


Abstract
Extracellular vesicles (EVs) are small, nonreplicating, lipid-encapsulated particles that contain a myriad of protein and nucleic acid cargo derived from their tissue of origin. The potential role of EV-derived biomarkers to the study of drug metabolism and disposition (DMD) has gained attention in recent years. The key trait that makes EVs an attractive biomarker source is their capacity to provide comparable insights to solid organ biopsy through an appreciably less invasive collection procedure. Blood-derived EVs exist as a heterogenous milieu of biologically distinct particles originating from different sources through different biogenesis pathways. Furthermore, blood (plasma and serum) contains an array of vesicular and nonvesicular contaminants, such as apoptotic bodies, plasma proteins, and lipoproteins that are routinely coisolated with EVs, albeit to a different extent depending on the isolation technique. The following minireview summarizes current studies reporting DMD biomarkers and addresses elements of EV isolation and quantification relevant to the application of EV-derived DMD biomarkers. Evidence based-best practice guidance aligned to Minimum Information for the Study of Extracellular Vesicles and EV-TRACK reporting standards are summarized in the context of DMD studies.
SIGNIFICANCE STATEMENT Extracellular vesicle (EV)-derived protein and nucleic acid cargo represent a potentially game-changing source of novel DMD biomarkers with the capacity to define within- and between-individual variability in drug exposure irrespective of etiology. However, robust translation of EV-derived biomarkers requires the generation of transparent reproducible evidence. This review outlines the critical elements of data generation and reporting relevant to achieving this evidence in a drug metabolism and disposition context.
Introduction
Extracellular vesicles (EVs) are small, nonreplicating, lipid-encapsulated particles that are released by all cells into biologic fluids, including blood, saliva, and urine (Hirsova et al., 2016; Devhare and Ray, 2018; Zhang et al., 2018). EVs mediate cellular communication within tissues and between organs (Sung et al., 2018). This function is important for the maintenance of homeostasis but is also implicated in disease states, including those affecting the liver, such as nonalcoholic fatty liver disease (Zhang et al., 2015; Eguchi and Feldstein, 2018; Newman et al., 2020). EVs contain an array of protein, lipid, and nucleic acid cargo that is derived from the cell of origin. Current evidence suggests that some EV cargo is explicitly packaged through defined pathways, whereas other cargo is passively incorporated as a byproduct of EV biogenesis (Russell et al., 2019; Abels and Breakefield, 2016).
The potential interplay between EVs and drug metabolism and disposition (DMD) was first described in 2010 (Conde-Vancells et al., 2010). The concept was largely based on pioneering data published 2 years earlier (Conde-Vancells et al., 2008) describing the proteomic profile of rat hepatocyte–derived EVs. The analysis identified that proteins involving metabolism accounted for 40% of the most abundant proteins detected. The analysis further specifically identified 10 drug metabolizing enzymes from the cytochrome P450 (P450) superfamily along with six members of the UDP-glucuronosyltransferase (UGT) superfamily. On the basis of these data, it was postulated that liver-derived EVs may play a role in spreading hepatic drug-metabolizing activity through the body and may contribute to extrahepatic drug metabolism. The potential direct or indirect role of circulating EVs containing P450 enzymes in extrahepatic drug metabolism continues to be explored (Gerth et al., 2019). More recently, the presence of multiple drug-metabolizing enzymes and transporters in EVs isolated from human biologic fluids has been reported (Table 1) (Kumar et al., 2017; Rowland et al., 2019; Achour et al., 2021; Rodrigues et al., 2021).
Identity of drug-metabolizing enzymes and transporters positively identified in EV isolated from human biofluids
Building on these observations, the potential role of EV-derived biomarkers in DMD research has gained attention in recent years (Rodrigues and Rowland, 2019), and there are several notable examples of EV-derived biomarkers characterizing variability in P450, UGT, and transporter-mediated pathways (Kumar et al., 2017; Rowland et al., 2019; Achour et al., 2021; Rodrigues et al., 2021). The key trait that makes EVs an attractive biomarker source is their capacity to provide comparable insights to solid organ biopsy through an appreciably less invasive collection procedure (Newman et al., 2021). Consistent with the increased attention to this field, there has been a notable surge in the array of techniques and commercial products marketed to facilitate EV isolation. Blood-derived EVs exist as a heterogenous milieu of biologically distinct particles originating from different sources through different biogenesis pathways (Yáñez-Mó et al., 2015). Furthermore, blood (plasma and serum) contains an array of vesicular and nonvesicular contaminants, such as apoptotic bodies, plasma proteins, and lipoproteins that are routinely coisolated with EVs, albeit to a different extent depending on the isolation technique (Jeppesen et al., 2019). Indeed, although different EV isolation strategies perform the same core function, the efficiency and specificity with which they work vary greatly.
The following minireview addresses elements of EV isolation and quantification relevant to the application of EV-derived DMD biomarkers. Evidence-based best practice guidance aligned to Minimum Information for the Study of Extracellular Vesicles (MISEV) and EV-TRACK reporting standards is summarized in the context of DMD studies.
Nomenclature
The generic term “extracellular vesicle” is used to describe a heterogenous mixture of membrane-encapsulated particles that are secreted into biologic fluids by various organs and tissues of the body (Van Niel et al., 2018; Doyle and Wang, 2019). “Extracellular vesicle” is the generic term endorsed by the International Society for Extracellular Vesicles (ISEV) to describe particles that are naturally released from cells that are encapsulated by a lipid bilayer and cannot replicate (i.e., do not contain a functional nucleus). In many disciplines, the terms “extracellular vesicles” and “exosomes” are largely used interchangeably; however, in reality “exosomes” (50–150 nm in diameter) are just one of three major subpopulations of EVs, the others being microvesicles (MVs) (100–1000 nm in diameter) and apoptotic bodies (50–5000 nm in diameter) (Yáñez-Mó et al., 2015). The different EV populations can be partially distinguished on the basis of their size, refractive index, and the presence of specific surface markers (Fig. 1). Indeed, recently the population of EVs historically considered “exosomes” has been revised to describe classic exosomes, which contain an array of three tetraspanins (CD9, CD63, and CD81), and nonexosomal small EVs (sEVs), which are comparable to exosomes in terms of size and density but lack the tetraspanins surface markers, instead containing annexin A1 and A2 on their surface (Pluchino and Smith, 2019; Jeppesen et al., 2019). Although some EV isolation techniques will preferentially enrich for vesicles of a particular size range or density, in general when isolating EVs from biologic fluids, such as the blood, the final sample will comprise a mixture of these vesicle populations (Xu et al., 2016). Understanding the differences in EV populations, in particular their different biogenesis pathways, can have important implications for EV biomarker discovery and development, as it can inform the optimal sample type and isolation technique to maximize the sensitivity and specificity of the marker.

Extracellular vesicle subtypes defined by particle size and density.
EV Isolation
There are four major global EV isolation strategies upon which most commercial isolation products are based; these are resin precipitation (e.g., ExoQuick), membrane affinity chromatography (e.g., ExoEasy), size exclusion chromatography (SEC; e.g., qEV) and ultracentrifugation (with or without density gradient). The yield, purity, and usability of EVs produced by each of these techniques can be enhanced by inclusion of low-speed centrifugation and ultrafiltration steps prior to or after the core isolation procedure. Currently there is no optimal isolation method, and selection of an isolation technique should be based on compatibility with downstream applications and the scientific question. The most recent MISEV guidelines (Théry et al., 2018) published in 2018 classify isolation methods into one of four theoretical categories based on the yield (recovery) and purity (specificity). These are:
- High recovery, low specificity: methods that recover the greatest yield of EVs; however, also retain a substantial amount of vesicular and nonvesicular contaminants (e.g., lipoprotein complexes).
- Intermediate recovery, intermediate specificity: methods that recover a mixture of EV subpopulations and contaminants, which may include free proteins, ribonucleoproteins, and lipoproteins, depending on the matrix.
- Low recovery, high specificity: methods that recover a subtype (or few subtypes) of EVs with minimal nonvesicular contaminants.
- High recovery and high specificity: to date, not yet been achieved by any published method.
When considering the application of EV-derived biomarkers, a recent review published in the New England Journal of Medicine (Shah et al., 2018) highlighted the importance of defining the tissue of origin for an EV to understanding the relevance of a marker to the disease. Extending this principle to the study of drug metabolism, the capacity to selectively isolate liver-derived EVs from blood is crucial to understanding the relative importance of factors affecting hepatic and extrahepatic pathways involved in drug clearance (e.g., the magnitude and time course of induction of hepatic and intestinal CYP3A4 by rifampicin). Notably, the novel potential to distinguish hepatic and extrahepatic pathways through selective isolation of liver-specific EVs cannot be achieved using any existing endogenous or exogenous phenotype measure except for paired tissue biopsy. Although oral versus intravenous midazolam dosing has used to discriminate hepatic and intestinal cytochrome P450 3A4 activities, it is now generally accepted that clearance of intravenous (liver-centric) midazolam dosing is contaminated with an element of intestinal clearance. To date the capacity to isolate tissue-specific EVs from blood has only been demonstrated by a small number of research groups (Park et al., 2016; Goetzl et al., 2017; Vella et al., 2017; Rodrigues et al., 2021), and no commercial product is readily available. Selective isolation of liver-derived EVs from blood has been demonstrated using an immunoprecipitation-based approach that selectively captures EVs expressing asialoglycoprotein receptor 1 (ASGR1) (Newman et al., 2021; Rodrigues et al., 2021) (Fig. 2). As with cell-based immunoprecipitation approaches, the selective isolation of ASGR1+ liver-derived EVs requires the use of an antibody with an epitope against the extracellular facing domain (amino acids 62–291). Downstream applications requiring release of the EV from the immunocapture bead can be facilitated using a biotin-labeled antibody and streptavidin-coated magnetic bead (e.g., M280 Dynabeads).

Idealized workflow for the isolation of global (multitissue) and tissue-specific (immunocaptured) extracellular vesicles from human blood and possible application to various DMD studies. kdeg, first order degradation rate constant; Kpuu, free drug tissue-to-plasma partition ratio; miR, specific microRNAs; PK, pharmacokinetics; TDI, time-dependent inhibition.
Composition of EV Membranes
Unlike single-layered high- and low-density lipoprotein complexes, which also circulate in blood, the EV membrane is comprised of a lipid bilayer resembling that of the cell membrane. Exosomes are formed through a process of endosomal maturation whereby the early endosome matures into a multivesicular body (MVB), the membrane of which undergoes invagination to form intraluminal vesicles (ILVs). MVBs fuse with the plasma membrane and release their contents into the extracellular environment. At this point, the ILVs are considered exosomes. As a result of this specific biogenesis pathway, exosomes comprise a distinct membrane structure, which compared with cells and MVs exhibits higher abundances of cholesterol, sphingomyelin, gangliosides, and disaturated lipids, whereas their abundance of phosphatidylcholine and diacylglycerol is decreased relative to the originating cell membrane (Laulagnier et al., 2004). The increased abundance of sphingomyelin and disaturated lipids in exosome membranes results in greater membrane rigidity compared with cell membranes (Parolini et al., 2009). The comparatively greater rigidity of EV membranes is hypothesized to contribute to their resistance to degradation and stability as transporters of biomolecules between cells (Ridder et al., 2014). Exosomes also contain a greater abundance of phosphatidylserine (Fitzner et al., 2011), which is believed to facilitate their internalization by recipient cells (Zaborowski et al., 2015). As MVs are formed by direct budding of the cell membrane, the lipid content and morphologic characteristics of MV membranes more closely resemble that of the originating cell. Both exosome and MV membranes contain an array of cell surface proteins, including vesicle markers (e.g., CD9, TSG101) and tissue-specific markers [e.g., ASGR1, glycoprotein A33].
These characteristics of EV membranes can have important implications for substrate, inhibitor, and cofactor uptake when undertaking ex vivo functional assays (for both cytochromes P450 and UGT enzymes) in a manner analogous to the use of human liver microsomes (HLMs). Appropriate mechanisms to remove latency (preincubation at 37°C or addition of alamethicin) should be considered when establishing a functional P450 or UGT assay using EVs as the protein source. Additionally, the different lipid profile and requirement for a higher protein content in EV assays compared with HLMs may result in increased nonspecific binding of substrates and inhibitors to the EV membrane compared with HLM incubations and should be accounted for, particularly for compounds that exhibit moderate to high nonspecific binding in HLM systems, such as propofol (Rowland et al., 2008) and tyrosine kinase inhibitors (Burns et al., 2015). Similarly, within a cell transporter protein, such as the organic anion transporter peptides (OATPs) and ATP-Binding Casette (ABC), transporters are typically exclusively expressed on either the apical or basolateral cell membrane. Accordingly, the biologic fluid containing the greatest abundance of EVs expressing specific transporters will depend on the cellular localization of the transporter and the biogenesis pathway (direct budding on endosomal maturation and invagination) of the vesicle.
EV Cargo
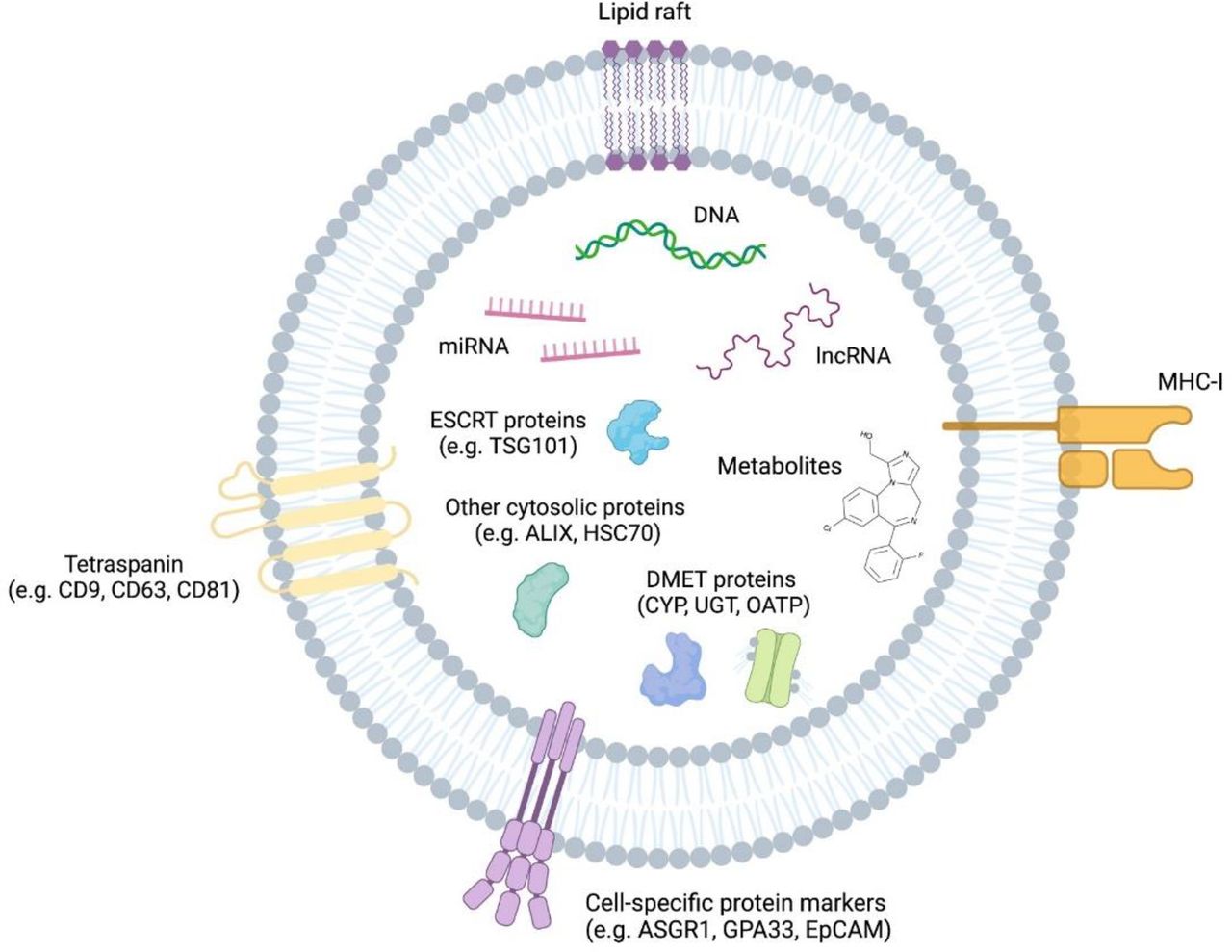
As described previously, EVs contain an array of proteins and nucleic acids that are derived from their cell of origin (Fig. 3).

Overview of extracellular vesicle cargo relevant to general characterization and DMD analysis. GPA33, glycoprotein A33.
EV-Derived Nucleic Acids
The nucleic acid content of EVs and methods to analyze them have been reviewed extensively (Hill et al., 2013; Momen-Heravi et al., 2018; Turchinovich et al., 2019). Extracellular vesicles contain a broad array of nucleic acids ranging from microRNAs and other small noncoding RNAs through to mRNA and DNA. Sequencing of EV-derived RNA in serum and urine demonstrates that small non-coding micro RNA (miRNA) is the dominant species in serum (accounting for 30% to 75% or total RNA), with transfer RNA (tRNA) (15% to 30%) and mRNA (10% to 20%) as other major species. In contrast, ribosomal RNA (rRNA) (35% to 60%) and tRNA (30% to 50%) are the dominant RNA species in urine, with miRNA and mRNA each accounting for <10% of total RNA (Li et al., 2014). The identity of specific nucleic acid cargo contained within EVs is collated in various online databases, including Vesiclepedia (Pathan et al., 2019) and EVmiRNA (Liu et al., 2019). Importantly to the role of EVs as DMD biomarkers, expression of P450 and UGT mRNAs in global plasma-derived EVs has been demonstrated by multiple groups (Rowland et al., 2019; Achour et al., 2021). Although Achour et al. (2021) did not selectively isolate liver-derived EVs from blood, they did demonstrate that by applying a scaling factor (“shedding factor”) to account for the proportion of hepatic EVs in blood, it was possible to achieve a sound correlation describing between-subject variability in EV-derived mRNA and liver-derived protein (n = 29) for a panel of eight P450s (r2 = 0.49–0.76), four UGTs (r2 = 0.36–0.64), and four transporters (r2 = 0.44–0.55). This observation is consistent with an earlier report that EV-derived CYP3A4 mRNA expression robustly described both the between subject variability in CYP3A4 activity in a cohort of healthy individuals before and after 7 days dosing of rifampicin (r2 = 0.79) and the within subject change in CYP3A4 activity prerifampicin and postrifampicin (r2 = 0.88) (Rowland et al., 2019).
EV-Derived Proteins
The proteomic profile of EVs has been extensively studied and reviewed (Simpson et al., 2008; Simpson et al., 2009; Welton et al., 2010; Schey et al., 2015). There are multiple examples wherein the proteomic profile of EVs has been studied to identify markers for diseases, including bladder (Welton et al., 2010), lung (Vykoukal et al., 2017) and ovarian (Zhang et al., 2019) cancers, gestational diabetes (Jayabalan et al., 2019), and coronary artery disease (Boulanger et al., 2017). More recently (Vagner et al., 2019) the proteomic profile of EVs has been used to distinguish EV subtypes. Indeed, MISEV guidance endorses a protein-content–based characterization of EVs based on the presence and absence of positive and negative EV markers across five categories:
- Category 1 (positive markers): Transmembrane or glycosylphosphatidylinositol-anchored proteins associated with the plasma membrane or endosome. Includes category 1a, which are non–tissue-specific (e.g., tetraspanins CD63 and CD81) and 1b, which are tissue-specific (e.g., EPCAM for epithelial EVs).
- Category 2 (positive markers): Cytosolic proteins recovered in all EVs. Includes category 2a, which are proteins with lipid or membrane protein-binding ability (e.g., TSG101), and 2b, which are proteins that are promiscuously incorporated into EVs (e.g., glyceraldehyde-3-phosphate dehydrogenase).
- Category 3 (negative markers): Major components of non-EV coisolated structures. Includes category 3a, which are lipoproteins produced in the liver and abundant in plasma (e.g., albumin), and 3b, which are protein and protein nucleic acid aggregates (e.g., ribosomal proteins).
- Category 4 (negative markers): Transmembrane, lipid-bound, and soluble proteins associated with other intracellular components. This category includes proteins derived from the nucleus (4a), mitochondria (4b), secretory pathways (4c), and other pathways (4d).
- Category 5 (functional markers): Secreted proteins recovered with EVs. Includes category 5a, which are cytokines and growth factors and 5b (e.g., VEGFA), which are adhesion and extracellular matrix proteins (e.g., collagen).
Classification of particles as EVs requires the detection of at least one protein from category 1 (a or b) and one protein from category 2a (optionally 2b). The loss of a relevant protein from category 3a or 3b must be analyzed to demonstrate the EV nature and purity of an EV preparation (typically albumin for plasma/serum analyses). It is necessary to demonstrate the absence of proteins from category 4 when claiming specific analysis of sEVs, and a category 5 protein must be analyzed to document function activities. Traditionally each marker has been characterized independently by Western blotting; however, panel-based liquid chromatography mass spectrometry assays have been reported (Newman et al., 2021) to characterize panels of markers (e.g., CD9, CD81, TSG101, albumin, and calnexin). Reporting the analysis of EV marker proteins is a component of the EV-TRACK scoring matrix (evtrack.org), which is used by major journals in the field to establish the quality of experimental design (Van Deun et al., 2017).
Specifically considering the analysis of EV-derived proteins involved in DMD, multiple studies have reported the presence of drug-metabolizing enzymes from the P450 and UGT superfamilies in human biofluids (Moon et al., 2011; Cho et al., 2017; Kumar et al., 2017; Rahman et al., 2019; Rowland et al., 2019; Rodrigues et al., 2021) and analogous enzyme families in rat hepatocyte–derived EVs (CYP 2A1, 2B3, 2C11, 2D1, 2D3, 2D10, 2D18, and 2D2 and UGT 2B2, 2B3, and 2B5) (Conde-Vancells et al., 2008) (Table 1). The expression of cofactors NADPH–cytochrome P450 reductase and cytochrome b5 and ex vivo functional activity of these enzymes has been confirmed in multiple studies (Conde-Vancells et al., 2008; Kumar et al., 2017; Rowland et al., 2019; Rodrigues et al., 2021), as has the presence of renal and hepatic drug transporters (Table 1). Specifically in this regard, functional activity has been demonstrated for CYP2E1 (Kumar et al., 2017), CYP3A4 (Kumar et al., 2017; Rowland et al., 2019; Rodrigues et al., 2021), and nonspecifically for UGTs (Rowland et al., 2019). To date there is no report of functional DMD transporter activity in EVs. It is worth noting why there are similarities between EVs and HLMs in terms of size and general structure: Current evidence suggests that EVs are structurally and morphologically different to HLM and as such cannot be directly substituted into existing HLM assay systems. By way of example, current evidence suggests that substrate and inhibitor access to the EV cytoplasm is required and may be impeded by the robust EV membrane. Likewise, differences in the lipid profile of EV membranes are likely to result in different small-molecule substrate and inhibitor nonspecific binding profiles during ex vivo incubations.
Best Practice
Isolation of EVs and quantification of EV-derived cargo can be confounded by an array of preanalytical and analytical factors. As such, in 2014 ISEV released the first minimal reporting guidance regarding EV studies (Lötvall et al., 2014), and this guidance was updated in 2018 (Théry et al., 2018) to provide more nuanced insights and clarification. The 2018 MISEV guidance defines the current best practices in terms of EV sample collection, storage, isolation, characterization, and reporting. Elements of this guidance that are of greatest importance to the analysis and reporting of plasma/serum-derived EV DMD biomarkers are summarized in this section.
Preanalytical Factors
A myriad of preanalytical factors can affect the recovery of EVs from collected samples. As such, careful attention should be made to maintaining consistent practices and detailed records of the collection technique and characteristics of the source. The MISEV 2018 guidelines (Théry et al., 2018) recognize the complexity presented by differing biofluid compositions and thus provide a generalizable list of factors with additional considerations applicable to blood derivatives (plasma/serum). General characteristics, including age, sex, body mass index, race/ethnicity, smoking status, and current or previous pregnancy, must be recorded for all donors. The time of day at which samples are collected should be kept constant to control for circadian variations (Witwer et al., 2013; Coumans et al., 2017; Théry et al., 2018). By way of example, a recent report from our group showed that although no change in total abundance and size of EVs occurred from morning to afternoon, levels of generic EV marker CD81 were decreased, and the hepatocyte specific marker ASGR1 was increased (Newman et al., 2021). The potential for variability in molecular cargo should thus be accounted for in sampling protocols, particularly in the context of liver-derived markers, such as DMEs. Prandial state has also been suggested to impact circulating EV (Witwer et al., 2013). Although this work demonstrated no effect of fasting on EV abundance, particle size, and generic or liver-specific markers (Newman et al., 2021), others previously reported significant increases in fed compared with fasted samples (Mørk et al., 2016). Differences may be attributed to EV isolation methods selected in these studies, as increased postprandial lipoprotein levels can interfere with vesicle counting in less-pure EV isolates. Nonetheless, consistently collecting samples at a defined time after last meal and recording food intake history may be valuable for effective comparison of results. Other donor characteristics of importance to EV recovery that should be documented include level of physical activity and time since last intense exercise, recent or present illness, and current medications (Witwer et al., 2013).
Sample Collection and Storage
Beyond donor-related factors, several technical considerations should be made with respect to the blood draw and sample handling. The type of collection tube (serum or plasma) and choice of anticoagulant (e.g., EDTA, heparin, citrate) depend on intended downstream analyses; heparin, for example, is known to interfere with polymerase chain reaction. EVs may be more abundant in serum compared with plasma because of platelet vesiculation occurring during clot formation after collection of blood (Witwer et al., 2013). Studies also demonstrate that other factors relating to handling, such as centrifugation, storage conditions, and transportation, have varying effects on samples depending on the type of collection tube (Lacroix et al., 2012; Bæk et al., 2016). Further studies are required to determine the best choice of anticoagulant for EV studies, so, when practical, it may be useful to collect samples in different types of tubes.
Platelet activation and consequent EV release due to physical forces during the blood-draw procedure, including contact with tubing and pressure from the tourniquet, should be minimized (Witwer et al., 2013). This can be achieved by use of a large diameter (21-gauge) needle and removal of the tourniquet shortly after first blood accumulation (Coumans et al., 2017). The sampling site (e.g., cubital fossa) and blood collection apparatus should be kept standard, and blood should be drawn smoothly, with the first 2–3 ml discarded (Mullier et al., 2013). Tubes should be filled entirely to ensure consistent ratio of blood to anticoagulant and gently mixed by inversion (Coumans et al., 2017). Required time between collection and centrifugation depends on tube type; EDTA and citrate tubes should be processed as soon as possible, whereas serum tubes must be kept at room temperature for up to 30 minutes to allow for clotting (Lacroix et al., 2012; Bæk et al., 2016). In all cases, tubes should remain upright, agitation should be minimized, and any deviations from this best practice should be recorded and kept constant across samples. In all cases, horizontal transport of samples and strong agitation should be avoided. Agitation, for instance arising from transportation, can alter EV abundance and may disproportionately affect EDTA tubes compared with heparin and serum (Lacroix et al., 2012; Bæk et al., 2016). Centrifugation on-site is recommended, but if this is not possible, tubes should be immobilized in a vertical position (Mullier et al., 2013; Bæk et al., 2016). Most efficient platelet removal and EV recovery is achieved by two successive centrifugation steps each at 2500 × g for 15 minutes and is recommended to be performed at room temperature (Lacroix et al., 2012; Witwer et al., 2013; Coumans et al., 2017). These centrifugation conditions as well as rotor type and deceleration should remain constant. Separated plasma/serum should be aspirated carefully, ensuring the pellet is undisturbed and leaving 0.5–1cm in the tube to prevent contamination with buffy coat (Coumans et al., 2017). Analysis of hemolyzed samples should preferably be avoided, particularly for RNA analysis; however, if these must be included, degree of hemolysis should be recorded and caution should be taken in the interpretation of results (Witwer et al., 2013; Coumans et al., 2017). It is acknowledged that for archived samples, several of the details discussed here may not be available. This limitation should be reported and considered when comparing results of different studies.
In our hands, intact exosomes have been reproducibly isolated from plasma stored at −80°C for up to 3 years. This is consistent with literature showing that exosome integrity is unaffected when stored below −20°C over this period, with minor (<10%) degradation over 5 years (Ge et al., 2014). The robustness of EV-derived cargo facilitates the capacity for retrospective analyses of archived clinical trial samples, although there are caveats regarding sample storage that can impact outcomes. The most important factor in terms of the impact of storage on EV marker stability is freeze-thaw cycles; multiple studies (Yuana et al., 2015; Bæk et al., 2016; Cheng et al., 2019) have demonstrated that three or more freeze-thaw cycles significantly reduce EV membrane integrity and impair analysis of various EV cargo, including miRNAs and protein. Similarly, EVs are comparatively unstable at room temperature compared with cold storage. Current evidence (Lacroix et al., 2012) suggests that along with strong agitation and horizontal transport, delaying EV isolation and cold storage for 4 hours has the greatest impact on particle abundance measured by flow cytometry and thrombin (contaminant) production in the sample. The impact of delaying cold storage by 4 hours on EV integrity was double that of delaying cold storage by 1 hour.
Sample Isolation
MISEV 2018 position statement provides step-by-step guidelines for cell culture conditioned media preparation, including culture and harvesting conditions, culture medium composition, and preparation information; however, with more than 30 types of biofluids available in mammals, detailed review is not provided on preanalytical variables or isolation techniques related to all biofluids (Théry et al., 2018).
For EV isolation from predominantly used biofluids such as serum and plasma, several ISEV position papers have outlined isolation and reporting requirements (Witwer et al., 2013; Mateescu et al., 2017). Since then, there was a dramatic surge in extracellular vesicle-based studies, indicating their significance in the scientific community (Srivastava et al., 2020). With increased interest in EV research, the number of approaches of EV isolation increased substantially, resulting in high numbers of methodology-orientated studies comparing and contrasting different methods of isolation and their suitability for a particular sample origin.
Density gradient ultracentrifugation (DG-UC) is considered the gold standard method for isolation of highest purity EVs, providing a valuable platform for quality EV separation in research (Zhou et al., 2020). It is also the only method of EV isolation accepted as gold standard by EV-TRACK self-reporting platform (Van Deun et al., 2017); however, because of lengthy, labor-intensive protocol and low-throughput approach, it is not suitable for diagnostic use in a clinical setting (Useckaite et al., 2020). Ultracentrifugation (UC) of samples at high speed is the most commonly used method of EV isolation based on findings from worldwide survey of ISEV members (Gardiner et al., 2016) despite the need of expensive equipment; low throughput, low recovery of EVs; significant contamination with soluble proteins; and aggregation of vesicles during pelleting (Cvjetkovic et al., 2014; Linares et al., 2015; Sódar et al., 2016). To improve the purity of EV isolates, UC is often coupled with SEC approach (Morán and Cubero 2018; Wei et al., 2020).
Commercially available EV isolation approaches are not outlined in MISEV guidelines; however, they are predominantly used to reduce hands-on time and to increase throughput (Table 2). Commercial approaches, such as SEC, are used for isolation of EVs from a large variety of sample matrices. Size exclusion chromatography columns, such as qEV from Izon Science, offer higher throughput than ultracentrifugation, and even though some lipoprotein contamination is present in SEC-isolated EV samples from human plasma or serum (Takov et al., 2018; Liangsupree et al., 2021), it is suitable for drug metabolism studies, especially when SEC is coupled with immunocapture of liver-specific EVs (Rodrigues et al., 2021).
Comparison on major EV isolation strategies
Filter, affinity, and polymer-based techniques have become more popular in the recent years; however, because of high range of availability, they are not outlined in MISEV guidelines. Different commercial isolation approaches promise pure EV isolates, but published data show that an increase in throughput and a reduction in isolation time often result in EV loss, morphologic changes, protein contamination, and aggregation (Rood et al., 2010; Stranska et al., 2018; Lucchetti et al., 2019; Tian et al., 2019; Useckaite et al., 2020; Liangsupree et al., 2021).
Reporting
ISEV endorses the EV-TRACK, a self-reporting knowledge base facilitating comprehensive and transparent reporting (Van Deun et al., 2017). Experimental parameters related to EV characterization include analysis of EV-enriched and non-EV proteins, specifics of antibodies used in the study, and methodology used for EV lysate preparation. For particle analysis, qualitative and quantitative parameters reported are electron microscopy, nanoparticle tracking analysis, and high-resolution flow cytometry. For quantitative reporting, particle concentration is expected (Welsh et al., 2020). The EV-TRACK platform is self-reporting and voluntary; however, it only accepts the DG-UC isolation method as the gold-standard approach. Reported studies that are not using DG-UC as their method of isolation are not able to report on EV density and do not achieve the maximum EV-METRIC score of 100%. High EV-METRIC score implies well annotated published data, but it is not a standalone measure relating to the quality of a study (Van Deun et al., 2017).
In 2020, in addition to MISEV guidelines, Minimal Information about a Flow Cytometry experiment standard in an EV Flow Cytometry specific reporting framework (MIFlowCyt-EV) was published by a working group of researchers from ISEV, International Association for the Advancement in Science, and International Society on Thrombosis and Haemostasis (Van Deun et al., 2017). EV Flow Cytometry working group platform facilitates sharing EV flow cytometry results without prescribing specific protocols and is constantly evolving. The MIFlowCyt-EV framework is based on MISEV guidelines for reporting of preanalytical factors (Théry et al., 2018) and MIFlowCyt-EV guidelines for reporting of flow cytometry experimental design and variables (Lee et al., 2008). The MIFlowCyt-EV reporting framework, which is available from http://www.evflowcytometry.org/, addresses seven domains:
Preanalytical variables and experimental design
Sample preparation
Assay controls
Instrument calibration and data acquisition
EV characterization
Flow cytometry data reporting
Flow cytometry data sharing
This framework provides a set of consistent criteria for reporting experiments and should be used for all work involving the use of flow cytometry for the analysis of single EVs. Examples of the rationale, components to report, and objectives for each domain of the framework have been described in detail (Welsh et al., 2020).
Major Challenges
Matching Isolation Strategy to Downstream Application.
As discussed throughout this review, there are an array of methods that can be applied to isolate EVs from biologic matrices (e.g., SEC, membrane affinity chromatography, resin precipitation) and an array of analytical platforms that can be used to quantify EV-derived cargo (e.g., polymerase chain reaction, liquid chromatography mass spectrometry). The major challenge in undertaking EV biomarker research for DMD is matching an appropriate isolation strategy to the downstream application. In making this determination it is important to consider the relative strengths and weakness of each isolation approach and their suitability to major analytical platforms (Table 2). By way of example, resin precipitation is a high-recovery, low-specificity method for EV isolation that applies polymer [typically polyethylene glycol (PEG)] precipitation to isolate EVs. Commercial kits based on this technology, such as ExoQuick, have been applied to DMD research (Achour et al., 2021) and are an effective mechanism to isolate a high yield of EVs but also coisolate any solid particles within the sample matrix, such as lipoproteins and protein complexes. Resin precipitation also introduces a substantial fraction of PEG (typically 8%) to the sample matrix. The high EV yield achieved by resin precipitation can make this a viable approach to detect low abundance nucleic acid cargo. However, caution must be applied when attempting to quantify EV-derived mRNA and miRNA using this approach, as the majority of these nucleic acid species isolated by resin precipitation come from coisolated non-EV sources in lipoprotein complexes bound to argonaut protein (Meister, 2013; Nik Mohamed Kamal and Shahidan, 2020). The substantial contamination can necessitate complex normalization strategies that may be avoided by using an alternate high-specificity isolation strategy (Ding et al., 2018; Macías et al., 2019). Similarly, the substantial matrix protein (typically albumin when isolating from blood) and PEG contamination preclude the use of resin precipitation as a viable isolation strategy for downstream proteomic applications (Helwa et al., 2017).
In contrast, DG-UC is recommended by ISEV as the definitive approach to give the highest certainty that a marker is truly EV-associated. However, DG-UC is the most labor-intensive isolation strategy and among the lowest-throughput. As such, although this approach forms a key element of EV-TRACK reporting and should be performed on a subset of samples when reporting EV markers, it is not suited to routine sample analysis or clinical applications. Additionally, as a low-recovery approach, DG-UC is unlikely to be well suited to the detection of low-abundance cargo.
Isolating EV from the Appropriate Sample Type.
The second major challenge to undertaking EV biomarker research for DMD is ensuring that an appropriate sample type is used. To make this determination, it is important to understand elements of EV biogenesis and the cellular/subcellular localization of the marker of interest.
In this regard, exosomes are formed through a process of endosomal maturation whereby the early endosome matures into an MVB, and this membrane undergoes invagination to form ILVs. MVBs are transported to and fuse with the plasma membrane, subsequently releasing their luminal contents into the extracellular environment. At this point, the ILVs are considered exosomes (Hessvik and Llorente, 2018; Morán and Cubero, 2018). In contrast, MVs are not of endosomal origin; rather their release occurs by outward budding of the cell membrane. At the site of MV shedding, lipid domains, such as phosphatidylserine, are redistributed in the plasma membrane; the local cytoskeletal network is reorganized; and the actin-myosin machinery contracts to alter membrane curvature and promote budding and detachment of vesicles (Hirsova et al., 2016; Greening and Simpson, 2018; Newman et al., 2020). As a result of these biogenesis pathways, MVs and exosomes may be differentially released into biofluids. By way of example, considering transporter proteins within the nephron, as organic anion and cation transporters are expressed on the basolateral (blood-facing) membrane of renal proximal tubule epithelial cells, when incorporated into MVs they are likely to be more abundant in the blood than the urine. In contrast, renal apical transporters, such as P-glycoprotein (P-gp), are theoretically more likely to be incorporated in MVs that are released into the urine. As such, when undertaking EV biomarker research to study the abundance and function of renal P-gp, it is likely that more robust results may be achieved by sampling from urine than blood. Indeed, in the case of MV-derived P-gp, it is likely that the major sources of this marker in blood-derived EVs will originate from the blood brain barrier. Similar considerations should be applied when considering the analysis of hepatic proteins associated with blood- and biliary-facing membranes.
When determining the appropriate sample type (blood, urine, cerebrospinal fluid) for EV biomarker analysis, it is also important to consider the ongoing logistical feasibility of the sampling strategy (e.g., blood vs. cerebrospinal fluid sampling) and the potential sources of the EV marker in the selected sample type. By way of example, cytochrome P450 3A4 is abundantly expressed in both the liver and intestine, and expression can be differentially perturbed by factors, such as drug interactions (Kapetas et al., 2019). As global circulating EVs in blood are derived from both the liver and intestine, in the absence of selectively isolating EVs from one source, interpretation of the resulting output is limited to a “net effect” that will be determined by the impact of the perturbation on each organ, the relative expression of the marker in each organ, and the relative abundance of circulating EVs derived from each organ. We recently reported for the first time a method for the selective isolation of EVs from the liver (Rodrigues et al., 2021). As the liver is the major source of most drug-metabolizing enzymes and transporters, application of this approach can minimally discriminate the hepatic and net extrahepatic sources of the marker.
Extrapolation of EV Data.
The third major challenge to the clinical translation of EV-derived DMD biomarkers is the absolute extrapolation of EV parameters to whole organ values. This challenge is of particular importance to the application of EV biomarkers to assessment of between-subject variability, whereby understanding absolute abundance of the target protein is crucial. By way of example, although studies have reported relatively consistent abundances for EV-derived CYP3A4 in healthy males in the range 800–1400 fmol/ml (Rowland et al., 2019; Rodrigues et al., 2021), it is unclear how this value relates to the corresponding whole-organ CYP3A4 abundance. The only way to definitively correlate EV-derived marker abundance to whole-organ abundance is through the analysis of matched EV and solid-organ biopsy samples. This is particularly challenging in the context of healthy donors. Notably in this regard, Achour et al. (2021) did correlate EV-derived mRNA expression to tissue protein abundances for a panel of DMD proteins in a cohort of cancer subjects, although this analysis was limited to evaluation of EV-derived mRNA (not protein). The absolute extrapolation of EV-derived data is less challenging in the context of assessment of within-subject variability, whereby the marker ration before and after the intervention has been demonstrated to robustly correlate with observed in vivo trait measures (Rowland et al., 2019; Rodrigues et al., 2021).
Conclusion and Envisioned Potential.
There is an emerging body of evidence to support the use of EV-derived proteins and mRNAs as novel and unique biomarkers of DMD capacity. To date, the strongest evidence supporting the role of EVs in DMD analyses has been in the form of defining within-subject changes in DMD protein function and abundance resulting from factors, such as induction-based metabolic drug interactions and genotype (e.g., CYP2D6 and CYP3A5) (Rowland et al., 2019; Rodrigues et al., 2021). Indeed, limited existing evidence suggests that by comparison the use of EV-derived markers to characterize between-subject differences in DMD protein function and abundance may be more challenging (Achour et al., 2021; Rodrigues et al., 2021). The key difference between the application of EVs to define these sources of variability in DMD pathways is that evaluation of between-subject differences in EV cargo may require some degree of normalization to account for differences in EV abundance. Although it is likely that the need for complex normalization, such as the “shedding factor” proposed by Achour et al. (2021), may be negated by isolating EVs through higher-specificity global isolation or (ideally) tissue-specific isolation, some normalization (e.g., particle count measured by nanoparticle tracking analysis) will likely still be required to account for between-subject differences in EV abundance. By contrast, provided that the best practice is applied to mitigate preanalytical factors, it is likely that assessment of within-subject changes in DMD protein function and abundance will be less susceptible to changes in EV abundance within an individual.
As described in our recent review (Rodrigues and Rowland, 2019), while still in their infancy, EV-derived DMD markers have the potential to redefine assessment of variability in drug exposure. Potential applications, such as delineation of changes in hepatic and nonhepatic protein function, cannot be robustly addressed by any existing phenotyping measure. As such, it is possible that application of EV-derived biomarkers during early clinical drug development (e.g., multiple ascending dose studies) may provide complementary evidence to inform design of phase one drug interaction studies or ultimately may replace these studies. Critical to ensuring the robustness of evidence regarding EV-derived DMD markers is consideration of the myriad of preanalytical and isolation factors that may impact study outcomes. For this reason, it is important that as new data are generated and published, efforts are made both by researchers and editorial staff to ensure adherence with the reporting and study design requirements described in MISEV 2018 and EV-TRACK guidance.
Authorship Contributions
Participated in research design: Useckaite, Rodrigues, Hopkins, Newman, Johnson, Sorich, Rowland.
Performed data analysis: Useckaite, Newman, Rowland.
Wrote or contributed to writing of the manuscript: Useckaite, Rodrigues, Hopkins, Newman, Johnson, Sorich, Rowland.
Footnotes
- Received February 10, 2021.
- Accepted July 28, 2021.
Zivile Useckaite is supported by a postdoctoral fellowship from Pfizer Inc. Andrew Rowland and Michael J. Sorich are supported by Beat Cancer Fellowships from Cancer Council South Australia.
Andrew Rowland and Michael J. Sorich are recipients of unrelated grant funding from Pfizer Inc. A. David Rodrigues and Jillian Johnson are employees of Pfizer Inc.
Abbreviations
- ASGR1
- asialoglycoprotein 1
- DG-UC
- density gradient ultracentrifugation
- DMD
- drug metabolism and disposition
- EV
- extracellular vesicle
- HLM
- human liver microsome
- ILV
- intraluminal vesicle
- ISEV
- International Society for Extracellular Vesicles
- MIFlowCyt-EV
- Minimal Information about a Flow Cytometry experiment standard in an EV Flow Cytometry specific reporting framework
- MISEV
- Minimum Information for the Study of Extracellular Vesicles
- MV
- microvesicle
- MVB
- multivesicular body
- OATP
- organic anion transporter peptide
- P450
- cytochrome P450
- PEG
- polyethylene glycol
- P-gp
- P-glycoprotein
- SEC
- size exclusion chromatography
- sEV
- small extracellular vesicle
- TSG101
- tumor susceptibility gene 101
- UC
- ultracentrifugation
- UGT
- UDP-glucuronosyltransferase
- Copyright © 2021 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}