


Abstract
Drug metabolism is a biotransformation process of drugs, catalyzed by drug-metabolizing enzymes (DMEs), including phase I DMEs and phase II DMEs. The aberrant expression of DMEs occurs in the different stages of cancer. It can contribute to the development of cancer and lead to individual variations in drug response by affecting the metabolic process of carcinogen and anticancer drugs. Apart from genetic polymorphisms, which we know the most about, current evidence indicates that epigenetic regulation is also central to the expression of DMEs. This review summarizes differentially expressed DMEs in cancer and related epigenetic changes, including DNA methylation, histone modification, and noncoding RNAs. Exploring the epigenetic regulation of differentially expressed DMEs can provide a basis for implementing individualized and rationalized medication. Meanwhile, it can promote the development of new biomarkers and targets for the diagnosis, treatment, and prognosis of cancer.
SIGNIFICANCE STATEMENT This review summarizes the aberrant expression of DMEs in cancer and the related epigenetic regulation of differentially expressed DMEs. Exploring the epigenetic regulatory mechanism of DMEs in cancer can help us to understand the role of DMEs in cancer progression and chemoresistance. Also, it provides a basis for developing new biomarkers and targets for the diagnosis, treatment, and prognosis of cancer.
Introduction
With an increasing incidence and mortality every year, cancer is a major public problem worldwide and is one of the most deathful diseases for both men and women. In the United States, prostate, lung, and colorectal cancers are three major cancers in men, whereas the three most common cancers in women are breast, lung, and colorectal cancers (Siegel et al., 2019). The high cancer mortality rate is because of a combination of factors, including the lack of reliable biomarkers for cancer diagnosis, drug resistance, and deficiency in effective targeted treatment.
Drug metabolism is a biotransformation process of drugs that is usually mediated by specific enzymes (Almazroo et al., 2017). The drug-metabolizing pathways mediated by drug-metabolizing enzymes (DMEs) are classified into phase I (functionalization) and phase II (conjugation) reactions. Phase I reactions are the redox or hydrolysis process of the drug to activate or detoxify it, which are mainly mediated by phase I DMEs, including cytochrome P450 enzymes (P450s), Flavin-containing monooxygenases, alcohol dehydrogenases (ADHs), and aldehyde dehydrogenases. P450s comprise 70%–80% of all phase I DMEs (Nebert and Dalton, 2006). The P450 superfamily can be divided into two parts; CYP1-4 families are responsible for the biotransformation of most xenobiotic compounds, whereas CYP7-51 families are mainly involved in the metabolism of endogenous substances in a substrate-specific manner. Most P450s are located in the liver, resulting in its strong detoxification effect. The most abundantly expressed P450 isoforms in the liver are CYP3A4, 2C9, 2C8, 2E1, and 1A2, whereas CYP2A6, 2D6, 2B6, 2C19, and 3A5 are less abundant (Zanger and Schwab, 2013). P450s, including CYP2C9, 2C19, 3A4, and 3A5, are also distributed in mature intestinal epithelial cells and are responsible for intestinal metabolism. Besides, some P450s such as CYP1A1 and 1B1 mainly express extrahepatically, which are in accordance with their metabolic roles of environmental pollutants and endogenous compounds. In phase II reactions catalyzed by phase II DMEs, the products from phase I pathways conjugate with a hydrophilic endogenous compound. After conjugation, the substances are converted into water-soluble products, which are easy to excrete. Most of phase II DMEs consist of transferases, including UDP-glucuronosyltransferases (UGTs), sulfoctransferases (SULTs), glutathione S-transferases (GSTs), and N-acetyltransferases (NATs) (Almazroo et al., 2017). Phase II DMEs are mostly located in the liver and kidney. UGTs are major members of phase II DME, which mediate glucuronidation and elimination of a variety of endogenous and exogenous substances and are considered integral parts of detoxification enzymes in the human body (Pathania et al., 2018). GSTs catalyze the conjugation reactions of nucleophilic glutathione with various electronic xenobiotics, thus facilitating their elimination and protecting cells from oxidative stress and other stimuli (Pljesa-Ercegovac et al., 2018). The abnormal expression of DMEs may lead to changes in the metabolism of drugs or procarcinogens, thus causing diseases. It also brings a considerable challenge for individualized treatment by affecting the metabolic process and adverse effects.
Epigenetics is the study of heritable changes in gene expression without any alternation in the DNA sequence. The main contents of epigenetics include DNA methylation, histone modification, and noncoding RNAs (ncRNAs) (Ivanov et al., 2012; Zanger et al., 2014). Many factors can influence Epigenetic regulation, such as age, diet, lifestyle, environment, and disease. Accumulating evidence demonstrates that epigenetic modification changes a lot during tumorigenesis. DNA methylation and histone modification patterns of some genes, as well as the expression of ncRNAs, are expected to be biomarkers for early detection, diagnosis, prognosis, drug disposition, and clinical response of cancer (Chen et al., 2013; Xu et al., 2017; Tan et al., 2018). Reversing gene expression in cancer by changing the abnormal epigenetic modification also provides a new train of thought for the treatment of cancer (Lachenmayer et al., 2012; Liu et al., 2016). This review will give a brief summary of abnormal expression of DMEs in cancer and epigenetic regulation of differential expression of DMEs.
Differential Expression of DMEs in Cancer
P450s are the most abundant family of DMEs, expressing in almost all organs. P450s are involved in the metabolic inactivation of endogenous and exogenous compounds. However, in some instances, they also mediate the metabolic activation of many carcinogens, which increase the risk of cancer. CYP1A1 and CYP1B1 are causally implicated in activation of procarcinogens such as polycyclic aromatic hydrocarbons (PAHs). CYP2A6, CYP2A13, and CYP2E1 can metabolize nitrosamines into unstable metabolites, which can form diazonium ions. CYP2E1 is also involved in the metabolic activation of tetrachloromethane, accompanying the production of free radicals (He and Feng, 2015). CYP3A4 participates in the metabolic activation and detoxification of hepatic carcinogen aflatoxin B1 and is tightly related to the carcinogenesis of hepatocellular carcinoma induced by aflatoxin B1 (Kamdem et al., 2006).
Emerging evidence indicates that DMEs play an essential role in the formation, prevention, metastasis, and treatment of cancer (Alzahrani and Rajendran, 2020). The high expression of some DMEs is commonly considered a reason for carcinogenesis, metastasis, and chemoresistance because of the increased activation of procarcinogens and inactivation of anticancer drugs. For instance, the high expression of CYP1A1 promotes the activation of PAH, and the active metabolites covalently bond to DNA and produce DNA adducts, which eventually leads to DNA damage and tumorigenesis (Moorthy et al., 2015). The overexpression of CYP2J2 in cancer cell lines brings on increased four regioisomeric epoxyeicosatrienoic acids, which promote cancer metastasis (Jiang et al., 2007). Besides, dihydropyrimidine dehydrogenase is pivotal to the catabolism of Fluorouracil, so the upregulation of dihydropyrimidine dehydrogenase can reduce the activity of cyclophosphamide (CTX) greatly (Pathania et al., 2018). The abnormal low expression of DMEs is also a risk factor for tumor initiation. Some DMEs act on carcinogen and play a detoxifying role, so the repression of them cause tumor growth. For prodrugs that require DMEs for metabolic activation, the repression of these DMEs can also cause drug resistance. CTX, a broad-spectrum antineoplastic prodrug, is converted to its active form by CYP2B6 and 3A4 (Lindley et al., 2002). Therefore, the suppression of these P450s will materially affect the efficacy of CTX. The aberrant expression of DMEs occurs in several cancer types, including liver, prostate, and lung cancers. A summary of the differentially expressed DMEs and their functions is listed in Table 1.
List of differentially expressed DMEs during carcinogenesis and their functions
Liver Cancer.
The liver is the most vital organ for drug biotransformation and is rich in DMEs. The expression and activity of DMEs can be modulated by several factors, such as genetic polymorphisms and disease states. It has been confirmed that the metabolism activities of P450 isoforms are severely impaired in patients with hepatocellular carcinoma (HCC) by investigating the activities of major P450s in microsomes from normal and HCC liver tissue samples (Yan et al., 2015b).
Several studies represent a series of evidence implicating that the expression of DMEs changed in HCC samples. Because of the decrease of functional hepatocytes in HCC, most of the phase I and phase II metabolizing enzymes are expressed at lower levels compared with noncancerous liver tissues, including CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4, CYP3A5, CYP3A7, CYP4A11, NAT1, NAT2, UGT1A1, UGT1A4, UGT1A9, and UGT2B7 (Chen et al., 2014; Lu et al., 2015; Yan et al., 2015a,b). The silence of CYP2C19 expression in hepatitis B virus–infected patients with HCC is reported to be regulated by e-box methylation of the constitutive androstane receptor (Tang et al., 2016). The dysregulation of DMEs in HCC is a pivotal reason for clinical chemotherapy failure (Ul-Islam et al., 2018). The expression of DMEs is also associated with the risk of liver cancer. CYP2E1 is related to the activation of many toxicants. The high expression of CYP2E1 is recognized as a risk factor for hepatic fibrosis (Guo et al., 2019). The research revealed that hepatic fibrotic rats with higher CYP2E1 activity develop a more severe form of HCC (Gao et al., 2018). Besides, CYP2E1 also participates in the formation of etheno-DNA adducts, which are potent carcinogens of liver cancer (Linhart et al., 2014). GSTs are detoxifying enzymes and play a predominant role in cell protection. A meta-analysis suggested that the inactivation of GSTP1 in HCC correlates with the hepatocarcinogenesis (Li et al., 2018).
Lung Cancer.
The lung is the main organ exposed to the inhaled chemical toxicants and carcinogens, so the metabolizing enzymes that participate in xenobiotic metabolism are essential for respiratory protection (Leclerc et al., 2010). In some instances, they can convert procarcinogens to active metabolites. These active intermediates can form DNA adducts, cause gene mutation, and eventually lead to cancer (Castell et al., 2005). The increase of CYP1A1 activity contributes to the metabolic activation and carcinogenicity of PAHs. The researchers designed a liposome-based CYP1A1 silencing nanomedicine, showing the potential for the treatment of lung cancer (Zhang et al., 2019). CYP1B1 catalyzes the activation of N-nitrosamines such as 4-methylnitrosamino-l-3-pyridyl-butanone (NNK). NKK can also induce CYP1B1 expression, thus accelerating lung cancer progression (Li et al., 2015b). GSTM2 is a detoxifying enzyme that expressed a low level in lung cancer cells (Tang et al., 2011). The mRNA expression level of DMEs in pulmonary parenchyma, bronchial mucosa, and tumoral lung tissues were detected using a high throughput quantitative real-time reverse-transcription polymerase chain reaction method. It is demonstrated that ADH1B, CYP3A7, and CYP4B1 show decreased mRNA levels in lung cancer (Leclerc et al., 2011).
Prostate Cancer.
Prostate cancer is the most common cancer that occurs in men, which is mostly androgen-dependent. Androgen deprivation therapy (ADT) is still the first-line treatment of metastatic prostate cancer (Litwin and Tan, 2017). UGT2B15 and UGT2B17 are involved in androgen inactivation in prostate cells (Pâquet et al., 2012). Some studies have found that UGT2B17 deletion polymorphism is related to prostate cancer susceptibility (Karypidis et al., 2008). UGT2B15 and UGT2B17 differentially expressed during prostate cancer progression (Pâquet et al., 2012). Besides, P450s are key inactivators of testosterone in the prostate. It was reported that decreased CYP2B6 and CYP3A4 are significantly related to the development and poor prognosis of prostate cancer (Kumagai et al., 2007; Fujimura et al., 2009). After continuous ADT, these cancers may become androgen-independent and resistant to ADT. In this process, the expression of GST-π increases, indicating its role in the development of prostate cancer (Hokaiwado et al., 2008).
Kidney Cancer.
In the kidney, UGT expression is related to the clearance of many xenobiotics. A proteomic study reported that UGT1A6, UGT1A9, and UGT2B7 are the most abundant UGT subtypes in the kidney. The mRNA and protein levels of UGT1A9 and UGT2B7 are significantly downregulated in tumor kidneys, accompanied by decreased glucuronidation capacity (Margaillan et al., 2015).
Esophageal Cancer.
A study revealed the potential clinical relevance between the expression of CYP2C9 and esophageal cancer. The expression of CYP2C9 in esophageal adenocarcinoma and adjacent esophageal mucosa was higher compared with esophageal squamous cell carcinoma. CYP2C9 is likely to promote esophageal cancer proliferation (Schmelzle et al., 2011).
Other Hormone-Induced Cancers.
Some DMEs are relevant to the metabolism of hormones, so the aberrant expression of these DMEs may occur in hormone-induced cancers. CYP1B1 is mainly responsible for the metabolism of estradiol, forming carcinogens 4-hydroxy estradiol. The enrichment of 4-hydroxy estradiol in breast, ovarian, and prostate is considered an increased risk for developing cancers (Yager, 2000). CYP1B1 is causally implicated in the carcinogenesis of breast cancer, ovarian cancer, prostate cancer, and lung cancer. CYP1B1 expression increases in estrogen-related tumors but is very low in normal tissues (McFadyen et al., 1999; Carnell et al., 2004; Gajjar et al., 2012). The CYP11 subfamily is responsible for steroid biosynthesis (Thomas, 2007). The downregulation of CYP11A1 in cancers may affect the biosynthesis of steroids. Data obtained from The Cancer Gene Atlas database revealed that CYP11A1 is significantly downregulated in six cancers types, including colon adenocarcinoma, renal clear cell carcinoma, hepatocellular carcinoma, lung squamous cell carcinoma, prostate adenocarcinoma, and uterine corpus endometrial carcinoma (Fan et al., 2016).
Epigenetic Regulation of Differentially Expressed DMEs in Cancer
DNA Methylation
DNA methylation is a dynamic process involving methylation and demethylation. DNA methyltransferase (DNMT) 1, 3A, and 3B can transfer a methyl group to the cytosine at cytosine-phosphoric acid-guanine (CpG) motif to form 5-methylcytosine. Passive or active DNA demethylation can reverse DNA methylation patterns. Passive DNA demethylation is likely to be because of the reduction or inhibition of DNA methyltransferase, so the DNA methylation status cannot maintain during DNA replication (Piccolo and Fisher, 2014). Active DNA demethylation is mainly mediated by activation-induced cytidine deaminase/apolipoprotein B mRNA-editing enzyme complex or ten-eleven translocation (Tet) enzymes Tet1, Tet2, and Tet3. Methyl-CpG binding proteins can recognize 5-methylcytosine, which has a high affinity for 5-methylcytosine. Methyl-CpG binding proteins cause chromatin structure modification and remodeling by recruiting corepressor complexes such as histone deacetylase (HDAC) to methylated promoter regions, thus reducing gene expression (Clouaire and Stancheva, 2008). DNA methylation plays a critical role in gene expression and chromatin remodeling. It can repress gene expression by changing the chromatin structure directly, hindering transcription factor, or coactivator binding to the promoter region of the target gene (Moore et al., 2013).
It has been widely reported that DNA methylation is involved in the regulation of differentially expressed DMEs in tumors. In tumors, the methylation status of CpG islands in the gene promoter regions is closely related to the expression level of the target gene. Abnormal DNA methylation in tumors and normal tissues can be detected in body fluids such as blood and urine, indicating that DNA methylation is expected to be a biomarker for liquid biopsy, which can be used for diagnosis and monitoring of cancer. Also, the abnormal expression of DNA methyltransferase may occur in the process of cancer development and affect the expression level of DMEs, thus promoting the development of cancer. Therefore, DNA methyltransferase is a potential therapeutic target of cancer. Changing the expression of DNA methyltransferase may reverse the expression of DMEs in cancer. DNA methylation inhibitor decitabine (5-aza-2'-deoxycytidine) has been approved by the US Food and Drug Administration for the treatment of hematologic malignancies (Nie et al., 2014).
Besides, considering the importance of DMEs toward personalized medicine, Genome-wide integrative analysis was used to analyze the DNA methylation and mRNA expression profiles of human tissues and hepatoma cells, which revealed that some DME genes, including CYP1A2, CYP2C19, CYP2D6, GSTA4, GSTM5, GSTT1, and SULT1A1 are regulated by DNA methylation, potentially leading to individual differences in drug metabolism (Habano et al., 2015).
Hypomethylation Status of DME Genes in Cancer
DNA hypomethylation is always considered the main reason for high-level gene expression. Some DMEs are responsible for the metabolic activation of environmental toxicants and carcinogens; their high expression can contribute to cancer progression. Meanwhile, some DMEs can metabolically inactivate anticancer drugs, thus causing chemoresistance (Fig. 1).
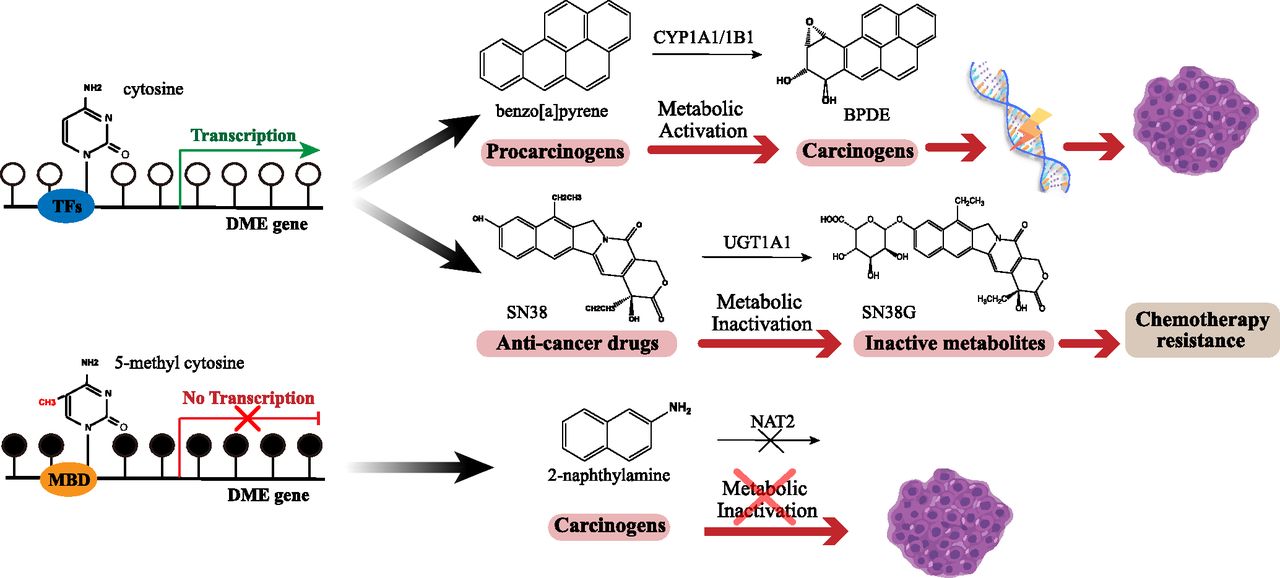
The methylation status of DME genes contributes to cancer progression and chemoresistance. White dots represent cytosine, and black dots represent 5-methyl cytosine.
CYP1A1 can activate multiple carcinogens and therefore promote cancer progression. The expression and methylation status were detected in prostate cancer cells. CYP1A1 expression is higher in cancer cells compared with normal cells. When treated with decitabine, the expression of CYP1A1 become much higher (Mitsui et al., 2016). In breast cancer cell line MCF-7 and T47D, estrogen receptor α can repress the expression of CYP1A1 through recruiting DNMT3b(Marques et al., 2013).
Aberrant DNA methylation status of CYP1B1 has been observed in several hormone-related cancer types, such as prostate cancer and breast cancer. The methylation status of CYP1B1 was analyzed in prostate cancer tissues and benign prostatic status hyperplasia samples; the results revealed that methylation of its promoter/enhancer region was much lower in prostate cancer, which may play a role in cancer development. It is well known that CYP1B1 is induced by the aryl hydrocarbon receptor (AhR) and AhR nuclear translocator directly. When treated with DNA methyltransferase inhibitor, 5-Aza-dC, no cell line showed a significant change of the expression of AhR and AhR nuclear translocator, indicating that CpG methylation of CYP1B1 promoter is key to its expression (Tokizane et al., 2005). Moreover, considering the function of metabolizing estradiol and tamoxifen, CYP1B1 hypomethylation is perceived as a carcinogenic factor as well as predictive markers for response to tamoxifen therapy in breast cancer (Widschwendter et al., 2004).
UGT1A1 is a critical phase II metabolizing enzyme involved in the metabolic inactivation of SN38, the active metabolite of irinotecan. Irinotecan is a first-line drug for the treatment of metastatic colorectal cancer (Hahn et al., 2019), so the hypomethylation status of UGT1A1 may accelerate the inactivation of irinotecan to reduce the efficacy of irinotecan. Bisulfite sequencing of UGT1A1 observed the abnormal methylation modification of specific CpG islands in UGT1A1-negative cells such as HCT-116, HCT-15, and COLO-320DM, whereas in HT-29, HT-115, and LOVO cell lines with high expression of UGT1A1, these sites were in the hypomethylation states. Methylation of the UGT1A1 promoter can repress its transcriptional activity completely. A combination of DNA methyltransferase inhibitor and histone deacetylase inhibitor can reverse the hypermethylation and restore the expression of UGT1A1 in UGT1A1 negative cells (Gagnon et al., 2006). Another research investigated the correlation between UGT1A1 expression and its sensitivity to irinotecan in seven colorectal cell lines. The cell lines with low UGT1A1 expression are more sensitive to irinotecan. The methylation status of UGT1A1 can obviously affect the cytotoxicity of irinotecan (Xie et al., 2014).
Hypermethylation Status of DME Genes in Cancer
In general, DNA hypermethylation suppresses the expression of DMEs, which are involved in detoxification. It is causally implicated in the occurrence and development of cancer (Fig. 1).
As we mentioned before, CYP1B1 hypomethylation was observed in several hormone-related cancers. However, the hypermethylation status of the CYP1B1 promoter can be found in colon cancer (Habano et al., 2009), bladder cancer (Putluri et al., 2011), and adolescents with acute lymphocytic leukemia(DiNardo et al., 2013), indicating a worse outcome. Metabolic profiling revealed that CYP1B1 hypermethylation could also be found in body fluids such as urine, suggesting it might be a potential biomarker for distinguishing benign bladder and bladder cancer (Putluri et al., 2011).
GSTs are significant phase II detoxification enzymes. GST-M2, a member of GST subfamily μ-class GST, has special clinical features. The activity of GST-M2 in human normal embryonic lung fibroblast MRC-5 is significantly higher than in lung cancer cell line H1355. The catalytic activity of GST-M2 is closely related to DNA damage induced by carcinogens (Weng et al., 2005). The low expression of GST-M2 in lung cancer cell lines can be reversed after treatment with DNMT inhibitor decitabine. The CpG islands on the GST-M2 promoter are highly methylated. It is demonstrated that in lung cancer tissues, the low expression of GST-M2 is accompanied by high expression of DNMT3b, indicating a close relationship between DNA methylation and GST-M2 expression. GST-M2 expression in lung cancer cell lines can be induced after silencing DNMT3b. Consequently, the expression of GST-M2 in lung cancer cells is negatively regulated by DNA methylation. CpG hypermethylation of GST-M2 blocks the binding of transcription factor specificity protein to GST-M2 promoter, thus inhibiting the transcription of GST-M2 (Tang et al., 2011). GST-M2 hypermethylation is also investigated in Barrett’s adenocarcinoma and pancreatic cancers (Peng et al., 2009; Tan et al., 2009).
GSTP1, the gene encoding the π-class GST, is repressed in multiple cancer subtypes, including solid tumors such as prostate (Henrique and Jerónimo, 2004), breast (Fang et al., 2015), liver (Revill et al., 2013), lung cancers (Gao et al., 2009), and hematologic malignancies, because of CpG island hypermethylation in the promoter regions. The aberrant methylation status of the GSTP1 promoter is regarded as a specific marker for prostate cancer and can be found in at least 90% of prostate cancers (Nakayama et al., 2004). It has been reported that GSTP1 promoter methylation may increase the incidence and recurrence of prostate cancer (Maldonado et al., 2014; Zhou et al., 2019). Besides, GSTP1 CpG island hypermethylation can also be detected in the urine and plasma from patients with prostate cancer. This means that GSTP1 CpG island hypermethylation can be used as a biomarker for the diagnosis and prognosis of prostate cancer. In a recent study, a sensitive methylation-specific polymerase chain reaction assay was applied to detect the serum-free methylated GSTP1 DNA in patients with metastatic castration-resistant prostate cancer. It has been demonstrated that the expression of serum-free methylated GSTP1 is closely correlated with overall survival and response to docetaxel in metastatic castration-resistant prostate cancer (Mahon et al., 2019).
NAT1 is a phase II metabolizing enzyme that is responsible for the biotransformation of most arylamine and hydrazine substrates. Several studies have shown that NAT1 can influence the development and drug resistance of breast cancer (Rodrigues-Lima et al., 2010). The frequency of NAT1 methylation was significantly lower in the control group compared with the tamoxifen-resistant breast cancer group. The hypermethylation of the NAT1 gene may affect the initiation of tamoxifen resistance in breast cancer (Kim et al., 2010).
Histone Modification
Histones are the basic structural proteins of chromatin. Histone octamer consists of two copies each of histones H2A, H2B, H3, and H4, which are wrapped with 147 base pairs of DNA. Histone is a basic protein because it contains a high proportion of basic amino acids such as lysine and arginine. The N terminal of histone is dissociated from the nucleosome, so the specific amino acid residues can be modified by methylation, acetylation, phosphorylation, ubiquitination, and similar processes. The chromatin structure changed after modification, thus regulating the gene transcription (Luger et al., 2012). Aberrant histone modifications may lead to abnormal gene expression in cancer. Histone acetylation neutralizes its positive charge, thereby opening the chromatin structure and making it easier for transcription factors to bind to their target genes, so histone acetylation is always regarded as a transcriptional activation signal (Haberland et al., 2009). HDACs are potential targets for cancer therapy, and the US Food and Drug Administration has approved several HDAC inhibitors such as Vorinostat and Belinostat (PXD101) for the treatment of cutaneous T-cell lymphoma and peripheral T-cell lymphoma. Furthermore, bromodomain-containing proteins recognize the acetylated lysine residues of histone, which play an essential role in the process of cancer development. Thus, designing small-molecule bromodomain-containing protein inhibitors may be a promising strategy (Fu et al., 2015; Yu et al., 2015b). Histone methylation exhibits distinct functions of gene activation or repression with different modification sites (H3K4, H3K9, H3K27, etc.) and methylation states (mono-/di-/tri-methylation) (Barski et al., 2007). Enhancer of zeste homolog 2 is a methyltransferase that can add methyl groups to histone H3 at lysine 27 (H3K27), thus repressing gene transcription. Disruptor of telomeric silencing 1–like methyltransferase is responsible for H3K79 methylation. Now the inhibitors of enhancer of zeste homolog 2 and disruptor of telomeric silencing 1–like are in clinical trials and show potent anticancer capacity (Mohammad et al., 2019).
Although emerging evidence suggests that these histone modifications affect the expression of DMEs in the nontumor environment (Tang and Chen, 2015; Yan et al., 2017), the regulation of histone modification of DMEs in the tumor has not yet been reported. Considering the extensive aberrant histone modifications in cancer and their essential roles, we need to clarify the relationship between histone modification and differential expression patterns of DMEs to provide references to personalized medicine.
1α, 25-dihydroxyvitamin D3 (1, 25-D3), the active form of vitamin D, is antiproliferative in lung adenocarcinoma. 1, 25-D3 is catabolized by CYP24A1, which is overexpressed in multiple types of cancer. CYP24A1 mRNA was elevated 8- to 50-fold in lung adenocarcinoma compared with normal tissues. The overexpression of CYP24A1 is much more significant in poorly differentiated cancers, accompanied by lower survival rates (Chen et al., 2011b). In lung adenocarcinoma cells, combined treatment with DNMT inhibitor 5-Aza-dC and HDAC inhibitor Trichostatin A (TSA) increases the CYP24A1 expression and enzyme affinity to its substrate 1, 25-D3. The chromatin immunoprecipitation coupled by quantitative polymerase chain reaction (ChIP-qPCR) assay revealed that TSA enriched H3K4me2 and H3K9ac and simultaneously decreased H3K9me2 at the CYP24A1 promoter, thus activating the transcriptional expression of CYP24A1 (Ramnath et al., 2014). In human neuroblastoma cells, histone deacetylase inhibitors such as valproic acid and TSA affect the expression of CYP1A1, CYP1B1, and CYP3A4 (Hřebačková et al., 2009). Another study found that inhibition of the β-catenin signaling pathway induced the CYP1A1 expression through histone H2AX phosphorylation (Kabátková et al., 2015).
Noncoding RNA
The term “noncoding RNA” refers to RNA molecules that are transcribed from genome but not translated into proteins, including microRNA (miRNA), long noncoding RNA (lncRNA), and circular RNA (circRNA) (Klingenberg et al., 2017). They can give full play to the function of gene regulation at the transcriptional and post-transcriptional levels. Mature miRNAs are single-stranded ncRNAs of 22–25 nucleotides in length, which are derived from primary miRNA transcripts (Li and Rana, 2014). The mature miRNA must assemble into the RNA-induced silencing complex to target its complementary mRNAs for translational repression or target gene degradation (Li et al., 2014). lncRNAs are transcripts longer than 200 nucleotides that have little or no protein-coding capacity, and they are transcribed by RNA polymerase II, capped, spliced, and polyadenylated. lncRNAs can regulate gene expression at different levels, including chromatin modification, transcription, and post-transcriptional processing (Mercer et al., 2009). Emerging evidence indicates the translation potential of lncRNAs with open reading frames, which has been overlooked over a long period (Matsumoto et al., 2017). These polypeptides encoded by lncRNAs may also play a crucial role in cancer occurrence and development, it was reported that a peptide encoded by lncRNA HOXB-AS3 suppresses colon cancer growth through a complex regulatory mechanism (Huang et al., 2017). circRNAs are a novel type of RNA molecule that are different from traditional linear RNAs. They have a closed-loop structure and exist in a lot of eukaryotic transcriptomes (Qu et al., 2015). Most circRNAs are composed of exon sequences, which are conserved in different species and have regulatory potency (Memczak et al., 2013). circRNAs are not sensitive to nuclease, which makes them more stable than linear RNA, owing to their closed-loop structures. Therefore, circRNAs have more potential to become biomarkers in the screening of cancer (Li et al., 2015a,c). The most common regulatory mechanism of lncRNA and circRNA is acting as the “sponge” of miRNA, which can regulate the target gene through changing miRNA expression (Wang et al., 2010; Yu et al., 2016).
With the development of RNA Sequencing, researchers have obtained the expression profiles of miRNA, lncRNA, and circRNA in different types of cancer and their matched paracancerous normal tissues, such as liver cancer, kidney cancer, and breast cancer. They screen the ncRNAs, which are closely related to the occurrence, development, and prognosis of cancer, providing a new biomarker or target for the diagnosis or treatment of cancer (Xie et al., 2013; Li et al., 2015a,c). At present, the regulation of ncRNA on differentially expressed DMEs in the tumor is still limited in the field of miRNA. The regulation of lncRNA and circRNA needs to be further explored.
Phase I DMEs.
Abnormal miRNA expression occurs in lung cancer tissues compared with normal tissues. To clarify the functions of these miRNA, researchers established a tobacco-induced cancer rat model to investigate the relationship between miRNA and the occurrence of early lung cancer. It has been demonstrated that carcinogen could reduce the expression of miR-101, miR-126*, miR-199, and miR-34. These miRNAs overlap with previously published reports on altered miRNA expression in human lung cancer samples, suggesting these four miRNAs may be involved in lung cancer development. Treatment with NNK inhibits miR-126* but induces CYP2A3 expression, an essential enzyme to activate NNK, indicating that miR-126* has the possibility of regulating CYP2A3 (Kalscheuer et al., 2008).
The low expression level of miR-27b may contribute to the high expression of CYP1B1 in the mammary gland(Tsuchiya et al., 2006), thus causing the accumulation of 4-hydroxy estradiol and increasing the risk of breast cancer. In HCC, a report has established a negative correlation between the level of hsa-miR-128-3p, hsa-miR-143-3p, and CYP2C9 expression based on in silico analysis and a series of biochemical assays (Yu et al., 2015a).
Recently, RNA-interfering miRNA materials have been designed to interfere with the expression of DMEs. It was reported that a newly established bioengineered RNA agent (BERA), BERA/miR-27b-3p, can be processed into mature miR-27b-3p in human cells, thus decreasing the expression and metabolic capability of CYP3A4 (Li et al., 2019b).
Phase II DMEs.
In prostate cancer, miRNA is causally implicated in post-transcriptional regulation of UGTs. Androgen plays a vital role in the development of prostate cancer. UGT2B15, UGT2B17, and UGT2B28 mediate the biotransformation of androgen in vivo. Reporter gene assays validated that miR-376c, miR-409, and miR-494 could interact with UGT2B17, and miR-331-5p and miR-376c could bind to UGT2B15 (Margaillan et al., 2016). miR-376c can effectively repress UGT2B15 and UGT2B17 expression, accompanied by a consequent decrease in dihydrotestosterone glucuronidation. The expression of UGT2B15 and UGT2B17 are negatively related to miR-376c expression but positively correlated to metastasis rate in advanced prostate cancer (Wijayakumara et al., 2015). miR-331-5p is also confirmed to reduce the UGT2B15 mRNA level by targeting its 3′-UTR via canonical and noncanonical pairing (Wijayakumara et al., 2018).
Furthermore, UGT2A1 is responsible for the detoxification of PAHs found in cigarette smoke and exhibits high expression in the lung. A recent study suggested that the UGT2A1 expression level can be regulated by both miR-196a-5p and miR-196b-5p (Sutliff et al., 2019).
In breast cancer, miR-1290 is confirmed to target the 3′-UTR of NAT1 directly, which is positively correlated with the overall survival of patients with breast cancer (Endo et al., 2014). The expression of NAT10 is dysregulated in colorectal cancer, which is validated to be inhibited by miR-7616-5p (Liu et al., 2019). Published studies have identified that hsa-miR-486-5p and hsa-miR-495-3p decrease the mRNA stability of phase II detoxification enzymes SULT2A1 in HepG2 human hepatocellular carcinoma cell line (Li et al., 2019a).
Summary and Prospect
DMEs are implicated in the metabolic activation or inactivation of xenobiotics, which are strongly associated with the occurrence and development of cancer. During the development of cancer, the expression level of DMEs changes, thus reducing the detoxification ability of DMEs and promoting cancer progression. In the treatment of cancer, the differentially expressed DMEs may influence the efficacy of anticancer drugs or cause adverse effects by affecting the metabolic process of these drugs.
Epigenetics, especially DNA methylation, plays a critical role in the regulation of differentially expressed DMEs in tumors. DNA methyltransferases are expected to be the target of antitumor drugs. Aberrant DNA methylation modifications are also promising biomarkers in liquid biopsy. A growing body of research suggests that histone modification and ncRNA can regulate the expression of DMEs under nontumor conditions. Histone modification and miRNA have already been proven to participate in the transcriptional regulation of differentially expressed DMEs in lung, liver, prostate, and breast cancer, and the regulatory mechanism in other cancer types needs to be further studied.
Emerging studies showed that the cross talk among various epigenetic mechanisms is noteworthy and needs to be further explored in the regulation of DMEs. Histone modification and DNA methylation always work in concert to regulate gene expression. Methyl-CpG binding proteins recruit protein complex, which contains HDACs and/or histone methyltransferases, inducing the formation of repressive chromatin circumstance (Nan et al., 1998; Fuks et al., 2003). In some instances, the administration of HDAC inhibitors such as TSA and valproic acid reverse the hypermethylation status of certain genes (Ou et al., 2007; Gu et al., 2012). The combination of DNMT and HDAC inhibitors always led to better efficacy, which is considered a good strategy. Histone methylation displays closer ties with DNA methylation. S-adenosyl-l-methionine is a universal methyl group donor, so the content of S-adenosyl-l-methionine in cells affects the methylation status of DNA and histone simultaneously. Furthermore, the cross talk between DNA and histone methylation can be mediated by the interaction between DNA and histone methyltransferases (Cedar and Bergman, 2009). DNA hypermethylation status, increased repressive histone modification H3k9me2, decreased H3K9ac, and H3K4me2 modification at CYP24A1 promoter contribute to the suppression of CYP24A1 in prostate cancer. Combined treatment with DNMT inhibitor DAC and HDAC inhibitor TSA upregulated the CYP24A1 expression, accompanied by increased recruitment of vitamin D receptor to CYP24A1 promoter (Luo et al., 2010). The interplay between miRNA and DNA methylation in gene regulation is also widely reported. The transcription and synthesis of miRNAs can be repressed by DNA methylation, and certain miRNAs can change the DNA methylation status of the gene by targeting DNMTs in return (Fuso et al., 2020). In colorectal cancer, inflammatory factor cytokine interleukin-6 was reported to promote DNMT1 nuclear translocation, and then DNMT1 caused DNA methylation of CpG islands near miR27b, thus suppressing its transcription. Because of the reduced degradation by miR27b, CYP1B1 showed a high expression level (Patel et al., 2014).
Also, further research is required to clarify if the epigenetic regulations of DMEs contribute to the metabolism of endogenous substrates during cancer progression and its downstream impacts. Elucidating the epigenetic regulatory mechanism of DMEs in tumors can provide a basis for implementing individualized and rationalized medication as well as developing new biomarkers and targets for the diagnosis, treatment, and prognosis of cancer.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Wang, Yu, Jiang, Zheng, Zeng.
Footnotes
- Received March 12, 2020.
- Accepted June 1, 2020.
This work was supported by the National Key R&D Program of China [2017YFC0908600] and [2017YFE0102200], the National Natural Science Foundation of China [81702801]; and Leading Talent of "Ten Thousand Plan"- National High-Level Talents Special Support Plan.
Abbreviations
- ADH
- alcohol dehydrogenase
- ADT
- androgen deprivation therapy
- AhR
- aryl hydrocarbon receptor
- BERA
- bioengineered RNA agent
- BET
- bromodomain-containing proteins
- circRNA
- circular RNA
- CpG
- phosphoric acid-guanine
- CTX
- cyclophosphamide
- 1
- 25-D3
- 1α
- 25-dihydroxyvitamin D3
- DME
- drug-metabolizing enzyme
- DNMT
- DNA methyltransferase
- GST
- glutathione S-transferase
- HCC
- hepatocellular carcinoma
- HDAC
- histone deacetylase
- lncRNA
- long noncoding RNA
- miRNA
- microRNA
- NAT
- N-acetyltransferase
- ncRNA
- noncoding RNA
- NNK
- 4-methylnitrosamino-l-3-pyridyl-butanone
- P450
- cytochrome P450
- PAH
- polycyclic aromatic hydrocarbon
- SULT
- sulfotransferase
- Tet
- ten-eleven translocation
- TSA
- trichostatin A
- UGT
- UDP-glucuronosyltransferase
- Copyright © 2020 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}