S.D. of duplicate determinations. The expression of individual UGT was confirmed by Western blotting, and the relative UGT expression between UGT1A3 and UGT1A4 supersomes was comparable, consistent with one of our previous reports using these protein preparations (Benoit-Biancamano et al., 2009b). HIM, human intestine microsomes; HKM, human kidney microsomes.

Kinetic profiles for the formation of Gs of Abi, D4A, 5α-Abi, and Gal by HLM and recombinant UGT1A4 enzyme. (A, C) HLM and (B, D) UGT1A4 were incubated with concentrations ranging from 0 to 400 μM (A, B) Abi, D4A, 5α-Abi, or (C, D) Gal for 30 minutes, as described in Materials and Methods. Results are expressed as mean ± S.D. of triplicate determinations of one representative experiment.
Enzyme kinetics of Abi, D4A, 5α-Abi, and Gal glucuronidation by human liver and the UGT1A4 enzyme
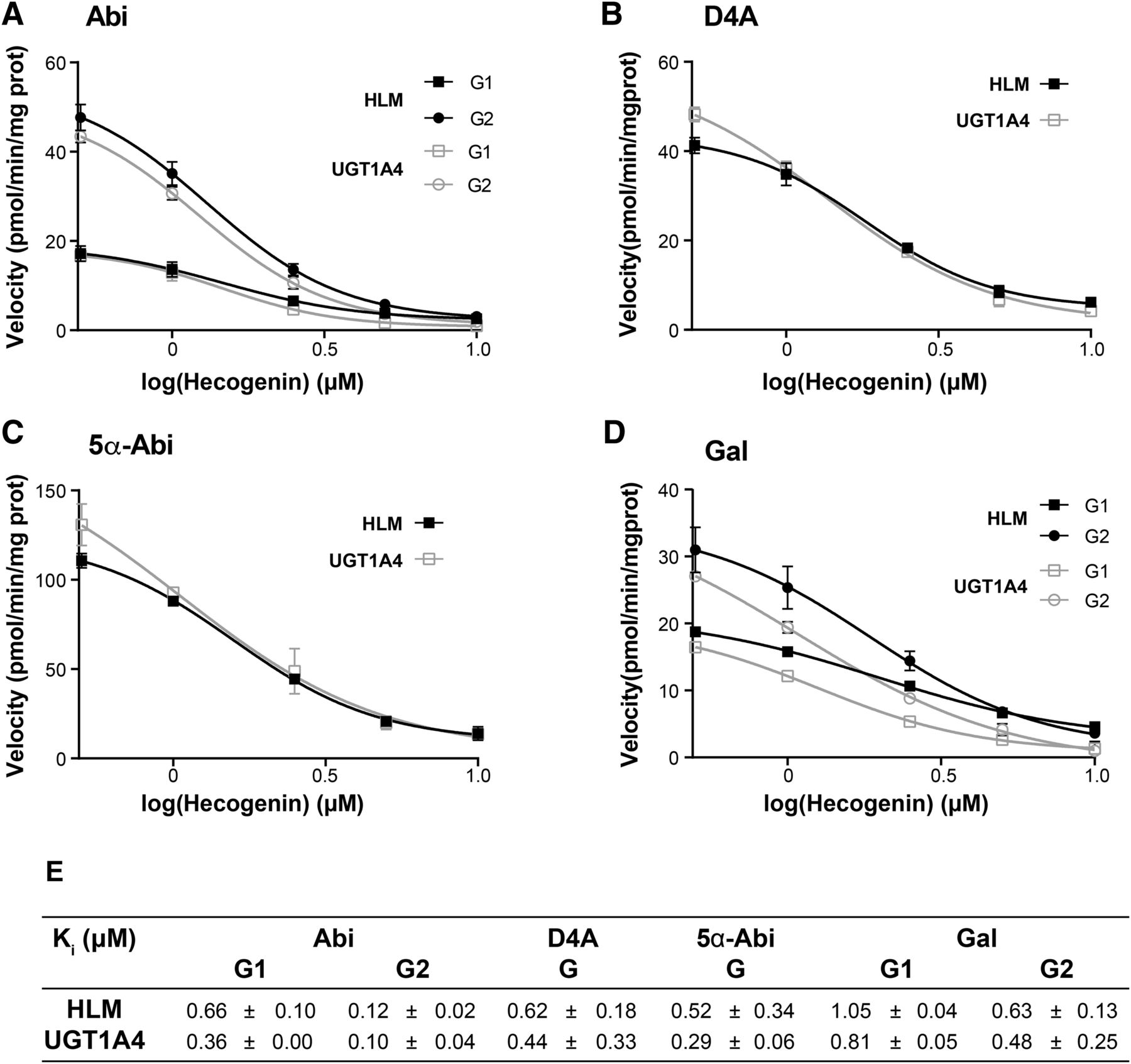
Dose-dependent inhibition of Abi-G1, Abi-G2, D4A-G, 5α-Abi-G, Gal-G1, and Gal-G2 formation by hecogenin in HLM and recombinant UGT1A4 enzyme. (A–D) Inhibition profiles of Abi (A), D4A (B), 5α-Abi (C), and Gal (D) glucuronidation by hecogenin. Activity was measured at constant concentration of substrate (2 μM Abi, 6 μM D4A, 5 μM 5α-Abi, or 0.5 μM Gal) with six concentrations of hecogenin ranging from 0 to 10 μM for 30 minutes, as described in Materials and Methods. In the control, no hecogenin was added. (E) Ki values of Abi, D4A, 5α-Abi, and Gal by hecogenin measured using three different concentrations of substrate ranging from 2 to 200 μM with six concentrations of hecogenin ranging from 0 to 10 μM for 30 minutes, as described in Materials and Methods. Results are expressed as mean ± S.D. of triplicate determinations of at least two experiments. Inhibition models observed were mixed.
The impact of common UGT1A4 coding region variations was then evaluated using recombinant UGT1A4 isoenzymes expressed in HEK293 human cells, namely UGT1A4*1 (R11P24L48), UGT1A4*2 (T24), UGT1A4*3 (V48), and UGT1A4*4 (W11) (Fig. 5). Compared with the UGT1A4*1 reference enzyme, the UGT1A4*4 isoenzyme presented a velocity reduced by nearly 50% for the conjugation of Abi, D4A, and 5α-Abi (Fig. 5D; Supplemental Table 1). UGT1A4*2 and UGT1A4*3 had slightly higher affinities. Additional rare variants presented a significantly reduced activity compared with UGT1A4*1 by up to 90% at concentration based on the apparent Km values, except for UGT1A4*9 (Fig. 5E). This suggests that collectively, these variants UGT1A4 allozymes with coding variations may be associated with reduced hepatic glucuronidation of Abi and its metabolites.

Formation of Gs of Abi, D4A, and 5α-Abi by UGT1A4 isoenzymes. Kinetic profiles for the formation of Abi (A and B), D4A (C), and 5α-Abi (D) Gs by UGT1A4 common isoenzymes (UGT1A4*1 R11P24L48, UGT1A4*2 T24, UGT1A4*3 V48, and UGT1A4*4 W11). UGT1A4 microsomes were incubated with increasing concentrations (up to 200 μM) of substrate for 30 minutes, as described in Materials and Methods. Results are expressed as mean ± S.D. of triplicate determinations. (E) Formation of Abi, D4A, and 5α-Abi Gs by additional UGT1A4 isoenzymes for 30 minutes using Abi (2 μM), 5α-Abi (5 μM), and D4A (6 μM). Results are expressed as mean ± S.D. of triplicate determinations of at least two experiments. UGT1A4*1 corresponds to R3R11P24L48E50H56H68I176S250I276. The level of UGT protein assessed by Western blot was used to calculate relative glucuronidation activities.
Inhibition of Steroid Glucuronidation by Abi, D4A, 5α-Abi, and Gal.
The ability of Abi, its metabolites, and Gal to inhibit glucuronidation of androgens (Testo and DHT) and their adrenal precursors (DHEA and A5-diol) was also evaluated. Conjugation of these steroids is significant in HLM and by the UGT1A4 enzyme, except for Testo, which is a poor substrate of this enzyme. Steroid glucuronidation was inhibited by ≥40% in the presence of 2 μM Abi and reached nearly 90% at 200 μM (Fig. 6). The inhibition pattern by D4A, 5α-Abi, and Gal was similar for HLM and UGT1A4 (Supplemental Fig. 6). This inhibition potency was comparable to the effect of Abi on the hepatic conjugation of tacrolimus, a specific substrate of the UGT1A4 enzyme, whereas no inhibitory effect was observed using 2 μM Abi for bilirubin-G formation performed by the UGT1A1 enzyme in HLM (data not shown). The estimated inhibitory Ki values for these drugs were in the low micromolar range, between 0.17 and 3.59 μM for HLM and UGT1A4 (Supplemental Fig. 8; Table 3). Inhibition was with mixed characteristics according to goodness-of-fit statistics and the Lineweaver–Burk plot graphical representation of experimental data. We observed similar inhibitory effects for Testo-G formation in assays with the androgen-conjugating recombinant UGT2B15 and UGT2B17 enzymes, and the PCa cell line LNCaP, expressing UGT2B15, UGT2B17, and UGT2B28 involved in local androgen inactivation. Ki values observed in the presence of Abi for Testo-G formation were 0.10 ± 0.04, 0.73 ± 0.53, and 3.19 ± 3.74 μM for LNCaP, UGT2B17, and UGT2B15, respectively (Table 3).

Dose-dependent inhibition of adrenal precursors (DHEA and A5-diol) and androgens (Testo and DHT) glucuronidation by Abi in HLM expressing a variety of steroid-conjugating UGTs (A) and recombinant UGT1A4 enzyme (B). Activity was measured at constant concentration of substrate (5 μM) with two concentrations of Abi (2 and 200 μM) for 30 minutes, as described in Materials and Methods. In the control, no Abi was added. Ki values were derived from an additional set of experiments performed using three different concentrations of substrates and three different concentrations of inhibitor ranging from 0 to 200 μM. Results are expressed as mean ± S.D. of triplicate determinations of at least two experiments. Inhibition models observed were mixed.
Abi inhibits glucuronidation of adrenal precursors (DHEA and A5-diol) and potent androgens (Testo and DHT)
Discussion
Our study establishes that Abi, a steroidal CYP17A1 inhibitor, and its downstream active metabolites are conjugated with GlcA in the liver, and that only the UGT1A4 enzyme expressed in human liver is involved. The structurally related androgen receptor axis–targeted agent Gal is also a substrate of the UGT1A4 enzyme. Their glucuronidation in vivo is sustained by the detection of G derivatives of Abi, D4A, and 5α-Abi in plasma samples of PCa patients treated with AA, with a predominance of Abi-G and 5α-Abi-G over D4A-G. We also demonstrate that a number of common and rare UGT1A4 variant allozymes with nonsynonymous substitutions exhibit significantly lower activity for these molecules compared with the reference UGT1A4 enzyme. Based on the results of our in vitro investigations, these pharmacological compounds further emerge as potent inhibitors of their conjugation in the liver by a mixed inhibition model. Abi, D4A, and 5α-Abi also proficiently inhibit steroid glucuronidation catalyzed by UGT1A4 and other UGTs, namely UGT2B15 and UGT2B17, expressed in the liver and PCa cells. This suggests that CYP17A1 inhibitors are conjugated by the glucuronidation pathway, with the potential to affect the UGT-associated drug-metabolizing system and potentially drug-endogenous molecule interactions.
Mass spectral analysis of purified G derivatives from these drugs suggests the formation of O- and N-G products of Abi, and N-G products of D4A and 5α-Abi, but this will require confirmation by NMR. Our work reveals that UGT1A4 catalyzes predominantly their glucuronidation with a potential minor role for UGT1A3. UGT1A4 is a main hepatic drug elimination pathway that participates in the N-glucuronidation of primary, secondary, and aromatic amines, which include many pharmacologically important drugs such as lamotrigine, tamoxifen, and tacrolimus (Argikar and Remmel, 2009; Zhou et al., 2010; Laverdière et al., 2011). UGT1A4 is also capable of conjugating steroidal compounds with hydroxyl groups such as hecogenin (Green and Tephly, 1996), a steroidal saponin aglycone found in the leaves of species from the Agave genus and a selective inhibitor of UGT1A4 (Paik et al., 2005). The glucuronidation efficiency of UGT1A4 for several steroids such as 3α-diol (5α-androstane-3α,17β-diol) and pregnanediol (5α-pregnane-3β,20α-diol) was demonstrated with Km values of 16 and 7.3 µM compared with 18 µM for hecogenin (Green and Tephly, 1996). Zhou et al. (2010) also showed that UGT1A4 displays significant activity for the potent androgen DHT, also revealing the existence of multiple aglycone substrate-binding sites in UGT1A4. In this study, we observed inferior Km values for Abi and its metabolites in the low-micromolar range varying from 0.61 to 7.4 µM. Based on highly similar kinetic parameters observed for HLM and the UGT1A4 enzyme, we conclude that UGT1A4 is the major UGT contributing to the formation of both N- and O-Gs of Abi, its metabolites, and Gal in vitro. This is also supported by the important loss of activity caused by the selective UGT1A4 inhibitor hecogenin, which hinders drug–G formation by over 50% at low-micromolar concentration (e.g., at Km value). The extent of the inhibition was comparable for UGT1A4 and liver microsomes, reinforcing the notion that this is likely the primary enzyme involved. Because UGT1A4 is not expressed in the prostate (Ohno and Nakajin, 2009), this reaction would take place primarily in the liver with a minor implication of the intestine. UGT1A3 is potentially responsible for the low activity observed in the human intestine microsomes based on its weak activity and its expression in this tissue (Ohno and Nakajin, 2009; Margaillan et al., 2015). Moreover, the lack of inhibition for UGT1A1-mediated bilirubin conjugation is also consistent with the low occurrence of hyperbilirubinemia observed in clinical trials (Fizazi et al., 2012, 2019; Smith et al., 2017). Besides, the microsomal assays exhibited complex glucuronidation kinetics, including substrate inhibition. Previous report showed that UGTs exhibit atypical kinetics in vitro, potentially explained by existence of two binding sites or substrate-induced changes in enzyme conformation (Kaivosaari et al., 2008; Uchaipichat et al., 2008; Zhou et al., 2010).
UGT1A4 displays significant variability in its coding sequence with three common reported allozymes, UGT1A4*2, UGT1A4*3, and UGT1A4*4, presenting frequencies of 1.8%, 13.8%, and 1.4%, respectively, according to 1000 genomes. These variants have been functionally characterized for different drugs in vitro and in vivo (Mori et al., 2005; Benoit-Biancamano et al., 2009a; Erickson-Ridout et al., 2011; Laverdière et al., 2011; Zhou et al., 2011; Edavana et al., 2013; Reimers et al., 2016; Sutiman et al., 2016; Smith et al., 2018). These variant allozymes displayed kinetic parameters comparable to those of the reference UGT1A4*1 enzyme, except UGT1A4*4, which had a significantly lower velocity for Abi and its metabolites, consistent with the reported impact of this specific variant on tamoxifen (Benoit-Biancamano et al., 2009a; Zhou et al., 2011). We also explored the impact of additional rare polymorphisms with frequencies below 1% that all presented much inferior activities, suggesting that collectively these UGT1A4 variant isoforms could potentially impact the hepatic glucuronidation of Abi and its metabolites. The impact of these UGT1A4 germline variants on the overall hepatic metabolism, response, and hepatic toxicity will require additional studies.
The data reported in this work also provide insights into the potential inhibitory effect of Abi and its active metabolites on steroid glucuronidation. These observations are in line with the reported inhibitory activity of Abi and Gal on the sulfonation of DHEA (Yip et al., 2018). Abi, D4A, 5α-Abi, and Gal exhibited strong inhibition toward glucuronidation of adrenal precursors (DHEA and A5-diol) and potent androgens (Testo and DHT) with comparable inhibitory profiles. The apparent Ki values (<10 µM for Testo-G formation) were in order of magnitude similar to the plasma drug concentrations achieved in PCa patients (Alyamani et al., 2018). In comparison, the antiandrogen finasteride was shown to inhibit UGT1A4-mediated activity using trifluoperazine as a substrate with a Ki of 6.05 µM (Lee et al., 2015). Our results further suggest that Abi and its metabolites not only inhibit hepatic glucuronidation of androgens but also glucuronidation of androgens by UGT2B15 and UGT2B17 enzymes highly expressed in the prostate (Chouinard et al., 2004). This is reinforced by the inhibition of androgen glucuronidation in LNCaP cells that express UGT2B15, UGT2B17, and other UGTs. The inhibition potential R (R value = 1 + [I]/Ki) was calculated to estimate the potential of Abi to block androgen metabolism by UGTs, as previously done (Deb et al., 2014; Oda et al., 2015), using the peak plasma concentration (I) of 2.2 µM (Ryan et al., 2010), and Ki values from liver microsomes (Ki = 0.24–1.01 µM for Testo, DHT, DHEA, and A5-diol) and prostate cells (Ki = 0.10 μM for Testo). R values of Abi were all above the cutoff R value >1.1 for having the potential to cause drug–drug interactions in vivo (Yu et al., 2017). R values ranged from 3.2 to 23.0, suggesting that the levels of Abi attained in humans could potentially inhibit the catabolism of androgens in vivo. However, as the metabolism of Abi is complex and involves multiple steroid-biotransforming enzymes, also expressed locally in PCa cells, additional in vivo evidence is needed. Although Abi leads to a major depletion of circulating steroid hormones, it is tempting to speculate that this inhibitory impact on steroid inactivation pathways may potentially alter pre-receptor control of androgen metabolism in patients with incomplete suppression of adrenal androgens. It may also be relevant when PCa cells become Abi-resistant, where the glucuronidation inhibition of Testo and DHT might impact PCa growth.
In conclusion, we have demonstrated that the metabolism of CYP17A1 steroidal inhibitors continues downstream to the final glucuronidation steps as observed for steroid biotransformation pathways. We showed that the hepatic UGT1A4 enzyme is the primary enzyme involved in this conjugation process and that coding polymorphisms affecting this gene significantly reduced conjugation capacity in vitro. Additional translational pharmacogenomics studies are required to establish the potential clinical relevance of the UGT1A4 pathway to drug metabolism and clinical response.
Acknowledgments
We are thankful to all participating patients and clinical staff who have made this scientific contribution possible.
Authorship Contributions
Participated in research design: Lévesque, Guillemette.
Conducted experiments: Vaillancourt, Turcotte, Caron, Villeneuve.
Contributed new reagents or analytic tools: Turcotte, Caron.
Performed data analysis: Vaillancourt, Turcotte, Caron, Villeneuve, Guillemette.
Wrote or contributed to the writing of the manuscript: Vaillancourt, Turcotte, Caron, Villeneuve, Lacombe, Pouliot, Lévesque, Guillemette.
Footnotes
- Received June 7, 2019.
- Accepted November 8, 2019.
Part of this work was supported by the CHU de Québec – Université Laval Foundation, Québec, Canada (to E.L. and C.G.) and the Canada Research Chair Program (Grant 950-203962 to C.G.). E.L. holds a Canadian Institutes of Health Research (CIHR) Clinician-Scientist Award. C.G. holds the Canada Research Chair in Pharmacogenomics (Tier I). J.V. holds a Fonds de Recherche du Québec – Santé Studentship Award.
No conflict of interests was disclosed.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- A5-diol
- androstanediol
- AA
- Abi acetate
- Abi
- abiraterone
- CYP17A1
- cytochrome P450 17α-hydroxy/17,20-lyase
- D4A
- Δ4-Abi
- DHEA
- dehydroepiandrosterone
- DHT
- dihydrotestosterone
- G
- glucuronide
- Gal
- galeterone
- GlcA
- glucuronic acid
- HEK
- human embryonic kidney
- HLM
- human liver microsome
- Ki
- inhibitor constant
- Km
- Michaelis constant
- LC
- liquid chromatography
- PCa
- prostate cancer
- Testo
- testosterone
- UGT
- UDP-glucuronosyltransferase
- Copyright © 2020 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}