


Visual Overview
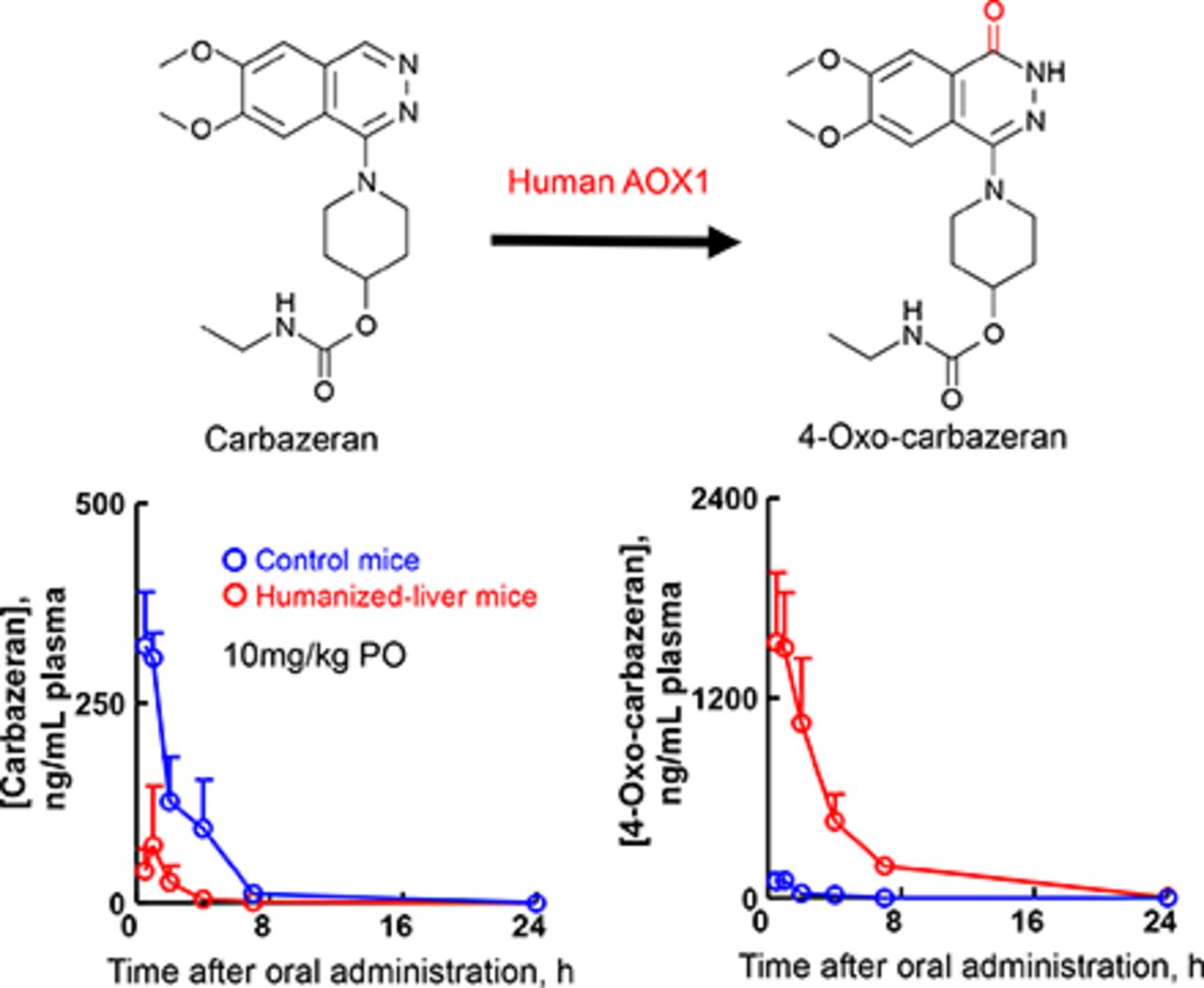
Abstract
Carbazeran is a potent phosphodiesterase inhibitor with species-dependent metabolic profiles in rats, dogs, and humans. In this study, we investigated the aldehyde oxidase (AOX)-mediated oxidation of carbazeran to 4-oxo derivatives in chimeric NOD/Shi-scid IL2 receptor gamma-null mice expressing a herpes simplex virus type 1 thymidine kinase transgene with humanized livers (humanized-liver mice). Liver cytosolic fractions from humanized-liver mouse effectively catalyzed carbazeran 4-oxidation with high affinity for the substrate, similar to those of the human liver cytosolic fractions and recombinant human AOX1 protein. Furthermore, hepatocytes prepared from humanized-liver mice and humans also exhibited substantial metabolism via carbazeran 4-oxidation. After a single oral administration of carbazeran (10 mg/kg), plasma levels of 4-oxo-carbazeran, N-desethyl-4-oxo-carbazeran, and 6,7-dimethoxy-1-[4-(hydroxy)-piperidino]-4-phthalazinone (three human metabolites formed via 4-oxidation) were higher in humanized-liver mice than in the control mice. In contrast, plasma levels of O-desmethylcarbazeran (a major metabolite in dogs) in control mice were higher than those in the humanized-liver mice. Relative excreted amounts of the three 4-oxidation–derived human-specific metabolites in the urine and feces were greater for humanized-liver mice than control mice, whereas the relative excreted amounts of O-desmethylcarbazeran were predominant in the urine and feces of control mice. Thus, the production of carbazeran 4-oxo derivatives was elevated in humanized-liver mice compared with control mice, in agreement with our in vitro enzyme-mediated oxidation data. These results suggest that hepatic human AOX1 functions in humanized-liver mice at the in vivo level and that humanized-liver mice may be useful for predicting drug metabolism in humans, at least with regard to human AOX1-dependent metabolism.
SIGNIFICANCE STATEMENT We found that the production of carbazeran 4-oxo derivatives was higher in humanized-liver mice than in control mice. These results were supported by the fact that carbazeran was rapidly metabolized to 4-oxo-carbazeran in humanized-liver mouse hepatocytes expressing human aldehyde oxidase 1. These results suggest that human aldehyde oxidase 1 is functional in humanized-liver mice in vivo and that chimeric NOD/Shi-scid IL2 receptor gamma-null mice expressing a herpes simplex virus type 1 thymidine kinase transgene transplanted with human hepatocytes may be a suitable model animal for predicting aldehyde oxidase–dependent biotransformation of drugs in humans.
Introduction
Species differences in drug metabolism make it difficult to evaluate the clearance, oral bioavailability, and half-life of a drug in vivo during preclinical testing, and this can lead to unpredictable toxicity and efficacy of drug candidates in first-in-human trials. A variety of experimental animals such as rodents, dogs, and nonhuman primates have been widely used in preclinical testing. However, predicting drug metabolism and pharmacokinetics is still difficult because species differences exist, particularly in the drug-metabolizing enzymes, including the cytochrome P450s (P450s), UDP-glucuronosyltransferases (UGTs), and aldehyde oxidase (AOX).
NOD/Shi-scid IL2 receptor gamma-null mice expressing a herpes simplex virus type 1 thymidine kinase transgene under the control of a mouse albumin promoter (TK-NOG) transplanted with human hepatocytes (termed humanized-liver mice) were previously developed at the Central Institute for Experimental Animals (Kawasaki, Japan) (Hasegawa et al., 2011). Humanized-liver mice have been shown to be useful as a preclinical model for estimating the pharmacokinetic profiles of drug candidates and their metabolite formations in humans (Kamimura and Ito, 2016). The major metabolic and excretory pathways of desloratadine in humanized-liver mice, which is mediated by the P450 and UGT enzymes, was reasonably similar to that reported for humans (Uehara et al., 2020). The time-dependent human plasma concentration profiles of five P450 probes extrapolated from pharmacokinetics in humanized-liver mice were consistent with human pharmacokinetics (Utoh et al., 2016). In addition, we recently reported the usefulness of hepatocytes isolated from humanized-liver mice (Hu-Liver cells) for assays of in vitro P450 isoform induction (Uehara et al., 2019). However, AOX-mediated metabolic profiles of probe drugs in humanized-liver mice have not yet been fully characterized.
Marked interspecies differences in AOX activities have resulted in a lack of clinical translation of drug candidates metabolized by this enzyme; for example, rats reportedly exhibit low AOX activity, and dogs have no AOX gene (Garattini and Terao, 2012; Terao et al., 2016). Carbazeran, i.e., 6,7-dimethoxy-l-[4-(ethylcarbamoyloxy)piperidino]phthalazine, was developed as a positive inotropic agent for the treatment of chronic heart failure; however, this compound failed clinical trials because of severe species differences in AOX-dependent drug oxidation, hampering the prediction of human pharmacokinetics (Beedham, 2020). The oral bioavailability of carbazeran was approximately 70% in dogs but less than 5% in humans, along with a considerable first-pass effect (Kaye et al., 1984). Furthermore, most of the carbazeran metabolites in humans were produced via oxidation at position 4 of the phthalazine ring, whereas in dogs, the major metabolic pathway was O-demethylation (Kaye et al., 1984). In vitro assays using liver cytosolic fractions or purified hepatic AOX enzymes showed substantial carbazeran metabolism in humans, marmosets, baboons, and guinea pigs but low to negligible metabolism in rats and dogs, respectively (Beedham et al., 1987). In human livers, the products of carbazeran 4-oxidation are now considered to be enzyme-selective catalytic markers of AOX-1 but not of the P450 enzymes (Xie et al., 2019).
Thus, based on the above studies, carbazeran was selected as a suitable substrate for investigating AOX-mediated drug-metabolizing activities in humanized-liver mice. In the present study, we investigated the conversion of carbazeran to the four human 4-oxo derivatives in humanized-liver mice using both in vivo and in vitro metabolic assays. We report herein that humanized-liver mice clearly display human AOX1-catalyzed carbazeran 4-oxidation, both in vivo and in vitro.
Materials and Methods
Chemicals and Reagents.
Carbazeran and 4-oxo-carbazeran were obtained from Toronto Research Chemicals (Toronto, Canada). Dimethylsulfoxide (DMSO) and corn oil were purchased from Sigma-Aldrich (St. Louis, MO). Recombinant human aldehyde oxidase (AOX) 1 was purchased from Cypex, Ltd. (Dundee, Scotland, UK). Pooled liver cytosolic fractions from humans (30 males and 20 females, 5–73 years old), cynomolgus monkeys (four males, 3 to 4 years old), minipigs (Göttingen, two males, ∼4 months old), dogs (eight males, >6 months old), rabbits (eight males, >9 months old), guinea pigs (50 males, ∼9 weeks old), and rats (454 males, ∼8 weeks old) were purchased from Xenotech (Lenexa, KS). Human adult (female, 18 years old, Caucasian) and fetal (male, 24-week gestation, race unknown) liver total RNA were purchased from Cell Applications, Inc. (San Diego, CA). All other reagents used were of the highest quality commercially available.
Animals, Hepatocytes, and Liver Cytosolic Fractions.
This study was carried out in strict accordance with the Guide for the Care and Use of Laboratory Animals of the Central Institute for Experimental Animals (CIEA) in Japan in an exploratory manner. All experimental protocols were approved by the Animal Care Committee of the CIEA (Permit Number: 17017A). All possible efforts were carried out to reduce pain and suffering and minimize the number of animals used. The use of commercially available cryopreserved human hepatocytes was approved by the Institutional Ethics Committee of the CIEA (Permit Number: 13-05). Humanized-liver mice were generated by transplanting the cryopreserved hepatocytes of three human donors [donor A, 35-year-old Asian male, provided from Lonza Walkersville, Inc. (Walkersville, MD); donor B, 30-year-old African American female, from Lonza Walkersville, Inc.; donor C, male, obtained from ADMET Technologies (Durham, NC)] into TK-NOG mice, using the method previously described (Suemizu et al., 2018). The human hepatocyte replacement index in the mouse liver was estimated based on the human serum albumin concentrations, which were quantified with the Human Albumin ELISA Quantitation Set (Bethyl Laboratories, Montgomery, TX). Only humanized-liver mice with high replacement index values (>95%) were used in this study. All animals were housed with free access to tap water and food (CLEA Rodent Diet CA-1; CLEA Japan, Tokyo, Japan) and maintained in environmentally controlled rooms (ambient temperature, 24°C; humidity, 55%) with a 12-hour light/dark cycle. Hu-Liver cells were prepared from fresh hepatocytes isolated directly from two individual humanized-liver mice (18 weeks old) transplanted with the human hepatocytes of either donor A or donor B, using the two-step collagenase perfusion method previously described (Uehara et al., 2020). The purity of the Hu-Liver cells (i.e., lack of contamination with normal mouse hepatocytes) was determined by fluorescence-activated cell sorting, using a monoclonal anti-human leukocyte antigen antibody (clone G46-2.6; BD Biosciences, Franklin Lakes, NJ) and a monoclonal anti-mouse H-2kd antibody (clone SF1-1.1; BD Biosciences) on the BD FACSAria (BD Biosciences) as previously described (Higuchi et al., 2016). The percentage of human leukocyte antigen-positive cells was >95% among the Hu-Liver cells. The cell viability was determined by trypan blue exclusion and was greater than 75%. TK-NOG mouse hepatocytes (control mice) were also prepared with the collagenase perfusion method for comparison with Hu-Liver cells and cryopreserved human hepatocytes for the in vitro biotransformation experiments. For the preparation of pooled liver cytosolic fractions, livers from humanized-liver mice (four males, 18 weeks old) transplanted with human hepatocytes from donor A, as well as livers from control TK-NOG mice (four males, 16–20 weeks old), were excised and homogenized in three volumes of homogenizing buffer (0.1 M Tris-HCl, pH 7.4, 0.1 M KCl, 1 mM EDTA) under ice-cold conditions, followed by centrifugation at 9000g for 20 minutes at 4°C. The resultant supernatants were centrifuged at 105,000g for 1 hour at 4°C, and the supernatants collected were the liver cytosolic fractions. The protein concentrations in these fractions was determined with the Bradford method by using the TaKaRa Bradford Protein Assay Kit (Takara Bio, Shiga, Japan) with bovine serum albumin as the standard.
In Vitro Metabolic Study Using Liver Cytosolic Fractions.
The incubation mixtures (0.25 ml) consisted of either 20 pmol/ml recombinant human AOX1 protein or 0.2 mg/ml liver cytosolic fractions, 0.5–80 μM carbazeran, and 0.1 M potassium phosphate buffer (pH 7.4) with a final DMSO concentration of 0.5% (v/v). The incubation mixtures were preincubated at 37°C for 3 minutes in a low-speed shaking water bath. By the addition of the substrate, reactions were initiated at 37°C for 5 minutes and then terminated by adding an equal volume of ice-cold acetonitrile containing 50 nM tolbutamide (internal control). These samples were immediately mixed and then centrifuged at 20,000g for 10 minutes at 4°C. The supernatants were collected and subjected to high-performance liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis.
In Vitro Metabolic Study Using Hepatocytes.
To investigate species differences in the biotransformation to 4-oxo-carbazeran, carbazeran was incubated with these hepatocytes. Incubations were conducted at 37°C with 5% CO2 for 30 minutes on a shaker in 0.2 ml reaction mixtures [final DMSO concentration, 0.1% (v/v)] containing carbazeran (10 µM), hepatocytes (5 × 105 cells/ml), and Williams’ Media E (Gibco, Grand Island, NY). Reactions were initiated by substrate addition and terminated by the addition of an equal volume of ice-cold acetonitrile containing the internal standard (tolbutamide). Samples were centrifugated at 20,000g for 10 minutes at 4°C, and the supernatants were collected and subjected to LC-MS/MS analysis.
Quantitative Reverse-Transcription Polymerase Chain Reaction.
The human AOX1 mRNA levels in humanized-liver mice were analyzed by real-time quantitative reverse-transcription polymerase chain reaction (qRT-PCR). Total RNA was extracted from the livers of humanized-liver mice transplanted with the human hepatocytes of donors A and B, using an RNeasy Mini Kit (Qiagen, Valencia, CA). The total RNA was then used to synthesize cDNA with the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, Waltham, MA) and random hexamers in 20-µl reactions. qRT-PCR was subsequently performed with SYBR Green–based detection on an ABI PRISM 7300 sequence detection system (Applied Biosystems, Foster City, CA), using human AOX1-specific primers, hAOX1 (5qrt2) 5′-CGAATCATAAACATGTACAAGGAAA-3′ and hAOX1 (3qrt2) 5′-TCCACAGCAACTTTCCTCAAG-3′. The reaction mixtures contained 400 nM of each primer, 5 µl of Power SYBR Green PCR Master Mix (Applied Biosystems), and 2 µl of template DNA in a total volume of 10 µl. The PCR conditions were as follows: initial denaturation for 10 minutes at 95°C followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. High PCR efficiency (98%) was confirmed with a standard curve generated by using a 10-fold dilution series of template cDNAs. The expression level of AOX1 mRNA was normalized to that of human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) measured using the Human GAPDH Endogenous Control (Thermo Fisher Scientific).
Administration of Carbazeran to Mice.
Carbazeran was orally administered to control mice (four males, 16 weeks old) and humanized-liver mice (four males, 18 weeks old) transplanted with human hepatocytes from donor C in a single dose of 10 mg/kg dissolved in corn oil containing 10% (v/v) DMSO (1 mg/ml). Blood was collected from the orbital vein of the mice by using capillary tubes at 0.5, 1, 2, 4, 7, 24, and 48 hours after oral administration. Plasma was collected by the centrifugation of blood samples at 3500g for 20 minutes at 4°C. Urine and feces samples were collected from mice during the postdose period of 0–72 hours. Fecal samples were homogenized with 10 volumes of methanol and then centrifuged at 10,000g for 20 minutes; the resultant supernatants were collected.
Quantitation of Carbazeran and Its Metabolites.
Samples (5.0 µl each) from plasma and urine were mixed well with four volumes of acetonitrile containing 50 nM of tolbutamide, and the precipitated proteins were removed by centrifugation at 20,000g for 10 minutes at 4°C. Aliquots (2 µl) of each supernatant were then transferred to an autosampler vial and injected into the column (YMC-Triart C18 column, 3 µm, 3.0 × 100 mm; YMC, Kyoto, Japan). LC-MS/MS analysis was carried out using the Shimadzu Nexera ultra–high-performance liquid chromatography system (Shimadzu, Kyoto, Japan) coupled with a QTRAP 5500 mass spectrometer (AB Sciex, Darmstadt, Germany). The mobile phase was composed of 0.1% (v/v) formic acid in water (A) and 0.1% (v/v) formic acid in acetonitrile (B), and the flow rate was 0.4 ml/min. The chromatographic run consisted of a linear gradient of 10% B to 90% B at 10 minutes, a holding period of 5 minutes, and then a linear gradient back to 10% B and equilibration within 5 minutes. The column temperature was maintained at 40°C throughout the run. Analytes were detected in the multiple-reaction monitoring mode with a precursor-to-product mass transition of m/z 361.0 to m/z 272.1 for carbazeran (4.6 minutes), m/z 377.0 to m/z 288.1 for 4-oxo-carbazeran (M1) (6.2 minutes), m/z 349.0 to m/z 288.1 for N-desethyl-4-oxo-carbazeran (M2) (5.0 minutes), m/z 306.0 to m/z 288.1 for 6,7-dimethoxy-1-[4-(hydroxy)-piperidino]-4-phthalazinone (M3) (4.5 minutes), m/z 347.0 to m/z 258.0 for O-desmethylcarbazeran (M4) (4.1 minutes), and m/z 271.1 to m/z 74.0 for tolbutamide (7.9 minutes). The QTRAP 5500 mass spectrometer was operated in positive ion mode with a spray voltage of 5.5 kV and a capillary temperature of 550°C. Plasma concentrations of M2, M3, and M4 were expressed as the peak response because their standard reference compounds were not available.
Data Analyses.
Pharmacokinetics parameters, including half-life (t1/2), area under the concentration–time curve (AUC), and peak plasma concentration (Cmax) were calculated with a noncompartmental model, using Phoenix WinNonlin version 7.0 software (Certara, St. Louis, MO). Statistical analyses for the comparing plasma concentrations of carbazeran and its metabolites in control and humanized-liver mice were carried out using two-way ANOVA with the Bonferroni post hoc test (GraphPad Prism Software version 7.05; La Jolla, CA). Statistical analyses of the pharmacokinetic parameters and the urinary and fecal excretion levels of carbazeran and its metabolites in control and humanized-liver mice were performed with GraphPad Prism by using an unpaired Student’s t test with or without Welch’s corrections. Kinetic parameters for carbazeran 4-oxidation were estimated using the KaleidaGraph program version 4.0 (Synergy Software, Reading, PA) by fitting the data to the Michaelis-Menten equation.
Results
Carbazeran 4-Oxidation Activity of Humanized-Liver Mouse Liver Cytosol and Hepatocytes.
The reported primary metabolic pathway of carbazeran in humans is 4-oxidation of the phthalazine moiety to formation of three derivates (4-oxo-carbazeran, M1; N-desethyl-4-oxo-carbazeran, M2; and 6,7-dimethoxy-1-[4-(hydroxy)-piperidino]-4-phthalazinone, M3), whereas the formation of O-desmethylcarbazeran (M4) comprises the major pathway in dogs (Fig. 1) (Kaye et al., 1984). Carbazeran 4-oxidation is reportedly catalyzed by liver cytosolic aldehyde oxidase but not liver microsomal P450 (Xie et al., 2019). To investigate species differences in aldehyde oxidase activity, we investigated the carbazeran 4-oxidation activity in the liver cytosolic fractions from humanized-liver mice, humans, cynomolgus monkeys, minipigs, dogs, rabbits, guinea pigs, rats, and mice. Liver cytosol from humanized-liver mice, humans, cynomolgus monkeys, and guinea pigs displayed substantial carbazeran 4-oxidation at a low substrate concentration (10 μM) (Fig. 2), suggesting that the liver cytosol from humans, humanized-liver mice, cynomolgus monkeys, and guinea pigs had high affinity toward this substrate and high capacity for carbazeran 4-oxidation, in contrast to that of the liver cytosol from rats, dogs, rabbits, and control mice (Fig. 2). Indeed, humanized-liver mouse and human liver cytosol had low Km values (3.6 and 3.2 μM, respectively) and high Vmax values (0.67 and 0.63 nmol/min per milligram protein, respectively) for carbazeran 4-oxidation, unlike the control mouse liver cytosol (Km, 74 μM; Vmax, 0.22 nmol/min per milligram protein) (Supplemental Fig. 1; Table 1). In other words, human and humanized-liver mice liver cytosol efficiently catalyzed carbazeran 4-oxidation with a high Vmax/Km value (0.21 and 0.18 ml/min per milligram protein, respectively) that was distinct from that of the control mouse liver cytosol (0.0050 ml/min per milligram protein; Table 1). In addition, the Km value (3.6 μM) for carbazeran 4-oxidation by the humanized-liver mice was similar to that of human recombinant AOX1 (8.2 μM; Table 1). The carbazeran 4-oxidation activities of Hu-Liver cells and human hepatocytes were approximately similar, and their activity was at least ninefold higher than that of the control mouse hepatocytes (Table 2). In addition, human AOX1 mRNA levels in the humanized-liver mice livers were comparable to those in the adult human liver but not the fetal human liver (Fig. 3). These results suggested that humanized-liver mice had AOX catalyzed-carbazeran oxidation activity close to that of humans.

Metabolic pathways of carbazeran in humans and dogs. This figure is drawn based on the data of Kaye et al. (1984) with slight modifications.

Carbazeran 4-oxidation activities of liver cytosol from humanized-liver mice, humans, cynomolgus monkeys, minipigs, dogs, rabbits, guinea pigs, rats, and control mice. Carbazeran (10 and 100 μM) was incubated with pooled liver cytosol (0.20 mg/ml) at 37°C for 5 minutes. Data represent the mean ± S.D. ND, not detected.
Kinetic parameters of carbazeran oxidation by liver cytosol and recombinant human AOX1
Kinetic parameters were determined by nonlinear regression analysis (mean ± S.E., n = 12 substrate concentrations of 0.5–80 μM) employing the Michaelis-Menten equation, v = Vmax × [S]/(Km + [S]) or the substrate inhibition equation. Data represent the mean of duplicate determinations.
Carbazeran 4-oxidation activities of hepatocytes from humans, humanized-liver mice, and control mice
Carbazeran (10 μM) was incubated for 30 min with human hepatocytes, Hu-Liver cells, or TK-NOG mouse hepatocytes at 5.0 × 105 cells/ml. Data represent the mean ± S.D. of triplicate determinations.
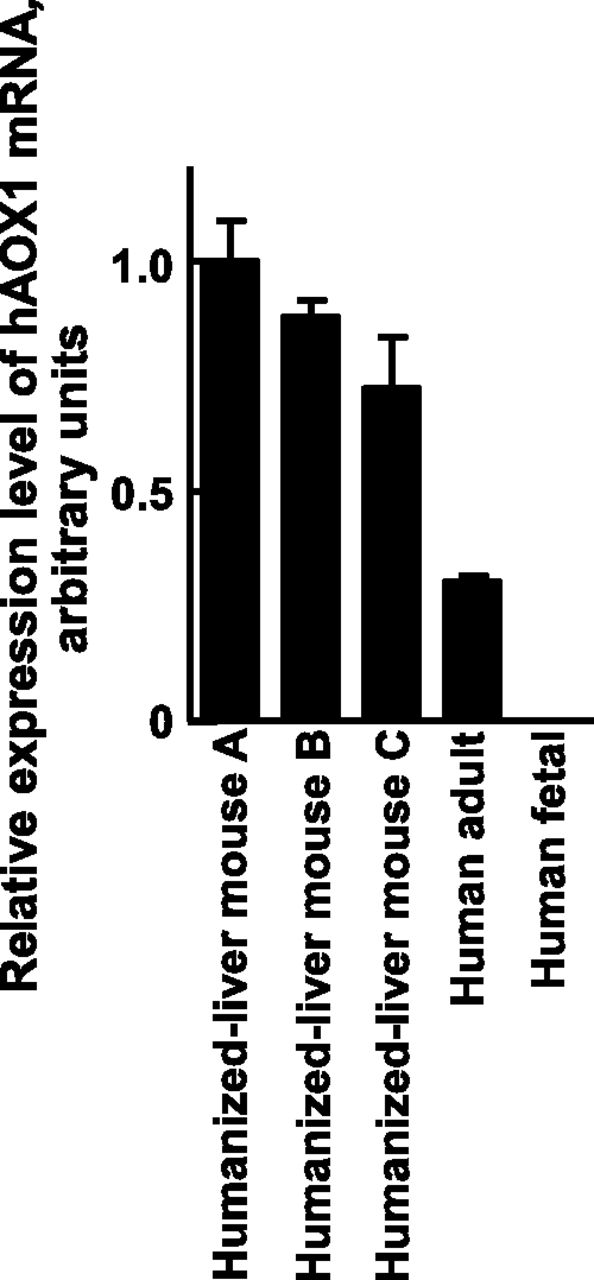
Expression level of human AOX1 in the livers of humans and humanized-liver mice. Expression levels of human AOX1 mRNA in the livers of humanized-liver mice were measured by qRT-PCR and normalized to human GAPDH expression levels. Data represent the mean ± S.D.; n = 3 animals. The expression level of human AOX1 mRNA in the livers of humanized-liver mice A (prepared using hepatocytes transplanted from donor A) was adjusted to 1 for relative comparisons.
In Vivo Metabolism of Carbazeran to 4-Oxo Derivatives in Humanized-Liver Mice.
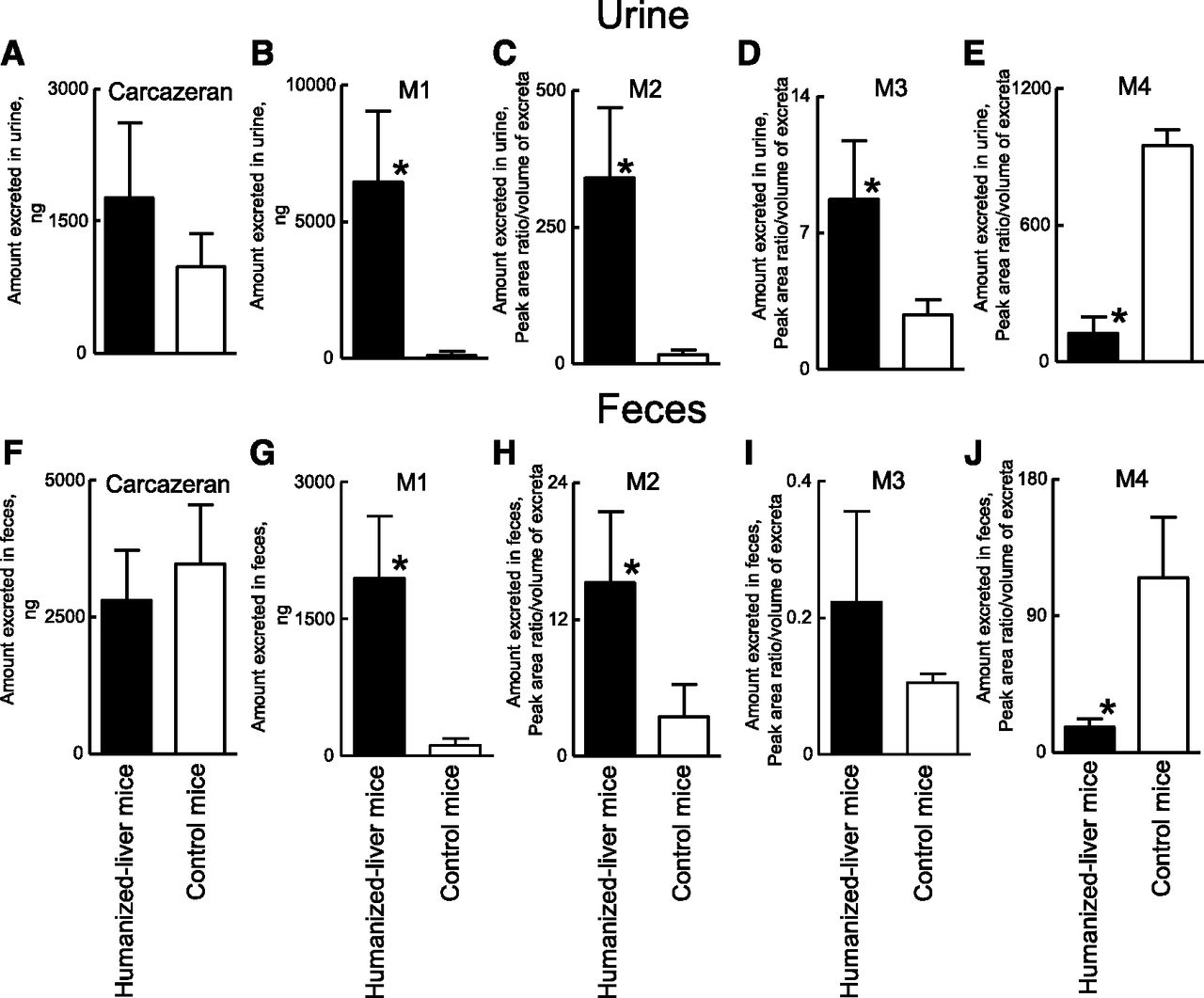
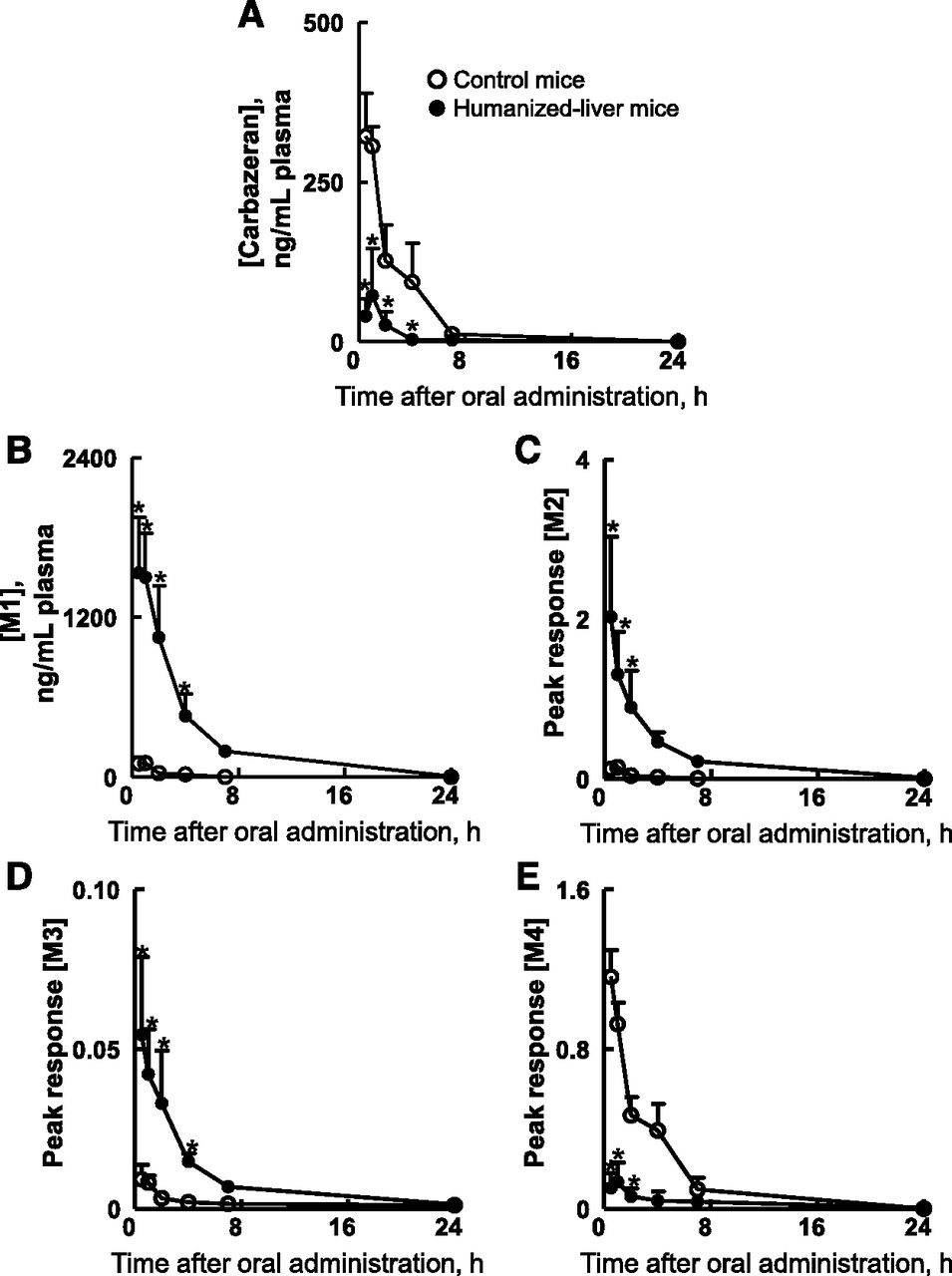
To investigate and compare the in vivo metabolic profile of carbazeran in humanized-liver mice and control mice, plasma, urine, and feces samples were collected until 72 hours after the oral administration of carbazeran at 10 mg/kg, and the unchanged drug and its metabolites were measured in each sample by LC-MS/MS. Urine samples from humanized-liver mice and control mice were analyzed by mass detection of m/z 361.0, 377.0, 349.0, 306.0, and 347.0 for the unchanged drug, M1, M2, M3, and M4, respectively. Five peaks with retention times of 4.6, 6.2, 5.0, 4.5, and 4.1 minutes, respectively, were observed in the urine of humanized-liver mice and/or control mice (Supplemental Fig. 2), and no interfering peaks in the region of these peaks were observed in the blank urine samples (data not shown). When the ions at m/z 349.0, 306.0, and 347.0 were subjected to collision-induced dissociation, product ions were obtained at m/z 234 and 288, m/z 234 and 288, and m/z 204 and 258, respectively (Supplemental Fig. 3). We considered that the peaks detected with m/z 349.0 at 5.0 minutes, m/z 306.0 at 4.5 minutes, and m/z 347.0 at 4.1 minutes corresponded to M2, M3, and M4, respectively (Supplemental Fig. 2). The accumulated amounts of M1/M2/M3 in the urine and M1/M2 in the feces were higher in the humanized-liver mice than in the control mice (Fig. 4). In contrast, the accumulated amounts of M4 in the urine and feces were lower in the humanized-liver mice than in the control mice (Fig. 4). The mean plasma concentrations of the unmetabolized carbazeran in samples from humanized-liver mice were lower than in those from control mice for 0.5–4 hours after oral administration (Fig. 5A). The Cmax (77 ng/ml), Tmax (0.75 hours), and AUC0-inf (134 ng·h/ml) in humanized-liver mice were lower than those in the control mice (Cmax, 340 ng/ml; Tmax, 0.88 hours; and AUC0-inf, 870 ng·h/ml) (Table 3). The plasma concentration versus time curves, AUC0-last, and AUCRatio (AUC in humanized-liver mice/AUC in control mice) for the four carbazeran metabolites after a single oral administration of carbazeran are shown in Fig. 5, B–E and Table 3. The AUC0-last values of M1 (6100 ng·h/ml), M2 (6.3 ng·h/ml), and M3 (0.21 ng·h/ml) in humanized-liver mice were higher than those in control mice (M1, 230 ng·h/ml; M2, 0.35 ng·h/ml; M3, 0.044 ng·h/ml) (Table 3). In contrast, the AUC0-last of M4 (0.64 ng·h/ml) in humanized-liver mice was lower than that in control mice (3.9 ng·h/ml) (Table 3). The AUCRatio values of M1, M2, M3, and M4 was 27, 18, 4.8, and 0.16, respectively (Table 4). These results confirmed that the in vivo metabolism of carbazeran closely reflected the in vitro metabolism of carbazeran in the liver cytosol of humanized-liver mice. These results further suggest that humanized-liver mice could be useful in predicting AOX-dependent drug metabolism in humans.

Cumulative amounts of carbazeran and its metabolites excreted in the urine and feces of humanized-liver mice and control mice after single oral administration of carbazeran at 10 mg/kg. Amounts of carbazeran (A), M1 (B), M2 (C), M3 (D), and M4 (E) excreted in the urine and feces up to 72 hours postdose were different (P < 0.05) between humanized-liver mice (closed) and control mice (open) based on the unpaired Student’s t test with Welch’s correction (*). Data represent the mean ± S.D.; n = 4 mice.

Plasma concentrations of carbazeran and its metabolites in humanized-liver mice and control mice after single oral administration of carbazeran at 10 mg/kg. Plasma concentrations of carbazeran (A), M1 (B), M2 (C), M3 (D), and M4 (E) were different (P < 0.05) between humanized-liver mice (closed circles) and control mice (open circles) based on two-way ANOVA followed by the Bonferroni post hoc test (*). Data represent the mean ± S.D.; n = 4 mice.
Pharmacokinetic parameters of carbazeran in humanized-liver mice and control mice after single oral administration of carbazeran at 10 mg/kg
AUC0-last of metabolites observed after single oral administration of carbazeran at 10 mg/kg in control and humanized-liver mice
Discussion
Several species differences in drug metabolism and disposition between humans and nonclinical species are known to be caused by differences in AOX activities (Kaye et al., 1984; Itoh et al., 2006; Dalvie et al., 2010; Isobe et al., 2016). To confirm whether humanized-liver mice could be the in vivo animal model for evaluating AOX-dependent oxidations in humans, we investigated AOX function in humanized-liver mice by using carbazeran as a probe drug for human AOX1 activity in both in vivo and in vitro biotransformation assays.
Human AOX1 mRNA was found to be expressed in humanized-liver mouse livers at a level comparable with than in the adult human liver. Human and humanized-liver mice liver cytosolic fractions efficiently catalyzed carbazeran 4-oxidation with a similarly high Vmax/Km (0.21 and 0.18 ml/min per milligram protein, respectively), distinct from that of the mouse liver cytosol (0.0050 ml/min per milligram protein). Humanized-liver mouse liver cytosolic fractions catalyzed carbazeran 4-oxidation with a similar affinity toward the substrate (Km = 3.6 μM) compared with recombinant human AOX1 (Km = 8.2 μM). However, the apparent Km of the mouse liver cytosol was about 60-fold higher than that of the humanized-liver mouse liver cytosol. As expected, the formation of 4-oxo-carbazeran was approximately ninefold higher in human hepatocytes and Hu-Liver cells than in the control mice because of the presence of human AOX1 in these cells. These in vitro results suggest that human AOX1 is a major catalyst for carbazeran 4-oxidation in humanized-liver mouse livers. Because the metabolism of carbazeran to 4-oxo-carbazeran in the humanized-liver mouse liver cytosol and Hu-Liver cells (hepatocytes from humanized-liver mice) is closer to that of humans, we hypothesized that the in vivo metabolic profile for carbazeran would also be similar between humans and humanized-liver mice. Our in vivo experiments in the current study using humanized-liver mice clearly indicated that the levels of carbazeran 4-oxo derivatives (M1, M2, and M3) in the plasma/urine/feces of humanized-liver mice were higher than those in the control mice. Although there were marked differences in overall exposure between humanized-liver mice and control mice, the t1/2 values were the same in both groups (Table 3). Interestingly, a similar phenomenon has been found in comparing pharmacokinetics after oral administration of deuterium and nondeuterated carbazeran to guinea pigs (Sharma et al., 2012). Oral administration of carbazeran to guinea pigs has reportedly resulted in large kinetic deuterium isotope effects on the AUC and Cmax, without a change of systemic half-life (Sharma et al., 2012). In addition, this increase in the kinetic deuterium isotope effects on the AUC has been higher than those for intravenously administered carbazeran, suggesting that oral first-pass metabolism via AOX enzymes in the guinea pig livers and intestines may contribute substantially to the clearance of carbazeran. The substantial carbazeran 4-oxidation activities have been observed in guinea pig hepatocytes similar to Hu-Liver cells and human hepatocytes (Sharma et al., 2012). Therefore, the low exposure of carbazeran in humanized-liver mice may be driven by high first-pass hepatic extraction by human AOX1. Overall, hepatic human AOX1 in humanized-liver mice showed typical functions as humanized-model in vivo and in vitro.
The quantity of 4-oxo derivatives found in the plasma of humanized-liver mice correlated with the in vitro carbazeran oxidation activity of their liver cytosol, suggesting that the formation of the 4-oxo derivative from carbazeran in humanized-liver mice in vivo can be ascribed to human AOX1 activity. However, the role of the mouse AOX enzyme activity in extrahepatic organs must also be considered. Although the AOX enzyme is a single protein in humans, four Aox isoforms (Aox1, Aox2, Aox3, and Aox4) are encoded by distinct genes in mice (Garattini et al., 2009). These four mouse Aox enzymes are expressed in various tissues, including the liver, in a tissue-specific manner (Kurosaki et al., 2013) and with different functions (Kucukgoze and Leimkuhler, 2018). The low activity of carbazeran oxidation we observed in the control mouse liver cytosol may be due to the different substrate specificity of the mouse Aox enzymes compared with that of human AOX1. In a preclinical study for predicting drug metabolism using humanized-liver mice, it would be necessary to consider the contribution of all of the hepatic human enzymes and extrahepatic mouse enzymes at the in vivo level.
Species differences in AOX-dependent drug metabolism are well known. Carbazeran is metabolized via 4-oxidation of the phthalazine ring in humans, whereas carbazeran O-demethylation is the major metabolic route in dogs (Kaye et al., 1984). Carbazeran is mainly metabolized to O-glucuronide of O-desmethyl-4-oxocarbazeran in guinea pigs, and this study suggests that AOX plays as key a role in the metabolism of carbazeran in guinea pigs as it does in humans (Critchley et al., 1994). Carbazeran oxidation activities in the dog and rat liver are negligible and low, respectively (Beedham et al., 1987). In contrast, carbazeran is rapidly metabolized to 4-oxo-carbazeran in the human and baboon liver cytosol (Kaye et al., 1985). Guinea pig and marmoset liver reportedly have the closest spectrum of activity to human liver enzymes, especially with regard to phthalazines and quaternary substrates (Beedham et al., 1987). In this study, the metabolic profile for carbazeran was very similar between humanized-liver mice and humans based on the similarity of their liver cytosolic AOX function. Therefore, humanized-liver mice appear to be a suitable animal model for analyzing species differences in AOX-dependent drug metabolism between humans and other experimental animals, but enzyme activity in extrahepatic organs needs to be taken into account.
Humanized-liver mice and human gene transgenic mice are emerging as promising in vivo models for studying xenobiotic metabolism and toxicity. Humanized-liver mice are prepared by the transplantation of human hepatocytes; thus, all genes expressed in the livers of these mice are of human origin, even though human gene transgenic mice models have genetically only incorporated a few humanized genes (Bissig et al., 2018). Indeed, the drug-metabolizing enzyme genes in donor human hepatocytes are reproducibly expressed at comparable levels in humanized-liver mouse livers (Hasegawa et al., 2011). Therefore, chimeric mice with humanized-liver potentially enable the prediction of complex drug metabolic pathways mediated by multiple drug-metabolizing enzymes (e.g., AOX, P450s, UGTs) in humans.
In conclusion, we demonstrated that the human AOX1 enzyme expressed in the humanized-liver mice liver is functional at the in vivo level. The observed differences in the biotransformation of carbazeran between humanized-liver mice and control mice reflected the functional differences in the AOX enzymes between humans and mice. The results of our study suggest that humanized-liver mice may be helpful for predicting human metabolism and disposition of drug candidates, at least in terms of human AOX1-dependent metabolism.
Acknowledgments
The authors thank Hiroaki Kato, Yasuhiko Ando, and Mizuho Noda for their technical help and Drs. Hidetaka Kamimura, Yasuyuki Ohnishi, and Mamoru Ito for their helpful advice. We also thank Natasha Beeton-Kempen from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Authorship Contributions
Participated in research design: Uehara, Suemizu.
Conducted experiments: Uehara, Yoneda, Higuchi.
Contributed new reagents or analytic tools: Yoneda, Higuchi.
Performed data analysis: Uehara.
Wrote or contributed to the writing of the manuscript: Uehara, Yamazaki, Suemizu.
Footnotes
- Received March 1, 2020.
- Accepted April 10, 2020.
This work was supported by the Platform Project for Supporting Drug Discovery and Life Science Research as part of the Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS) from Japan Agency for Medical Research and Development (AMED) [Grant 19am0101121j0003]; S.U. was also supported partly by the Japan Society for the Promotion of Science Grant-in-Aid for Young Scientists B [Grant 17K15520].
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AOX
- aldehyde oxidase (EC 1.2.3.1)
- AUC
- area under the concentration–time curve
- CIEA
- Central Institute for Experimental Animals
- Cmax
- peak plasma concentration
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- Hu-Liver cells
- hepatocytes freshly prepared from humanized-liver mice
- LC-MS/MS
- liquid chromatography–tandem mass spectrometry
- P450
- cytochrome P450
- qRT-PCR
- quantitative reverse-transcription polymerase chain reaction
- t1/2
- half-life
- TK-NOG mice
- NOD/Shi-scid IL2 receptor gamma-null mice expressing a herpes simplex virus type 1 thymidine kinase transgene
- UGT
- UDP-glucuronosyltransferases
- Copyright © 2020 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}