


Abstract
Intestinal permeability is a critical factor for orally administered drugs. It can be facilitated by uptake transporters or limited by efflux transporters and metabolic enzymes in the intestine. The present study aimed to characterize the Ussing chamber system incorporating human intestinal tissue as an in vitro model for investigating the impact of intestinal uptake/efflux transporters on the intestinal absorption of substrate drugs in humans. We confirmed the functions of major intestinal uptake/efflux drug transporters in freshly isolated human jejunum sections by demonstrating a significant decrease in the mucosal uptake of cefadroxil (peptide transporter 1) and methotrexate (proton-coupled folate transporter), mucosal-to-serosal permeability of ribavirin (concentrative nucleoside transporters/equilibrative nucleoside transporters), and serosal-to-mucosal permeability of P-glycoprotein and breast cancer resistance protein substrates in the presence of their typical inhibitors. The mucosal-to-serosal apparent permeability coefficients (Papp) of 19 drugs, including substrates of drug transporters and cytochrome P450 3A, ranged from 0.60 × 10−6 to 29 × 10−6 cm/s and showed a good correlation with reported fraction of an oral dose that enters the gut wall and passes into the portal circulation with escaping intestinal metabolism (FaFg) values in humans. Furthermore, the Papp values for cefadroxil, methotrexate, and ribavirin in the presence of the corresponding transporter inhibitors underestimated the FaFg of these drugs, which clearly showed that intestinal uptake transporters facilitate their intestinal absorption in humans. In conclusion, the functions of major intestinal uptake/efflux drug transporters could be maintained in freshly isolated human jejunum sections. The Ussing chamber system incorporating human intestinal tissue would be useful for evaluating the impact of intestinal uptake/efflux transporters on the intestinal absorption of various types of drugs in humans.
SIGNIFICANCE STATEMENT Although previous studies have predicted the intestinal absorption of drugs in humans using the Ussing chamber system incorporating human intestinal tissue, there is little systematic information about drug transport mediated by multiple transporters in this system. We confirmed the functions of major intestinal uptake/efflux transporters in freshly isolated human jejunum sections and demonstrated that the mucosal-to-serosal apparent permeability coefficient of various types of drugs showed a good correlation with reported human FaFg values.
Introduction
The intestinal absorption properties of compounds are a critical factor in the early stages of drug development to assist in identifying candidate drugs for oral administration that have sufficient bioavailability. In addition to passive diffusion, intestinal absorption mediated by uptake/efflux transporters and metabolic enzymes expressed in enterocytes determines the efficiency of oral drug absorption. For example, uptake transporters, such as peptide transporter 1 (PEPT1), proton-coupled folate transporter (PCFT), and concentrative nucleoside transporters (CNTs), can facilitate the uptake from the gut lumen to enterocytes of their physiologic substrates dipeptides/tripeptides, folate, and nucleosides and some drugs, such as cephalosporins, antifolates, and nucleoside analogs, that share similar structural moieties with these endogenous compounds (Gray et al., 2004; Visentin et al., 2014; Müller et al., 2017). Efflux transporters, such as P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP), present on the apical membrane of enterocytes are responsible for limiting the intestinal absorption of various substrate drugs by pumping them back from enterocytes to the gut lumen (Estudante et al., 2013). Previous clinical drug-drug interaction (DDI) and pharmacogenetic studies have demonstrated the importance of intestinal transporters for the bioavailability of drugs in humans (Zhang et al., 2006; Urquhart et al., 2008; Fenner et al., 2009). Therefore, it is essential to evaluate the quantitative impact of transporters on intestinal drug absorption.
Caco-2 cells, an immortalized cell line originating from human colon cancer, are widely used in drug development as an in vitro model for the prediction of human oral absorption. The fraction of oral drugs that enters the gut wall (Fa) in humans significantly correlates with the apparent permeability coefficient (Papp) across Caco-2 cell monolayers for drugs passing through enterocyte monolayers mainly via passive diffusion (Artursson and Karlsson, 1991). Several studies have reported that Caco-2 cells are not ideal to investigate transporter-mediated membrane transport and metabolism in the intestine because of their low expression of some transporter isoforms, such as PEPT1 and BCRP, and cytochrome P450 (CYP) 3A, the most important intestinal CYP isoform in terms of drug metabolism (Nakamura et al., 2002; Larregieu and Benet, 2013). With regard to the prediction of human oral absorption from preclinical animal studies, Chiou and Barve (1998) have demonstrated that although Fa values of drugs in humans are similar to those in rodents, a large species difference in the product of Fa and the intestinal availability (Fg) of drugs was observed even between humans and cynomolgus monkeys (Takahashi et al., 2009; Akabane et al., 2010). Therefore, it is difficult to predict intestinal drug absorption in humans from conventional approaches using Caco-2 cells or animal models, especially when drugs are substrates of transporters and/or metabolic enzymes.
Human-derived tissue samples, such as hepatocytes and kidneys, have facilitated the prediction of the pharmacokinetic properties of drugs (Nozaki et al., 2007; Maeda and Sugiyama, 2010). Human intestinal tissues mounted in Ussing chambers, an experimental system developed by Ussing and Zerahn (1951) to measure ion transport across epithelial tissues, have also been used as an in vitro system to investigate drug permeation across intestinal epithelial cells. Several studies that have investigated the intestinal permeation of drugs using freshly isolated human intestinal tissues mounted in Ussing chambers have observed a theoretical sigmoidal relationship between Papp values of drugs across human intestinal tissues and their Fa values in humans (Haslam et al., 2011; Rozehnal et al., 2012; Sjöberg et al., 2013) or their transport indices, a recently defined parameter (Miyake et al., 2013). P-gp–mediated efflux or CYP3A4-mediated metabolism of a limited number of compounds was also successfully assessed in this system (Makhey et al., 1998; Berggren et al., 2003; Rozehnal et al., 2012; Sjöberg et al., 2013). Keiser et al. (2017) recently carried out experiments focusing on the expression and function of organic anion transporting polypeptide (OATP) 2B1. However, because of the limited availability of human intestine, no study has comprehensively evaluated the impact of multiple uptake/efflux transporters on the intestinal absorption of their substrate drugs. To address this issue, we established a protocol to obtain frequently (typically twice a month) freshly isolated human small intestinal sections and to seamlessly perform Ussing chamber assays on the same site as the surgery.
In the present study, we evaluated the functions of major intestinal uptake/efflux drug transporters in freshly isolated human intestinal sections and demonstrated the usefulness of this system for evaluating the impact of transporters on the fraction of an oral dose that enters the gut wall and passes into the portal circulation with escaping intestinal metabolism (FaFg) values of transporter substrate drugs in humans.
Materials and Methods
Chemicals.
Cefadroxil, digoxin, enalapril, sulpiride, nadolol, sulfasalazine, valspodar (PSC833), nitrobenzylthioinosine (NBMPR), glycylsarcosine (Gly-Sar), and folate were purchased from Merck (Darmstadt, Germany). Midazolam, metoprolol, propranolol, ketoprofen, antipyrine, rosuvastatin, methotrexate, ranitidine, hydrochlorothiazide, atenolol, pravastatin, ribavirin, and lucifer yellow were purchased from Fujifilm Wako Pure Chemical (Osaka, Japan). Fexofenadine was purchased from Toronto Research Chemicals (North York, Canada). Ko143 and decitabine were purchased from ChemScene LLC (Monmouth Junction, NJ). All other chemicals used for analyses were commercially available and analytical-grade products.
Excision of Human Intestinal Sections from Patients.
Human small intestinal specimens used in this study were obtained from 16 clinical patients with pancreatic head tumors (nine men, seven women; median age 68 years, range 39–81 years) who underwent pancreatoduodenectomy at the University of Tsukuba Hospital (Ibaraki, Japan) between June 2019 and March 2020 (Table 1). Approximately 15–20 cm normal jejunum is usually coexcised at pancreatoduodenectomy, but it is heavily damaged with 2–4 hours ischemia at the timing of whole-specimen removal. Instead of such usual sampling, approximately 5 cm of jejunum, which is included within the planned removal region, was excised just after the ligation of the artery of jejunum at the early stage of surgery because of the minimization of the warm ischemic period (for 10 minutes at most). The excised jejunum was immediately put in ice-cold Dulbecco’s modified Eagle’s medium at the operating theater and transferred to the laboratory at University of Tsukuba within 30 minutes. All the experimental protocols with regard to these jejunum sections were approved by the ethics committees of both the Graduate School of Pharmaceutical Sciences in the University of Tokyo (Tokyo, Japan) and the Faculty of Medicine in University of Tsukuba (Ibaraki, Japan). Written informed consent prior to surgery was obtained from all patients.
Sex and age of individual subjects donating jejunum sections
Quantification of mRNA Expression in Human Intestinal Sections.
A small part of the excised freshly isolated human intestinal section was immediately put into RNAlater solution (Thermo Fisher Scientific, Waltham, MA). The part of the tissue was transferred on dry ice to the laboratory at the University of Tokyo and preserved at −80°C until analysis by reverse transcription–quantitative polymerase chain reaction (qPCR). After thawing the frozen tissue section, total RNA was isolated from the tissue section using ISOGEN II (NIPPON GENE, Tokyo, Japan), and cDNA was synthesized using ReverTra Ace qPCR RT Master Mix with gDNA Remover (TOYOBO, Tokyo, Japan) according to the manufacturer’s instructions. The mRNA expression levels of ABCB1 (P-gp), ABCG2 (BCRP), SLC15A1 (PEPT1), SLC46A1 (PCFT), SLC28A1 (CNT1), SLC28A2 (CNT2), SLC28A3 (CNT3), SLC29A1 [equilibrative nucleoside transporter (ENT) 1], SLC29A2 (ENT2), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were determined by qPCR analysis using THUNDERBIRD SYBR qPCR Mix (TOYOBO) and a LightCycler 480 System II (Roche Diagnostics, Mannheim, Germany). The linear range of crossing point (Cp) value of each gene was 13–30 (ABCB1), 14–27 (ABCG2), 15–29 (SLC15A1), 14–26 (SLC46A1), 13–26 (SLC28A1), 16–30 (SLC28A2), 17–31 (SLC28A3), 17–32 (SLC29A1), 14–29 (SLC29A2), and 13–27 (GAPDH). All the quantified data were confirmed to be within the range of linearity. The sequences of primers for the detection of the above-mentioned mRNAs are shown in Supplemental Table 1. The mRNA expression levels of transporter isoforms were normalized to the mRNA expression level of GAPDH (reference gene).
Preparation of Human Intestinal Sections for Ussing Chamber Assays.
We essentially followed the protocol for the preparation of human intestinal tissue sections for Ussing chamber assays previously published by Kisser et al. (2017) as kindly instructed by Dr. Katja Damme, Dr. Markus Keiser, and Dr. Veronika Rozehnal at Daiichi Sankyo Europe GmbH, Tissue and Cell Research Center Munich (Munich, Germany). The excised human intestinal section was opened with scissors and rinsed with ice-cold Krebs-Ringer bicarbonate (KRB) buffer (116 mM NaCl, 1.25 mM CaCl2, 3.6 mM KCl, 1.4 mM KH2PO4, 1.2 mM MgSO4, 10 mM D-glucose, 23.1 mM NaHCO3; adjusted to pH 7.4). The tissue section was fixed on a silicone mat by pins with the mucosal side facing downward and kept in ice-cold KRB buffer, which was continuously gassed with 95% O2/5% CO2 gas mixture. The muscle layers were removed by cutting the connective tissue between the muscle layer and the submucosa carefully with sharp scissors. After that, the fat and submucosa was also removed carefully with sharp scissors as much as possible. The prepared tissue section was mounted carefully using fine-tipped tweezers in Ussing chambers with an exposed area of 0.636 cm2 (Harvard Apparatus Inc., Holliston, MA). Five milliliters of KRB buffer (37°C, pH 7.4) was added to each compartment of the Ussing chambers, and the mounted tissue section was washed once and equilibrated for 20 minutes in KRB buffer. KRB buffer was continuously gassed with 95% O2/5% CO2 gas mixture before and during these processes, and the temperature of the KRB buffer was maintained at 37°C.
Measurement of the Mucosal-to-Serosal Transport and Mucosal Uptake of Cefadroxil and Methotrexate in Human Intestinal Sections Using the Ussing Chamber System.
Cefadroxil and methotrexate (each 10 μM) were selected as substrates to evaluate the functions of the uptake transport mediated by PEPT1 and PCFT, respectively (Yokooji et al., 2009; Posada and Smith, 2013). Gly-Sar (20 mM) and folate (200 μM), typical substrates of PEPT1 and PCFT, respectively, were used to saturate the functions of PEPT1 and PCFT. To evaluate the integrity of intestinal sections, lucifer yellow (100 μM) was chosen as a paracellular marker. Propranolol (10 μM) was used to evaluate the effects of transporter inhibitors on its passive transcellular transport.
Prior to the addition of test compounds, the buffer in both compartments was replaced with fresh KRB buffer (pH 7.4) in the presence or absence of PEPT1/PCFT inhibitor cocktail (added only to the mucosal side), and tissue sections were preincubated for another 20 minutes. Next, the experiment was initiated by replacing the buffer in both compartments with 5 ml of fresh KRB buffer (mucosal side, pH 6.5; serosal side, pH 7.4) with or without test compounds in the presence or absence of PEPT1/PCFT inhibitor cocktail (added only to the mucosal side). To determine the initial concentrations of test compounds, 10 μl of the buffer in the donor compartment was collected immediately after starting the transport study. One milliliter of the buffer in the receiver compartment was collected, and an equal volume of fresh KRB buffer was added to the receiver compartment at 30, 60, 90, and 120 minutes. After 120 minutes incubation, the tissue sections were taken out of the chambers and washed with ice-cold KRB buffer to determine the accumulation of drugs in the tissue section. The collected samples were preserved at −20°C until liquid chromatography–tandem mass spectrometry (LC-MS/MS) analyses.
Measurement of the Mucosal-to-Serosal Transport of Ribavirin across Human Intestinal Sections Using the Ussing Chamber System.
Ribavirin (10 μM) was selected as a CNTs/ENTs substrate to evaluate the mucosal-to-serosal vectoral transport mediated by CNTs and ENTs expressed on the apical and basolateral membrane of enterocytes, respectively (Moss et al., 2012). Decitabine (500 μM) and NBMPR (100 μM) were used as typical inhibitors of CNTs and ENTs, respectively. As above, lucifer yellow (100 μM) and propranolol (10 μM) were added to the buffer in the donor compartment.
Prior to the addition of test compounds, the buffer in both compartments was replaced with fresh KRB buffer (pH 7.4) in the presence or absence of each inhibitor and a mixture of these inhibitors (added only to the mucosal side), and the tissue sections were preincubated for another 20 minutes. Afterward, the experiment was initiated by replacing the buffer in both compartments with 5 ml of fresh KRB buffer (mucosal side, pH 6.5; serosal side, pH 7.4) with or without test compounds in the presence or absence of each inhibitor and a mixture of these inhibitors (added only to the mucosal side). The sampling of the buffer was carried out as described above.
Measurement of the Serosal-to-Mucosal and Mucosal-to-Serosal Transport of Digoxin, Fexofenadine, Sulfasalazine, and Rosuvastatin across Human Intestinal Sections Using the Ussing Chamber System.
Digoxin and fexofenadine (each 5 μM) were selected as P-gp substrates (Tahara et al., 2005; Fenner et al., 2009), and sulfasalazine and rosuvastatin (each 5 μM) were selected as BCRP substrates (Zhang et al., 2006; Urquhart et al., 2008) to evaluate the efflux transport mediated by P-gp and BCRP. PSC833 (15 μM) and Ko143 (15 μM) were used as typical inhibitors of P-gp and BCRP, respectively. As above, lucifer yellow (100 μM) and propranolol (10 μM) were added to the buffer in the donor compartment.
Prior to the addition of test compounds, the buffer in both compartments was replaced with fresh KRB buffer (pH 7.4) in the presence or absence of P-gp/BCRP inhibitor cocktail (added to both mucosal and serosal sides), and the tissue sections were preincubated for another 20 minutes. Afterward, the experiment was initiated by replacing the buffer in both compartments with 5 ml of fresh KRB buffer (mucosal side, pH 6.5; serosal side, pH 7.4) with or without test compounds in the presence or absence of P-gp/BCRP inhibitor cocktail (added to both mucosal and serosal sides). The sampling of the buffer was carried out as described above.
Measurement of the Mucosal-to-Serosal Transport of Compounds across Human Intestinal Sections Using the Ussing Chamber System.
We measured the mucosal-to-serosal transport across human intestinal sections of 19 test compounds, including compounds transported mainly passively via the transcellular route and/or the paracellular route (metoprolol, propranolol, ketoprofen, antipyrine, enalapril, ranitidine, hydrochlorothiazide, sulpiride, nadolol, atenolol, and pravastatin), transporter substrate drugs (cefadroxil, methotrexate, ribavirin, digoxin, fexofenadine, sulfasalazine, and rosuvastatin), and CYP3A substrate drug (midazolam). These compounds were used to investigate the relationship between the Papp in the mucosal-to-serosal direction of drugs in the Ussing chamber system and the reported FaFg values in humans. Donor concentrations of test compounds were initially set at 10 μM. As above, lucifer yellow (100 μM) was used as a paracellular marker to evaluate the integrity of the tissue sections.
Prior to the addition of test compounds, the buffer in both compartments was replaced with fresh KRB buffer (pH 7.4), and the tissue sections were preincubated for another 20 minutes. Afterward, the experiment was initiated by replacing the buffer in both compartments with 5 ml of fresh KRB buffer (mucosal side, pH 6.5; serosal side, pH 7.4) with or without test compounds. The sampling of the buffer was carried out as described above.
Analytical Method for Determining Drug Concentration.
For quantification of test compounds except for lucifer yellow by LC-MS/MS, an AB Sciex QTRAP 5500 tandem mass spectrometer (AB Sciex, Foster City, Canada) equipped with Nexera UHPLC (Shimadzu, Kyoto, Japan) was used.
After thawing, the sampled buffer from the receiver compartment (200 μl) was mixed with three volumes of acetonitrile containing an internal standard (5 nM talinolol), vortexed, and centrifuged (20,000g for 5 minutes at 4°C). The supernatant (400 μl) was dried using a centrifugal concentration device (CC-105; TOMY, Tokyo, Japan). The sample was redissolved in distilled water containing 0.1% formic acid/acetonitrile mixture (8:2, v/v) (100 μl) and injected into the LC-MS/MS system. The sampled buffer from the donor compartment was diluted 10 times with distilled water, mixed with three volumes of acetonitrile containing an internal standard (50 nM talinolol), vortexed, and centrifuged (20,000g for 5 minutes at 4°C). The supernatant was diluted five times with distilled water containing 0.1% formic acid and injected into the LC-MS/MS system. The sampled tissue section was homogenized in 5 ml of distilled water/acetonitrile mixture (1:3, v/v) using an ULTRA–TURRAX T25 (IKA, Staufen, Germany). The homogenate sample was mixed with three volumes of acetonitrile containing an internal standard (0.5 μM talinolol), vortexed, and centrifuged (20,000g for 10 minutes at 4°C). The supernatant was diluted 40 times with distilled water containing 0.1% formic acid and injected into the LC-MS/MS system.
The injection volume was 10 μl in each case. An Atlantis T3 C18 column (3 μm, 2.1 × 50 mm; Waters Corp., Milford, MA) was used as the analytical column for compounds except for ribavirin and a Gemini C6-Phenyl column (5 μm, 4.6 × 100 mm; Shimadzu GLC) was used for ribavirin. Distilled water containing 0.1% formic acid (solvent A) and acetonitrile containing 0.1% formic acid (solvent B) were used as the mobile phase. The mass spectrometer with electrospray ionization was operated in positive and negative multiple-reaction monitoring (MRM) mode. The details of the high-pressure liquid chromatography and MRM conditions are summarized in Supplemental Tables 2 and 3. Lucifer yellow was quantified using a fluorescence microplate reader (excitation wavelength 428 nm, emission wavelength 536 nm) (Infinite 200; TECAN, Männedorf, Switzerland).
Data Analysis.
The Papp (centimeters per second) was calculated according to eq. 1: (1)in which dQ/dt is the amount of the test compound transported to the acceptor compartment per unit time when the linearity of its time-dependent transport was apparently maintained, A is the exposed area of tissue section (0.636 cm2), and C0 is the initial concentration of test compound in the donor compartment.
(1)in which dQ/dt is the amount of the test compound transported to the acceptor compartment per unit time when the linearity of its time-dependent transport was apparently maintained, A is the exposed area of tissue section (0.636 cm2), and C0 is the initial concentration of test compound in the donor compartment.
The mucosal uptake (microliters per 120 minutes) in the tissue section was calculated according to eq. 2: (2)in which Q120 is the total amount of test compound transported to the acceptor compartment after 120 minutes incubation, X120 is the total amount of test compound accumulated in the tissue section after 120 minutes incubation, and C0 is the initial concentration of test compound in the donor compartment.
(2)in which Q120 is the total amount of test compound transported to the acceptor compartment after 120 minutes incubation, X120 is the total amount of test compound accumulated in the tissue section after 120 minutes incubation, and C0 is the initial concentration of test compound in the donor compartment.
Nonlinear regression analysis was performed using the Papp values of 11 test compounds transported mainly passively via the transcellular and/or the paracellular route (metoprolol, propranolol, ketoprofen, antipyrine, enalapril, ranitidine, hydrochlorothiazide, sulpiride, nadolol, atenolol, and pravastatin) and the reported FaFg values in humans of drugs based on a logistic nonlinear regression model (Karpinski et al., 1987) (eq. 3): (3)in which λ is Hill’s slope representing the steepness of the curve, and A is the Papp value at which FaFg = 50%. GraphPad Prism 8 (GraphPad Software, San Diego, CA) was used for this nonlinear regression analysis.
(3)in which λ is Hill’s slope representing the steepness of the curve, and A is the Papp value at which FaFg = 50%. GraphPad Prism 8 (GraphPad Software, San Diego, CA) was used for this nonlinear regression analysis.
Human FaFg values were cited from a previous report (Varma et al., 2010) or calculated by us based on the following equations using pharmacokinetic parameters from previous reports; (4)
(4) (5)
(5) (6)
(6) (7)
(7) (8)
(8) (9)
(9) (10)in which F, Fa, Fg, and Fh are bioavailability, the fraction of an oral dose that enters the gut wall, the fraction of drug that passes into the portal circulation with escaping intestinal metabolism (intestinal availability), and a fraction of a drug that escapes from first-pass metabolism in the liver (hepatic availability), respectively. CLtotal, CLrenal, and CLnon-renal represent total clearance, renal clearance, and nonrenal clearance, respectively. Aurine is the cumulative amount of unchanged drug excreted into urine, and furine is the fraction of unchanged drug excreted into urine. AUCpo and AUCiv are the area under the blood concentration–time curve after oral administration and intravenous administration of compounds. dosepo and doseiv are the oral dose and the intravenous dose. The RB value (blood-to-plasma concentration ratio) of all the drugs was assumed to be 1. Qh (hepatic blood flow rate) was set at a typical value of 23 ml/min per kilogram body weight.
(10)in which F, Fa, Fg, and Fh are bioavailability, the fraction of an oral dose that enters the gut wall, the fraction of drug that passes into the portal circulation with escaping intestinal metabolism (intestinal availability), and a fraction of a drug that escapes from first-pass metabolism in the liver (hepatic availability), respectively. CLtotal, CLrenal, and CLnon-renal represent total clearance, renal clearance, and nonrenal clearance, respectively. Aurine is the cumulative amount of unchanged drug excreted into urine, and furine is the fraction of unchanged drug excreted into urine. AUCpo and AUCiv are the area under the blood concentration–time curve after oral administration and intravenous administration of compounds. dosepo and doseiv are the oral dose and the intravenous dose. The RB value (blood-to-plasma concentration ratio) of all the drugs was assumed to be 1. Qh (hepatic blood flow rate) was set at a typical value of 23 ml/min per kilogram body weight.
Statistical Analysis.
Results are presented as the mean ± S.D. with n representing the number of replicates in each experiment (except for Table 2) or the total number of replicates, including intestinal sections originating from independent subjects (N = 3–6) (Table 2). Statistical significance of the differences in the mean values between the data for two groups and among the data for three or more groups was assessed by unpaired Student’s t test and ANOVA followed by Dunnett’s test, respectively. All analysis was performed using GraphPad Prism 8 (GraphPad Software). Statistical significance was defined at P < 0.05.
Summary of the Papp (centimeters per second) in the mucosal-to-serosal direction across human intestinal sections for test compounds
Papp values are presented as the mean ± S.D. (n = 12–29).
Results
Analysis of Expression Levels of mRNA for Intestinal Transporters in Human Intestinal Sections.
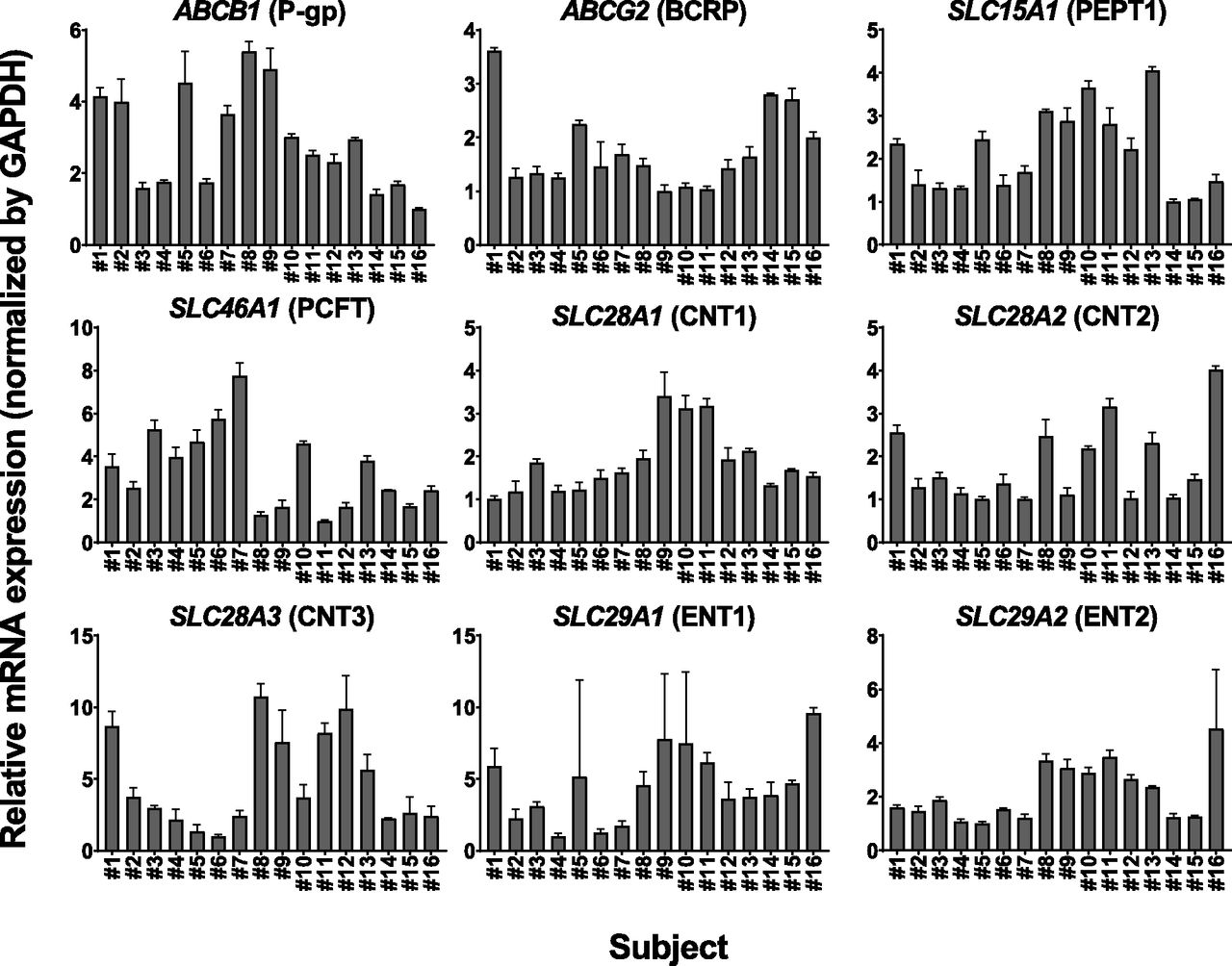
The mRNA expression levels of ABCB1, ABCG2, SLC15A1, SLC46A1, SLC28A1, SLC28A2, SLC28A3, SLC29A1, and SLC29A2, which were normalized to the expression level of GAPDH, were determined by reverse transcription–qPCR in human intestinal sections originating from 16 independent subjects (Table 1). The ratios between the minimum and maximum values of mRNA expression were 5.40 for ABCB1, 3.62 for ABCG2, 4.06 for SLC15A1, 7.75 for SLC46A1, 3.40 for SLC28A1, 4.01 for SLC28A2, 10.7 for SLC28A3, 9.59 for SLC29A1, and 4.53 for SLC29A2, respectively. The average mRNA expression level was 1.76 ± 0.81 for ABCB1, 2.83 ± 1.16 for ABCG2, 5.50 ± 2.37 for SLC15A1, 1.76 ± 0.95 for SLC46A1, 0.276 ± 0.107 for SLC28A1, 0.326 ± 0.159 for SLC28A2, 0.131 ± 0.088 for SLC28A3, 0.334 ± 0.177 for SLC29A1, and 0.204 ± 0.097 for SLC29A2 (Fig. 1).
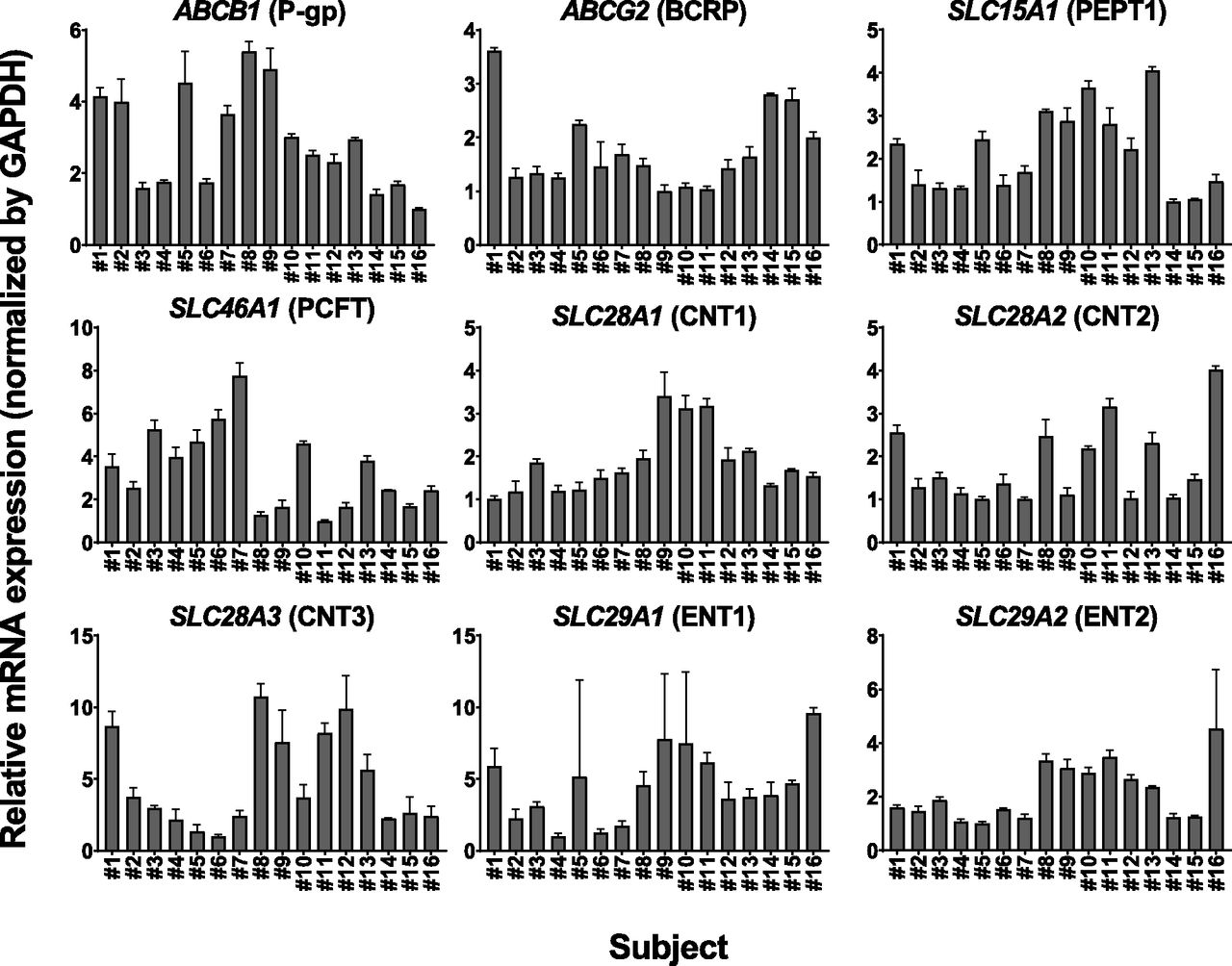
The relative expression levels of mRNAs for intestinal transporters in human intestinal sections originating from 16 different subjects (numbers 1–16). The mRNA expression levels of ABCB1 (P-gp), ABCG2 (BCRP), SLC15A1 (PEPT1), SLC46A1 (PCFT), SLC28A1 (CNT1), SLC28A2 (CNT2), SLC28A3 (CNT3), SLC29A1 (ENT1), and SLC29A2 (ENT2) were normalized to that of GAPDH. Data are presented as fold change relative to the minimum values of the expression level for each transporter gene among tested samples (ABCB1; number 16, ABCG2; number 9, SLC15A1; number 14, SLC46A1; number 11, SLC28A1; number 1, SLC28A2; number 5, SLC28A3; number 6, SLC29A1; number 4, SLC29A2; number 5). Each bar and error bar represent the mean value and S.D., respectively (n = 3).
Effect of Pre-Experimental Period on the mRNA and Protein Expression Levels of Transporters and Mucosal-to-Serosal Transport of Compounds across Human Intestinal Sections.
To investigate whether the expressions and functions of major intestinal uptake and efflux transporters were maintained in freshly isolated human intestinal sections, we evaluated the mucosal uptake or the permeability of the major uptake and efflux transporter substrates in the presence or absence of their inhibitors. In our preliminary studies, the mRNA and protein expression levels of intestinal transporters in the tissue sections kept in ice-cold KRB buffer for 4 hours after excision showed no major changes compared with those in the fresh tissue sections immediately after excision (Supplemental Figs. 2 and 3). However, during this period, a significant decrease was observed in the mucosal-to-serosal transport of cefadroxil (PEPT1 substrate) and methotrexate (PCFT substrate) across the tissue sections compared with freshly excised tissue sections (Supplemental Fig. 1, A and B). The 4-hour incubation of the excised tissue sections with ice-cold KRB buffer also led to a significant increase in the mucosal-to-serosal transport of lucifer yellow, which is a paracellular marker, but not of propranolol, which is a lipophilic compound that mainly passes passively via the transcellular route, indicating that even short-term incubation in ice-cold KRB buffer might affect the integrity of tissue sections (Supplemental Fig. 1, C and D). Therefore, all the following transport experiments were performed at the site where the tissue sections were excised to maintain their freshness as much as possible.
Effects of PEPT1 and PCFT Inhibitors on the Mucosal-to-Serosal Transport and Mucosal Uptake of Cefadroxil and Methotrexate in Human Intestinal Sections.
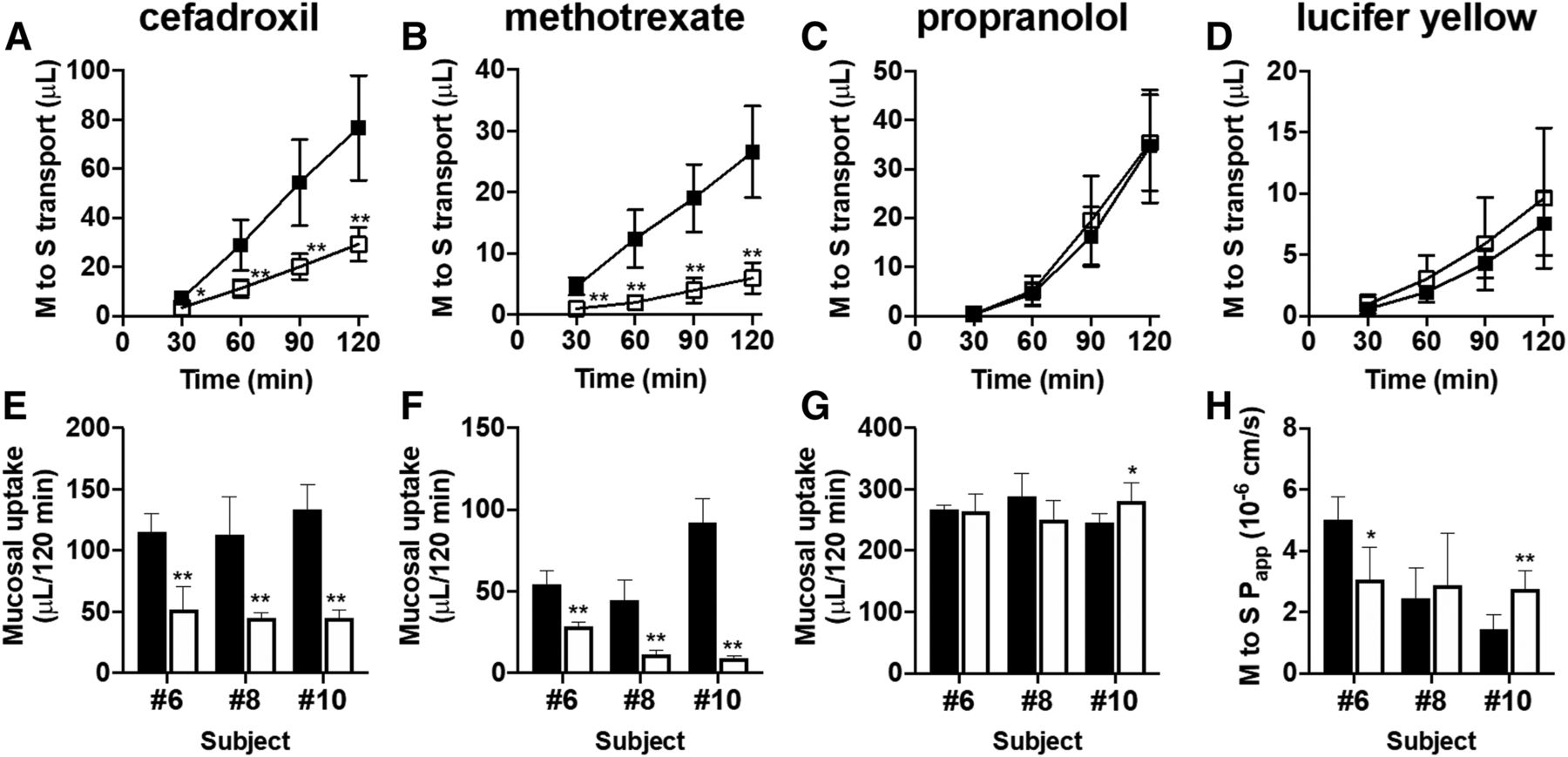
We used freshly isolated human intestinal sections originating from three independent subjects (numbers 6, 8, and 10) to evaluate the mucosal-to-serosal transport and mucosal uptake of cefadroxil (10 μM; PEPT1 substrate) and methotrexate (10 μM; PCFT substrate) in the presence or absence of a mixture of 20 mM Gly-Sar and 200 μM folate. In parallel, the mucosal uptake of propranolol (10 μM) and the mucosal-to-serosal permeability of lucifer yellow (100 μM) were evaluated to check the effect of PEPT1/PCFT inhibitor cocktail on passive transcellular transport and paracellular transport, respectively. The mucosal-to-serosal transport and mucosal uptake of cefadroxil and methotrexate were significantly decreased in the presence of PEPT1/PCFT inhibitor cocktail in all batches of tissue sections (Fig. 2, A, B, E, and F). In contrast to these PEPT1 and PCFT substrate drugs, although some batches of tissue sections showed significant differences in the mucosal uptake of propranolol and the mucosal-to-serosal Papp of lucifer yellow, the direction of that change was not consistent across batches (Fig. 2, C, D, G, and H), suggesting that transport of propranolol and lucifer yellow was not unambiguously altered by the presence of PEPT1/PCFT inhibitor cocktail.

Mucosal-to-serosal (M to S) transport and mucosal uptake of cefadroxil and methotrexate in the presence or absence of a cocktail of PEPT1/PCFT inhibitors. Typical data for mucosal-to-serosal transport of cefadroxil (A) (10 μM; PEPT1 substrate drug), methotrexate (B) (10 μM; PCFT substrate drug), propranolol (C) (10 μM; passive transcellular marker), and lucifer yellow (D) (100 μM; paracellular marker) across human intestinal sections (number 8) mounted in Ussing chambers in the presence (open squares) or absence (closed squares) of a mixture of 20 mM Gly-Sar (PEPT1 inhibitor) and 200 μM folate (PCFT inhibitor). Each symbol and error bar represent the mean value and S.D., respectively (n = 6). Mucosal uptake of cefadroxil (E) (10 μM), methotrexate (F) (10 μM), and propranolol (G) (10 μM) and the mucosal-to-serosal Papp of lucifer yellow (H) (100 μM) across human intestinal sections originating from three independent subjects (numbers 6, 8, and 10) mounted in Ussing chambers in the presence (open columns) or absence (closed columns) of a mixture of 20 mM Gly-Sar and 200 μM folate. Each bar and error bar represent the mean value and S.D., respectively (n = 5 to 6). *P < 0.05; **P < 0.01 vs. control.
Effects of CNTs and ENTs Inhibitors on the Mucosal-to-Serosal Transport of Ribavirin across Human Intestinal Sections.
In freshly isolated human intestinal sections originating from three independent subjects (numbers 14–16), the mucosal-to-serosal transport of ribavirin (10 μM; CNTs and ENTs substrate) in the presence of 500 μM decitabine (CNTs inhibitor), 100 μM NBMPR (ENTs inhibitor), or a mixture of 500 μM decitabine and 100 μM NBMPR was compared with that in the absence of any inhibitors. The mucosal-to-serosal transport of ribavirin was significantly decreased in the presence of each inhibitor, and a mixture of these inhibitors further suppressed that transport (Fig. 3, A and D). In contrast, no significant difference in the mucosal-to-serosal transport of propranolol and lucifer yellow was observed in the presence of CNTs/ENTs inhibitors (Fig. 3, B, C, E, and F).
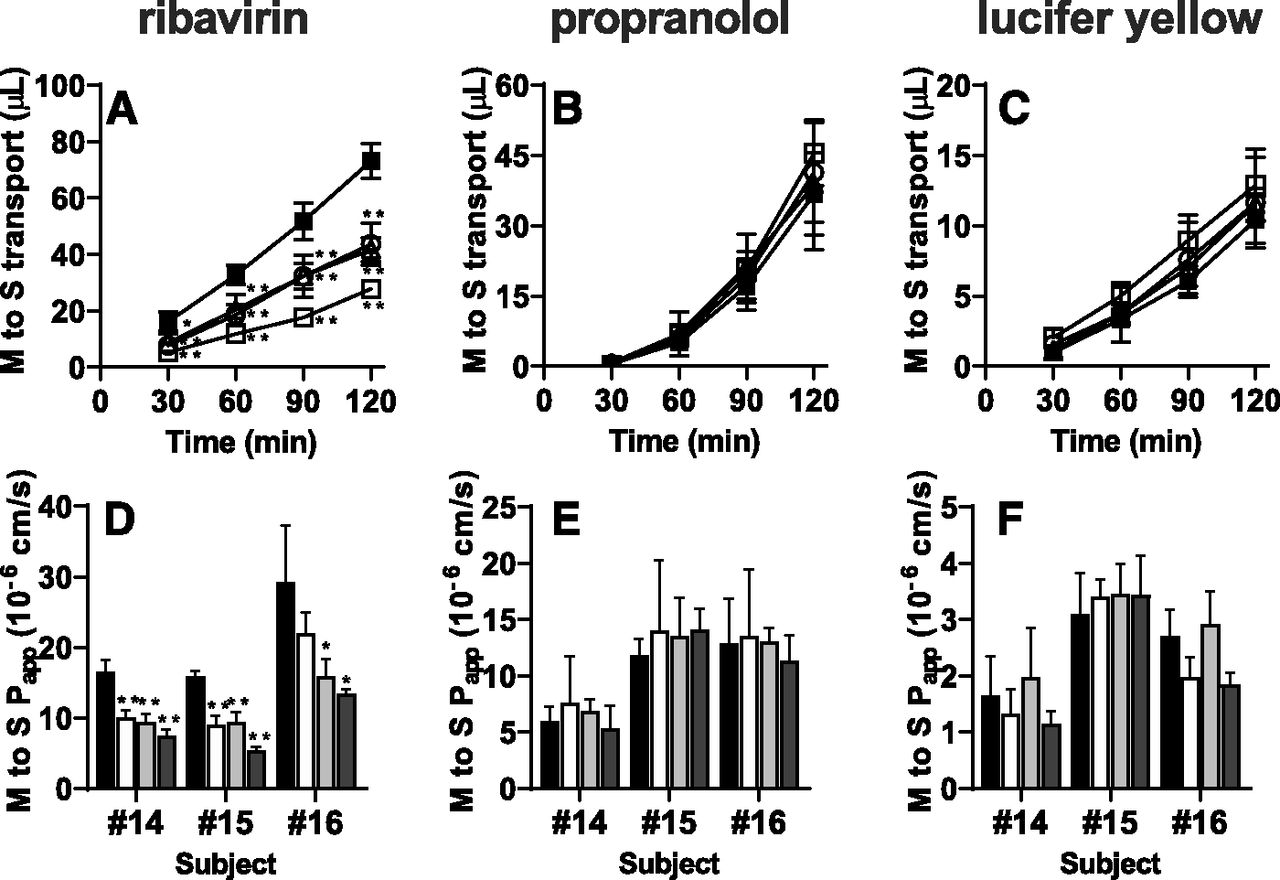
Mucosal-to-serosal (M to S) transport of ribavirin in the presence or absence of CNTs/ENTs inhibitors. Typical data for mucosal-to-serosal transport of ribavirin (A) (10 μM; CNTs and ENTs substrate), propranolol (B) (10 μM; passive transcellular marker), and lucifer yellow (C) (100 μM; paracellular marker) across human intestinal sections (number 15) mounted in Ussing chambers with 500 μM decitabine (open triangles), with 100 μM NBMPR (open circles), with a mixture of 500 μM decitabine and 100 μM NBMPR (open squares), and without any inhibitors (closed squares). Each symbol and error bar represent the mean value and S.D., respectively (n = 3). The Papp of ribavirin (D) (10 μM), propranolol (E) (10 μM), and lucifer yellow (F) (100 μM) across human intestinal sections originating from three independent subjects (numbers 14–16) mounted in Ussing chambers with 500 μM decitabine (white columns), with 100 μM NBMPR (gray columns), with a mixture of 500 μM decitabine and 100 μM NBMPR (dark gray columns), and without any inhibitors (black columns). Each bar and error bar represent the mean value and S.D., respectively (n = 3). *P < 0.05; **P < 0.01 vs. control.
Effects of P-gp and BCRP Inhibitors on the Serosal-to-Mucosal Transport of Digoxin, Fexofenadine, Sulfasalazine, and Rosuvastatin across Human Intestinal Sections.
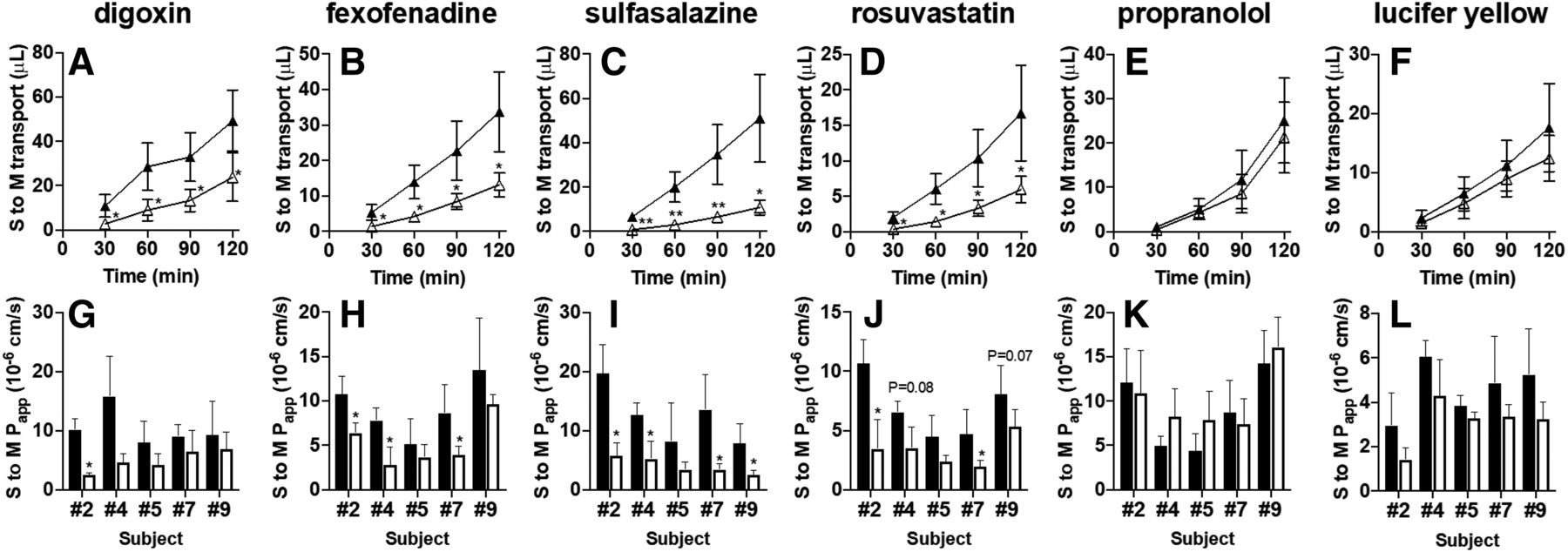
In freshly isolated human intestinal sections originating from five independent subjects (numbers 2, 4, 5, 7, and 9), we evaluated the serosal-to-mucosal transport of digoxin and fexofenadine (each 5 μM; P-gp substrates) and sulfasalazine and rosuvastatin (each 5 μM; BCRP substrates) in the presence or absence of a mixture of PSC833 (15 μM; P-gp inhibitor) and Ko143 (15 μM; BCRP inhibitor). Serosal-to-mucosal transport of all the tested P-gp and BCRP substrate drugs was significantly decreased or tended to decrease in the presence of P-gp/BCRP inhibitor cocktail (Fig. 4, A–D and G–J). In contrast, there was no significant difference in the serosal-to-mucosal transport of propranolol and lucifer yellow (Fig. 4, E, F, K, and L).
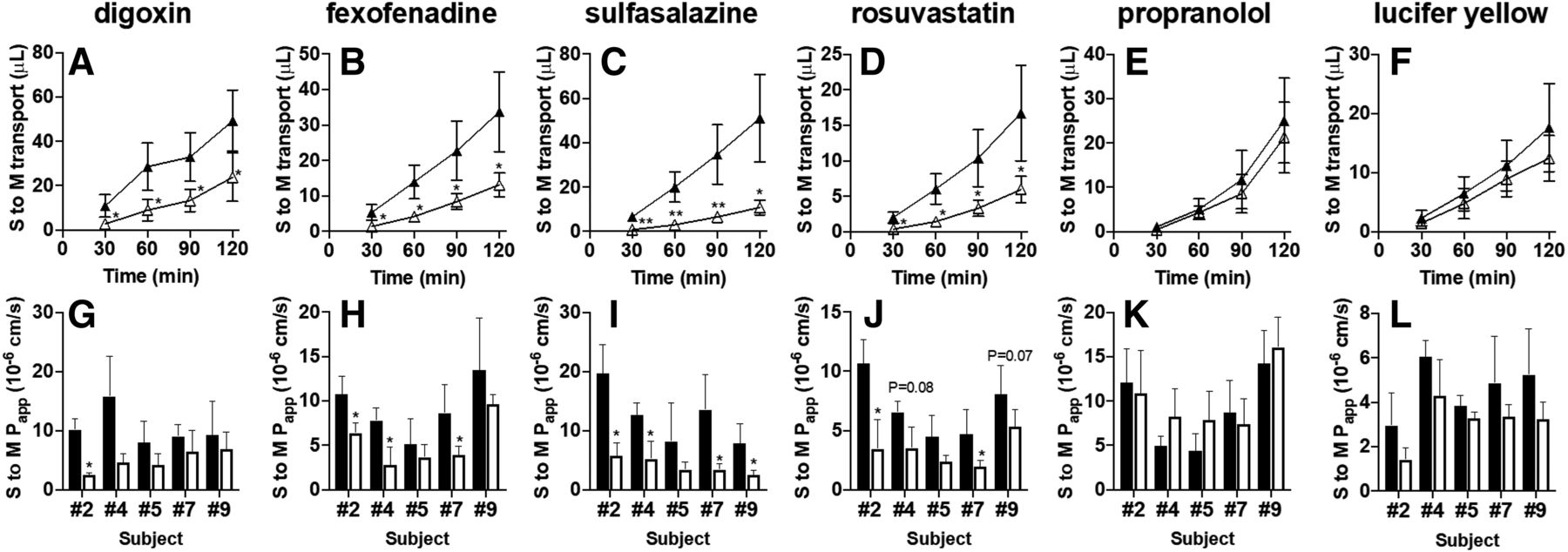
Serosal-to-mucosal (S to M) transport of P-gp and BCRP substrate drugs in the presence or absence of a cocktail of P-gp/BCRP inhibitors. Typical data for serosal-to-mucosal transport of digoxin (A) and fexofenadine (B) (each 5 μM; P-gp substrate drugs), sulfasalazine (C) and rosuvastatin (D) (each 5 μM; BCRP substrate drugs), propranolol (E) (10 μM; passive transcellular marker), and lucifer yellow (F) (100 μM; paracellular marker) across human intestinal sections (number 7) mounted in Ussing chambers in the presence (open triangles) or absence (closed triangles) of a mixture of PSC833 (15 μM; P-gp inhibitor) and Ko143 (15 μM; BCRP inhibitor). Each symbol and error bar represent the mean value and S.D., respectively (n = 4 to 5). The Papp of digoxin (G), fexofenadine (H), sulfasalazine (I), and rosuvastatin (J) (each 5 μM), propranolol (K) (10 μM), and lucifer yellow (L) (100 μM) across human intestinal sections originating from five independent subjects (numbers 2, 4, 5, 7, and 9) mounted in Ussing chambers in the presence (open columns) or absence (closed columns) of a mixture of PSC833 (15 μM) and Ko143 (15 μM). Each bar and error bar represent the mean value and S.D., respectively (n = 3–6). *P < 0.05; **P < 0.01. The actual P value is shown on each bar if it was in the range of 0.05–0.10 (for G–L).
Relationship between the Papp in the Mucosal-to-Serosal Direction of Drugs and Their Human FaFg Values.
To investigate whether the Ussing chamber system incorporating human intestinal sections would be a valuable in vitro tool for the quantitative evaluation of FaFg, the relationship between the Papp in the mucosal-to-serosal direction of drugs in the Ussing chamber system and their human FaFg values obtained from the literature was investigated. Table 2 shows a summary of the in vitro mucosal-to-serosal Papp values and human FaFg values for 19 tested drugs. Initially, nonlinear regression analysis was performed using the Papp values of 11 drugs that mainly pass through the intestinal epithelial cell layer via transcellular and/or paracellular route without the aid of uptake/efflux transporters (metoprolol, propranolol, ketoprofen, antipyrine, enalapril, ranitidine, hydrochlorothiazide, sulpiride, nadolol, atenolol, and pravastatin) to determine the theoretical curve relating the Papp values and human FaFg values of drugs. Then the data points for transporter substrate drugs (cefadroxil, methotrexate, ribavirin, digoxin, fexofenadine, sulfasalazine, and rosuvastatin) and a CYP3A substrate drug (midazolam) were overlaid on that figure (Fig. 5). The data points for transporter and CYP3A substrate drugs mapped close to the fitted curve. In addition, the data points for cefadroxil and methotrexate were shifted to the left away from the fitted curve in the presence of PCFT/PEPT1 inhibitor cocktail. In contrast, no clear shift in the data points for P-gp and BCRP substrate drugs was observed when P-gp/BCRP inhibitor cocktail was coincubated with substrate drugs.

Correlation between the Papp in the mucosal-to-serosal (M to S) direction across human intestinal sections and human FaFg values. Nonlinear regression analysis was performed using the Papp of 11 compounds that mainly pass through the intestinal epithelial cell layer via transcellular and/or paracellular routes without the aid of uptake/efflux transporters (•) in human intestinal sections originating from three independent subjects (numbers 11–13). The solid line represents a fitted curve based on the four-parameter logistic model equation (eq. 11) (R2 = 0.75). (11)
(11)
Discussion
The present study aimed to evaluate the impact of uptake/efflux transporters on the intestinal absorption of substrate drugs in humans using the Ussing chamber system incorporating freshly isolated human intestinal sections.
Limited availability of human intestinal tissue precludes systematic analysis of drug transport in the human intestine. Indeed, few studies have directly investigated drug permeation in human intestinal tissues. Fortunately, we were able to obtain human intestinal sections from 16 patients with pancreatic cancer over the past 9 months (approximately two samples per month). We paid particular attention to the maintenance of the freshness of intestinal tissue sections at the time of experiments.
First, with the great care taken by the physicians and surgeons, the ischemic period was strictly controlled to within a few minutes, which is important because ischemia/reperfusion injury affects P-gp mRNA and protein expression levels in the small intestine (Terada et al., 2014). Hence, we were able to maintain the interbatch variation in the mRNA expression levels of intestinal uptake/efflux transporters in the tissue sections within an 11-fold range (Fig. 1). Second, the tissue sections surprisingly lost the freshness required for transport experiments faster than expected. Even a 4-hour incubation in ice-cold KRB buffer reduced the transport activities of PEPT1 and PCFT (Supplemental Fig. 1, A and B). It should be noted that we cannot exclude the possibility of interbatch differences in the freshness required for the transport studies because we assessed it based on the data from the tissue section from a single subject (number 10), and we have not addressed the mechanism underlying the rapid loss of activity of these transporters. Consequently, we decided to conduct all the permeability experiments on site immediately after excision of the human intestinal sections.
We successfully demonstrated that the freshly isolated human intestinal sections maintained the functions of major intestinal uptake/efflux transporters, as indicated by significant decreases in the mucosal uptake of cefadroxil (PEPT1) and methotrexate (PCFT) (Fig. 2, E and F), the mucosal-to-serosal permeability of ribavirin (CNTs/ENTs) (Fig. 3D), and the serosal-to-mucosal permeability of P-gp and BCRP substrates in the presence of their typical inhibitors (Fig. 4, G–J). In our preliminary studies, 20 mM of Gly-Sar was required to saturate PEPT1 (unpublished data) although the Km of Gly-Sar was 0.33–3.1 mM (median: 0.64 mM) in an overexpression system (University of Washington Metabolism and Transport Drug Interaction Database). Intact intestinal tissues contain some barriers to diffusion, such as the mucus layer on the apical side or the submucosal layer on the basolateral side, which may partially interfere with the access of inhibitors to target transporters (Clarke, 2009). Thus, in the present study the concentration of Gly-Sar was finally set to 20 mM, and those of other transporter inhibitors were also set near the highest concentration achievable from their solubility in the buffer (see Supplemental Table 4).
As shown in Fig. 5, the permeability across the tissue sections mounted on Ussing chambers showed a sigmoidal curve with human FaFg as reported for Caco-2 cells and human intestine (Haslam et al., 2011). The Papp values obtained in the present study ranged from 0.60 to 29 × 10−6 cm/s and were comparable with those observed in previous studies using Ussing chamber systems incorporating human intestine (Haslam et al., 2011; Rozehnal et al., 2012; Sjöberg et al., 2013). Moreover, consistent with previous reports (Haslam et al., 2011; Rozehnal et al., 2012), the Papp values of drugs, such as atenolol, that are mainly transported via the paracellular route were about 10-fold higher than those across Caco-2 cell monolayers, possibly because of a difference in the tightness of tight junctions of cell layers. The Papp values for transporter/CYP3A substrate drugs were relatively close (within 3.6 times) to the theoretical fitted curve obtained from the nonlinear regression analysis using the data for 11 drugs that are thought to be mainly transported without the aid of any transporters. Furthermore, the Papp values for cefadroxil (PEPT1), methotrexate (PCFT), and ribavirin (CNTs/ENTs) in the presence of the corresponding transporter inhibitors underestimated the FaFg of these drugs. This system provided an appropriate estimation of the FaFg value of cefadroxil (93%) derived from the fitted curve and its Papp, which was very close to the actual FaFg value (96%), whereas Caco-2 cells underestimated that of cefadroxil (20%) because of their low expression level of PEPT1 (Balimane et al., 2007; Larregieu and Benet, 2013). These results suggested that the Papp values in this system might be suitable for predicting the human FaFg values of various types of drugs.
In contrast, P-gp and BCRP substrate drugs unexpectedly showed almost no change in their mucosal-to-serosal transport with the addition of P-gp/BCRP inhibitor cocktail at close to the highest concentration achievable from their solubility in the buffer (Fig. 5). These results are not consistent with previous clinical studies showing that the area under the plasma concentration-time curve after oral administration of these compounds could be increased by coadministration of P-gp/BCRP inhibitor drugs or by mutations of these genes in humans. Because these compounds showed a significant reduction in serosal-to-mucosal transport in the presence of P-gp/BCRP inhibitor cocktail (Fig. 4, A–D), it is unlikely that the inhibitor concentration did not reach the effective concentration for transporter inhibition because of nonspecific adsorption or rapid elimination by metabolism. Thus, we recommend evaluating the decreased serosal-to-mucosal transport of compounds in this system rather than the increased mucosal-to-serosal transport for detection of intestinal P-gp/BCRP substrates; otherwise, their impact may be underestimated for reasons that remain unclear. One possible underlying reason might be that the permeability determined in this study is not anatomically identical to that across a monolayer of intestinal epithelial cells (in vivo situation) (Yamashita et al., 1997). Although we surgically removed the muscle layers from the tissue section, the submucosal layer, which consists of an aggregate of connective tissues mainly composed of collagen, remained under the basolateral membrane of the enterocytes. This layer might partly hinder the sensitive detection of a change in transporter functions in mucosal-to-serosal transport. In contrast, the brush-border side of epithelial cells where P-gp and BCRP are expressed is directly exposed to the buffer, and thus, serosal-to-mucosal transport may be directly influenced by these transporter activities. For hydrophilic drugs, such as cefadroxil, methotrexate, and ribavirin, the mucosal-to-serosal transport was sensitive to the transporter activities. For such compounds, permeation across this layer might not be the rate-determining process, but rather permeation across the cell membrane of enterocytes. This is probably because the diffusion or binding to some components of the submucosa layer limited the permeation of lipophilic drugs, such as P-gp and BCRP substrate drugs, across this layer, whereas the same was not true for hydrophilic drugs.
To investigate the interexperimental variability of the paracellular transport and passive transcellular transport among intestinal mucosa preparations, the permeability of lucifer yellow and propranolol were measured in every experiment (See Supplemental Figs. 4 and 5). The mean ± S.D. of the Papp values of lucifer yellow and propranolol were 2.99 ± 1.53 × 10−6 cm/s (n = 133, 16 independent subjects) and 9.35 ± 5.07 × 10−6 cm/s (n = 133, 16 independent subjects), respectively. The coefficients of variation for lucifer yellow and propranolol were 51.1% and 54.2%, respectively. No obvious age-dependent and sex-dependent differences were observed in the Papp values of lucifer yellow. On the other hand, weak relationship between the age of subjects and Papp values of propranolol was observed, and the reasons for this are unclear. This study highlighted the application of human intestinal tissue to the study of drug transport mediated by uptake/efflux transporters. At present, the system has some restrictions to expansion of its use in drug discovery and development, of which the preservation of the tissue section is the most critical. In-depth and systematic investigations (e.g., intracellular ATP levels, localization of transporters), an increase in the number of experiments, and optimization of the storage conditions are required. Moreover, it was difficult to draw a clear conclusion about the correlation between mRNA/protein expression levels of transporters and their transport activities because of the limited number of samples at this moment (see Supplemental Figs. 5 and 6). Further accumulation of experimental data is needed. In addition, further studies are needed to demonstrate the usefulness of this system for understanding the mechanism of intestinal DDIs involving transporter-mediated uptake/efflux transport and/or metabolism (Fenner et al., 2009; Tachibana et al., 2010; Kusuhara et al., 2012; Tomaru et al., 2015). However, because it is possible that the IC50 values in this system are higher than those in overexpression systems, it is also necessary to accumulate cases of clinically relevant DDI to judge its applicability to the quantitative evaluation of DDIs. Moreover, we need to investigate the predictability of its FaFg of substrate drugs of metabolic enzymes, such as CYP3A, UDP-glucuronosyltransferase, and carboxylesterase.
In conclusion, we confirmed the functions of major intestinal uptake/efflux drug transporters in freshly isolated human intestinal sections. The Ussing chamber system incorporating human intestine would be a useful tool for evaluating the impact of intestinal uptake/efflux transporters on the intestinal absorption of various types of drugs in humans.
Acknowledgments
The authors thank Drs. Katja Damme, Markus Keiser, and Veronika Rozehnal at Daiichi Sankyo Europe GmbH, Tissue and Cell Research Center Munich, for their technical assistance with experiments using human intestinal sections. We also thank Drs. Tomoyo Takeuchi and Hiroyuki Nishiyama from the Tsukuba Human Tissue Biobank Center and Drs. Satoshi Suzuki and Katashi Fukao from the Human and Animal Bridging (HAB) Research Organization for their kind assistance with establishing the infrastructure to obtain freshly isolated human intestinal sections.
Authorship Contributions
Participated in research design: Michiba, Maeda, Oda, Kusuhara.
Conducted experiments: Michiba, Maeda.
Contributed new reagents or analytic tools: Kurimori, Enomoto, Shimomura, Oda.
Performed data analysis: Michiba, Maeda, Kusuhara.
Wrote or contributed to the writing of the manuscript: Michiba, Maeda, Oda, Kusuhara.
Footnotes
- Received June 8, 2020.
- Accepted October 2, 2020.
This study was supported by Grant-in-Aid for Scientific Research (B) [Grant 20H03402] (K.Ma. and T.O.) from the Ministry of Education, Culture, Sports, Science, and Technology in Japan.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- ABC
- ATP-binding cassette
- BCRP
- breast cancer resistance protein
- CNT
- concentrative nucleoside transporter
- CYP
- Cytochrome P450
- ENT
- equilibrative nucleoside transporter
- Fa
- fraction of oral drugs that enters the gut wall
- FaFg
- fraction of an oral dose that enters the gut wall and passes into the portal circulation with escaping intestinal metabolism
- Fg
- intestinal availability
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- Gly-Sar
- glycylsarcosine
- KRB
- Krebs-Ringer bicarbonate
- LC-MS/MS
- liquid chromatography–tandem mass spectrometry
- NBMPR
- nitrobenzylthioinosine
- OATP
- organic anion transporting polypeptide
- Papp
- apparent permeability coefficient
- PCFT
- proton-coupled folate transporter
- PCR
- polymerase chain reaction
- PEPT1
- peptide transporter 1
- P-gp
- P-glycoprotein
- qPCR
- quantitative PCR
- SLC
- solute carrier
- Copyright © 2020 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}