


Abstract
Linerixibat, an oral small-molecule ileal bile acid transporter inhibitor under development for cholestatic pruritus in primary biliary cholangitis, was designed for minimal absorption from the intestine (site of pharmacological action). This study characterized the pharmacokinetics, absorption, metabolism, and excretion of [14C]-linerixibat in humans after an intravenous microtracer concomitant with unlabeled oral tablets and [14C]-linerixibat oral solution. Linerixibat exhibited absorption-limited flip-flop kinetics: longer oral versus intravenous half-life (6–7 hours vs. 0.8 hours). The short intravenous half-life was consistent with high systemic clearance (61.9 l/h) and low volume of distribution (16.3 l). In vitro studies predicted rapid hepatic clearance via cytochrome P450 3A4 metabolism, which predicted human hepatic clearance within 1.5-fold. However, linerixibat was minimally metabolized in humans after intravenous administration: ∼80% elimination via biliary/fecal excretion (>90%–97% as unchanged parent) and ∼20% renal elimination by glomerular filtration (>97% as unchanged parent). Absolute oral bioavailability of linerixibat was exceedingly low (0.05%), primarily because of a very low fraction absorbed (0.167%; fraction escaping first-pass gut metabolism (fg) ∼100%), with high hepatic extraction ratio (77.0%) acting as a secondary barrier to systemic exposure. Oral linerixibat was almost entirely excreted (>99% recovered radioactivity) in feces as unchanged and unabsorbed linerixibat. Consistent with the low oral fraction absorbed and ∼20% renal recovery of intravenous [14C]-linerixibat, urinary elimination of orally administered radioactivity was negligible (<0.04% of dose). Linerixibat unequivocally exhibited minimal gastrointestinal absorption and oral systemic exposure. Linerixibat represents a unique example of high CYP3A4 clearance in vitro but nearly complete excretion as unchanged parent drug via the biliary/fecal route.
Significance Statement This study conclusively established minimal absorption and systemic exposure to orally administered linerixibat in humans. The small amount of linerixibat absorbed was eliminated efficiently as unchanged parent drug via the biliary/fecal route. The hepatic clearance mechanism was mispredicted to be mediated via cytochrome P450 3A4 metabolism in vitro rather than biliary excretion of unchanged linerixibat in vivo.
Introduction
Primary biliary cholangitis (PBC) is a rare, chronic autoimmune liver disease caused by immune-mediated destruction of the intrahepatic bile ducts. This results in impaired bile acid flow and retention in the liver, leading to hepatic scarring, fibrosis, and ultimately cirrhosis and liver failure (Boonstra et al., 2012; Gao et al., 2015). Cholestatic pruritus is a major symptom of PBC that significantly affects a patient’s quality of life (Gotthardt et al., 2014; Hegade et al., 2015a; Jin and Khan, 2016; Hegade et al., 2019a; Lindor et al., 2019). The currently recommended treatments for pruritus can lead to symptomatic improvement in a subset of patients; however, a sizable population remains refractory and requires the utilization of invasive experimental treatments, such as nasobiliary drainage or even liver transplantation (Trivella and Levy, 2021). As such, an effective antipruritic drug therapy is an unmet clinical need in PBC (Trivedi et al., 2017).
Several putative pruritogens have been proposed in the treatment of PBC, including circulating bile acids (Hegade et al., 2015b), suggesting that the ileal bile acid transporter (IBAT), a solute carrier family transporter that mediates bile acid uptake from the gut lumen into enterocytes, may be a potential target for pruritus therapy (Hegade et al., 2015b; Al-Dury and Marschall, 2018). Inhibition of IBAT decreases bile acid reabsorption from the gastrointestinal (GI) tract, leading to decreased bile acid levels in the circulation and increased fecal bile acid excretion (Hegade et al., 2017, 2019b). Alongside pruritus, IBAT inhibition could benefit diabetic dyslipidemia by decreasing bile acid reabsorption and forcing the liver to increase de novo bile acid synthesis from cholesterol (Beysen et al., 2012). More recently, it has also shown utility in the treatment of chronic idiopathic constipation (Khanna and Camilleri, 2021) by increasing bile acid concentrations entering the colon, thereby stimulating colonic secretion and motility (Bampton et al., 2002).
Linerixibat (GSK2330672) is an oral small-molecule IBAT inhibitor, which has been shown to be effective in reducing PBC-associated pruritus and serum bile acids in two phase 2 studies (Hegade et al., 2017; Levy et al., 2020). Linerixibat was intentionally designed for minimal absorption from the GI tract (Wu et al., 2013). The rationale for this approach was to restrict drug exposure to the pharmacologic site of action while minimizing systemic exposure that may cause adverse events and drug-drug interactions (DDIs).
Preclinical studies demonstrated oral bioavailability of <1% in mice, rats, and dogs (Wu et al., 2013). However, in rodents this was caused by the low fraction absorbed from the GI tract, whereas in dogs the fraction absorbed was higher, which was consistent with relatively leakier canine intestines (Lennernäs, 1997), and high hepatic extraction served as a secondary barrier to systemic exposure of linerixibat. Although clinical trials have demonstrated minimal systemic linerixibat exposure in humans (Hegade et al., 2017; Ino et al., 2019), oral pharmacokinetics (PK) alone cannot prove that minimal GI absorption is the underlying cause. Understanding the basis of minimal oral systemic exposure is key in the context of linerixibat’s drug development. Studying both intravenous and oral linerixibat PK in humans enables quantification of the contributions of the fraction absorbed from the GI tract and first-pass hepatic extraction to the minimal systemic exposure. Thus, the objectives of this study were to assess the PK, absorption, metabolism, distribution, and excretion of [14C]-linerixibat in humans after both intravenous and oral administration.
Materials and Methods
Materials.
The [14C]-linerixibat (GSK2330672D) intravenous and oral solutions were obtained from Hammersmith Medicines Research Ltd (London, UK). Linerixibat (GSK2330672B) oral tablets were obtained from Wuxi Apptec Co., Ltd (Shanghai, China). Reference standards were provided by GlaxoSmithKline: GSK2330672B (free acid/free base, batch C13090301-QF17602, chemical purity 99.5%), [13C]-radiolabeled GSK2330672C (free acid/free base, batch R18284/139/2, radiochemical purity 97.8%), and [14C]-radiolabeled GSK2330672D (free acid/free base, batch DN27404-056A1, radiochemical purity 97.6%). All other solvents and reagents were of laboratory grade and purchased from commercial suppliers.
Study Design.
This was a single-group, single-center, open-label, nonrandomized, two-period, single-sequence [14C]-linerixibat mass balance study (Clinical trials.gov identifier NCT03992014) conducted at Hammersmith Medicines Research Centre (London, UK) between July 8, 2019 and August 26, 2019. The study adhered to the Declaration of Helsinki and was approved by the Health and Social Care Research Ethics Committee (National Health Service, UK). Written informed consent was obtained from participants prior to any study-specific procedures. All analyses were performed according to a predefined protocol.
The study included a screening visit and two treatment periods separated by approximately 7 days (≥13 days for oral doses) (Fig. 1). During the treatment periods, participants resided in the unit from the morning of day –1 (the day before dosing) until all procedures were completed at 168 hours postdose (day 8). Participants were followed up 1–2 weeks after the last assessment in treatment period 2.

Study schematic.
All participants received a [14C]-linerixibat intravenous microtracer (100 µg; 3-hour infusion) concomitant with 90-mg linerixibat oral tablets during treatment period 1 and a 90-mg [14C]-linerixibat oral solution during treatment period 2; all participants received their doses on site. The 90-mg oral dose of linerixibat was determined from a previous phase 2a study (Hegade et al., 2017). The [14C]-linerixibat intravenous microtracer dosing regimen was extrapolated by allometry from preclinical in vivo data (see Supplemental Data) and in vitro–to–in vivo extrapolation (IVIVE) using data from in vitro analyses of human liver microsomes (presented in this paper). The relatively long (3-hour) intravenous infusion of the relatively high microtracer dose (100 μg) was selected to ensure adequate characterization of the intravenous concentration-time curve considering projected rapid human elimination of intravenous dose (projected half-life 0.8 hours, see Supplemental Data) as well as to reduce the maximal concentration expected at the end of intravenous infusion to ensure it does not exceed established safety at the low concentrations achieved after oral linerixibat administration in previous human studies (Hegade et al., 2017).
For both treatment periods, participants underwent an overnight fast of at least 8 hours. A blood sample was collected at 0 hours, which was immediately followed by either a 90-mg oral dose of linerixibat administered as two 45-mg tablets along with a 3-hour intravenous infusion of 100 μg of [14C]-linerixibat (treatment period 1) or a 90-mg [14C]-linerixibat dose administered as an oral solution (treatment period 2). Participants continued fasting for 2 hours after dosing. The last blood sample was collected on day 8 of treatment period 1 and on day 7 (completing on day 8) of treatment period 2. Urine and fecal samples were collected every 24 hours for a total of 168 hours after oral dosing for both treatment periods. Duodenal bile was collected on day 1 of treatment period 1. For both treatment periods, the last vital sign assessment, electrocardiogram, and brief physical examination were completed on day 8.
Each participant received 9.25 kBq (250 nCi) in treatment period 1 and approximately 4.96 MBq (134.1 μCi) in treatment period 2. The total amount of radioactivity administered to each participant in the study was 4.97 MBq (134.3 μCi). It was estimated that the combined total effective dose for the two treatment periods would be <1 millisievert. On this basis, the maximum administered activity complied with recommendation of 1 millisievert maximum for category IIa projects by International Commission on Radiologic Protection (ICRP, 1992).
Study Population.
Healthy male participants between 30 and 55 years of age with body weight ≥50 kg, body mass index 19–31 kg/m2, and a history of regular bowel movements were eligible for the study. The additional eligibility criteria were nonsmokers or smokers who had not regularly smoked for 6 months prior to screening, no history of drug abuse, and those not exposed to significant radiation in the 3 years prior to the study. Standard participant criteria for a human radiolabeled disposition study were used (see Supplemental Methods for full eligibility criteria).
Sample Collection and Processing.
Blood samples were collected at preselected time points up to day 7 after dosing and transferred into dipotassium EDTA tubes. The maximum amount of blood collected from each participant over the duration of the study did not exceed 600 ml. Plasma was separated by centrifugation. Bile samples were collected via an Entero-test noninvasive string device (Neo-Medical Inc., Sparks, NV) in treatment period 1 only. The Entero-test device was swallowed 3.5 hours before the oral dose or start of the intravenous infusion to allow peristaltic transit to the duodenum. A food cue was used to stimulate gall bladder emptying at 2 hours after the start of intravenous infusion, and the string was withdrawn 1 hour later. All samples were stored frozen prior to shipment for analysis.
Mass Balance and Excretion.
Total radioactivity excreted in urine and fecal samples was determined by using liquid scintillation counting (LSC) and accelerator mass spectrometry (AMS) for treatment period 1 (Pharmaron ABS, Inc., Germantown, MD) and by LSC for treatment period 2 (Covance Laboratories Limited, Harrogate, UK). For LSC, counting efficiency and quench correction were achieved automatically by an external standard ratio method. The lower limit of quantification (LLQ) for LSC was assigned as twice the mean background disintegration rate.
AMS instrument standards were analyzed alongside the samples for graphitization. For measurement of total radioactivity by AMS, urine and fecal samples underwent combustion (oxidation) and graphitization (reduction), and radioactivity was determined using a single stage accelerator mass spectrometer-250 (NEC, Middleton, WI). For treatment period 1, the LLQ value using AMS for urine samples was 2.97 pg linerixibat Eq/ml and for fecal samples was 17.2 pg Eq/ml. For the assessment of radioactivity in feces, one participant was a recovery outlier and was not included in the mean calculations.
Pharmacokinetic Assessments.
Plasma concentrations of [14C]-linerixibat (parent drug) were determined by liquid chromatography (LC) and AMS (LC+AMS), and plasma concentrations of [12C]-linerixibat were determined by LC with tandem mass spectrometry; the details of these assays are presented in Supplemental Methods.
Quality-control (QC) samples were analyzed with each batch of study samples against separately prepared calibration standards. For measurement of samples containing linerixibat using LC-MS/MS, four concentrations were used. For measurement of samples containing [14C]-linerixibat using LC+AMS, three concentrations were used. To pass acceptance, no more than one-third of the QC results were to deviate from the nominal concentration by >15% (LC-MS/MS) or >20% (LC+AMS), and ≥50% of the results from each QC concentration were required to be within 15% (LC-MS/MS) or 20% (LC+AMS) of nominal. The applicable analytical runs met all predefined run acceptance criteria.
The total radioactivity of plasma from treatment period 1 was determined with AMS and from treatment period 2 with LSC and AMS. For plasma total radioactivity measured by AMS, the mean LLQ was 16.1 pg Eq/ml for treatment period 1 and 25.2 pg Eq/ml for treatment period 2. Further details of bioanalytical methodologies are provided in the Supplemental Methods.
Quantification and Characterization of Metabolites.
To create pooled plasma samples at 1–3 hours after treatment period 1, individual participant plasma samples were vortexed, and equal volumes of 1-, 2-, and 3-hour samples were pooled across participants. Using an area under the plasma concentration-time curve (AUC) approach (Hamilton et al., 1981), individual participant plasma samples were vortexed and pooled across all participants for the preparation of plasma AUC0–12h samples after treatment period 2.
Individual participant urine (0–24 hours) and homogenized fecal (0–120 hours) samples from treatment period 1 were pooled based on a ratio of the total weight of the sample excreted at each time point and were made to represent ≥95% of the total excreted radioactivity across all participants (Penner et al., 2009). For treatment period 2, representative individual participant fecal samples were created according to excretion balance data; weighed aliquots of selected samples of feces (from time intervals containing >2% of the administered dose) were combined proportionally to total sample weight to give a single pooled representative sample per participant. The pooled fecal samples were extracted once with acetonitrile containing 0.2% formic acid and once with methanol containing 0.2% formic acid. Combined extracts were dried down and reconstituted with methanol containing 10% DMSO. Samples were diluted with 0.1% formic acid in water prior to LC-MS/MS analysis with quantitation by microtiter plates scintillation counting from collected fractions. The total radioactivity of pooled treatment period 2 (oral) feces samples was expressed as a percentage of the total administered radioactive dose. Calculations were based on the percentage of administered dose and the pooled weight ratio data of the individual samples.
Crossparticipant pools of all sample types [plasma AUC0–12h, urine (0–24 hours) and fecal (0–120 hours, treatment period 1) samples] were prepared after each individual participant pool was created by taking a constant proportion the participant pool. Predose plasma, urine, and fecal samples were created. These pools were used to obtain background values to subtract from the postdose pools for analysis by AMS. Prior to analysis by LC+AMS, plasma samples were extracted with acetonitrile and then methanol containing 0.2% formic acid, reconstituted in 10% DMSO in methanol, and diluted with 0.1% formic acid in water; fecal samples were extracted with acetonitrile containing 0.1% formic acid and then with methanol containing 0.2% formic acid (3×) and diluted with 0.1% formic acid in water, and urine was centrifuged. All the samples analyzed by AMS were spiked with [12C]-linerixibat reference standard.
Duodenal bile was first extracted with acetonitrile from the bile-string samples, and this was followed by a second extraction with water. Aliquots of the bile extracts were mixed with scintillation fluid and measured separately by LSC; vials were individually counted for 15 minutes. After analysis and review of each sample, certain extracts were combined prior to further analysis by LC+AMS.
Radiochromatograms of plasma (treatment periods 1 and 2) and urine, homogenized feces, and bile string (treatment period 1) were generated by collecting LC fractions, which was followed by graphitization and analysis of fractions by AMS. Column recovery was calculated based on total amount of radioactivity (14C) injected and the amount recovered. Metabolites were quantified in plasma, urine, fecal (treatment period 1), and bile-string extracts using AMS. Further details are provided in the Supplemental Methods.
Metabolite identification for the treatment period 2 feces was conducted using high-resolution mass spectrometry, wherein the chromatographic retention time, accurate mass, and multi stage mass spectrometry fragmentation pattern were used for structure elucidation of metabolites. Metabolites and linerixibat assignments in the AMS radiochromatograms were made by retention time matching with either a corresponding linerixibat reference standard UV peak, metabolites from treatment period 2 feces, or metabolite peaks from prior metabolite studies (unpublished data).
Pharmacokinetics and Statistical Analysis.
No formal sample size calculation was performed for this study. As the objective was to gain a better understanding of the PK, excretory routes, and metabolic profile of linerixibat, inclusion of 4–6 participants was deemed sufficient (Penner et al., 2009).
Plasma linerixibat, [14C]-linerixibat, and total radioactivity concentration-time data were analyzed by noncompartmental methods with PhoenixWinNonlin Version 6.3 (Certara USA, Inc., Princeton, NJ) using the actual sampling times for derivation of PK parameters. Linerixibat plasma kinetics in terms of Cmax, time to Cmax, AUCo‒t, AUC0‒24 and AUC0‒inf, terminal phase rate constant, apparent terminal phase half-life, clearance, volume of distribution at steady state, absolute bioavailability, fraction of drug escaping first-pass hepatic clearance, hepatic extraction ratio, and fraction absorbed were calculated from plasma concentration-time data.
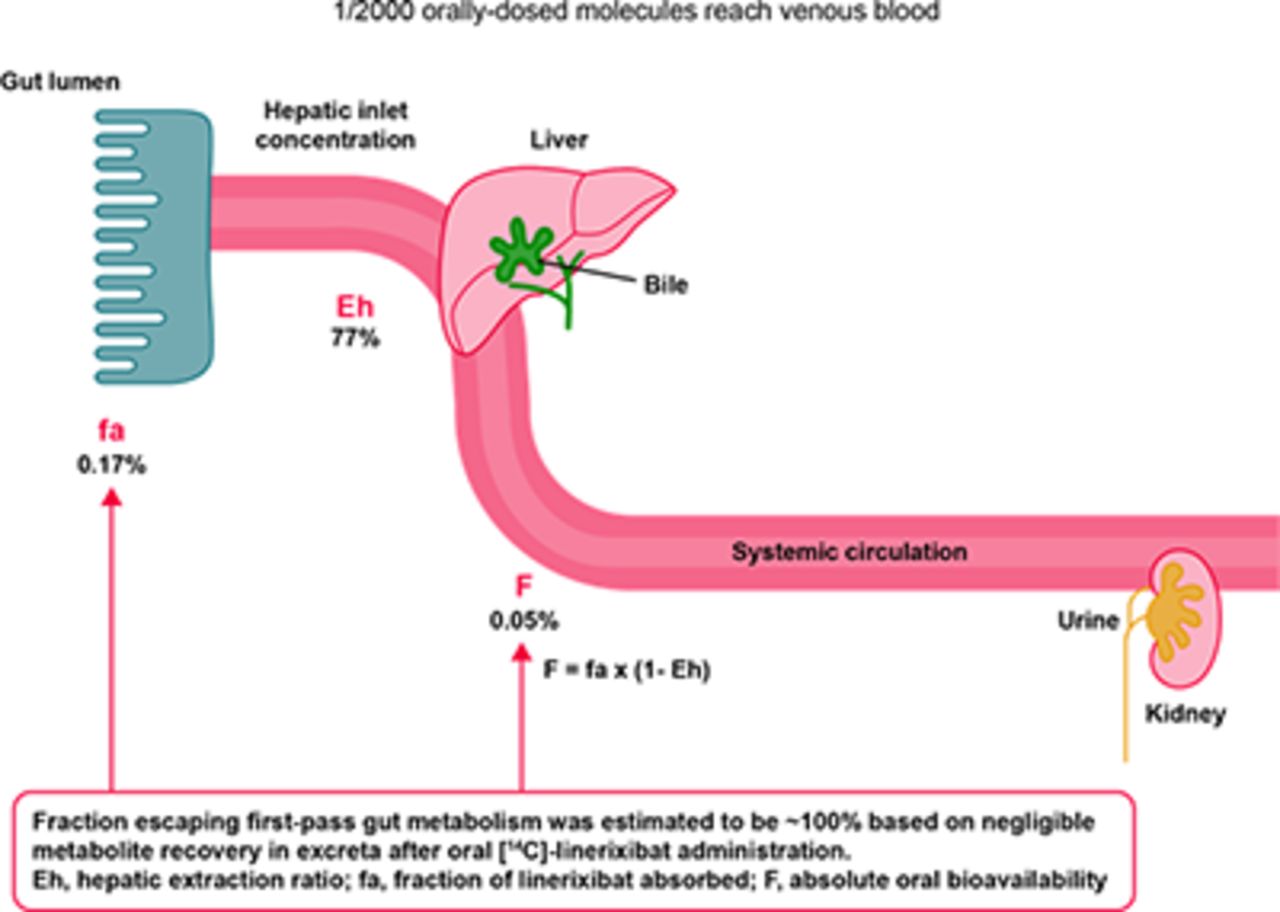
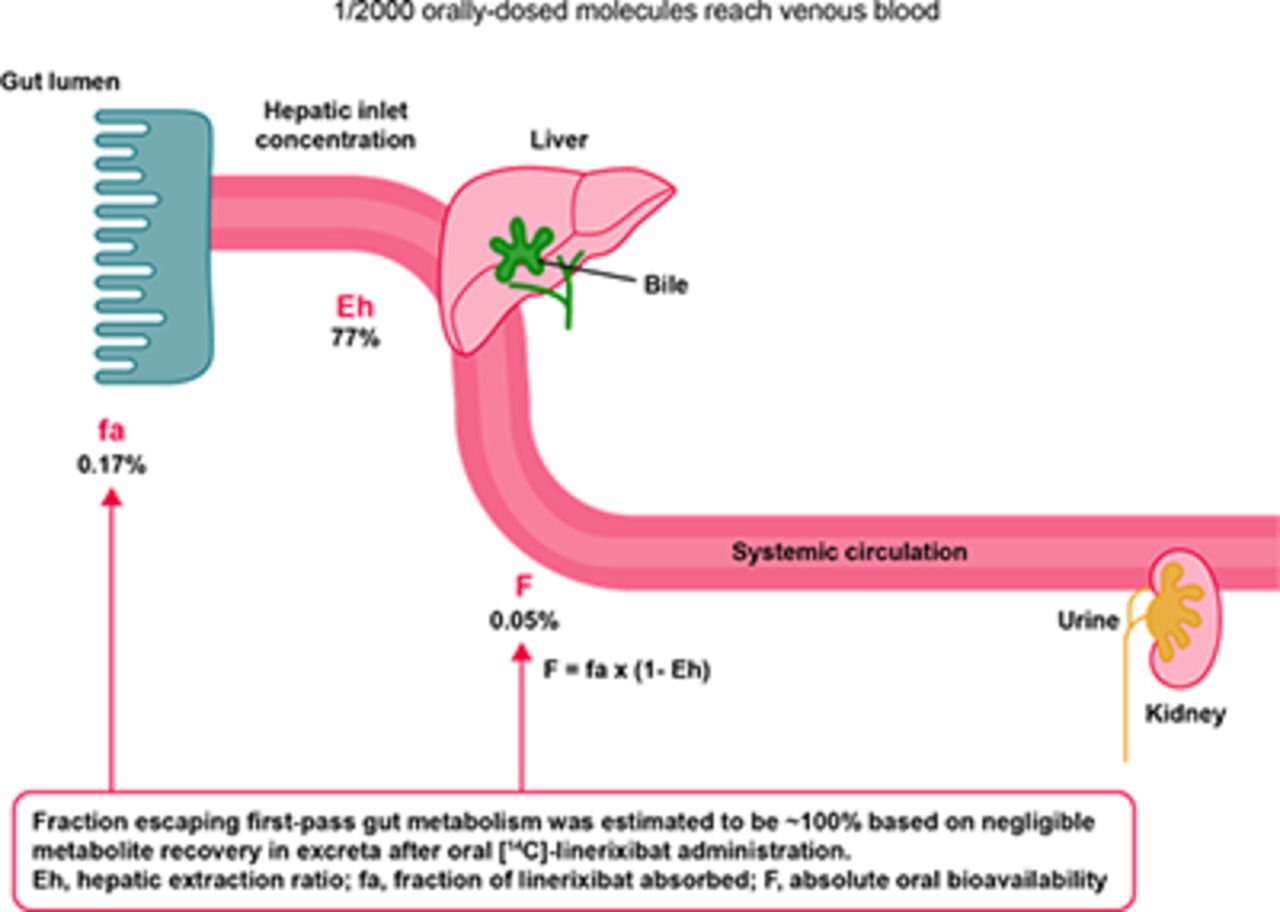
The hepatic plasma clearance was calculated as the difference between linerixibat total plasma clearance and renal clearance. Hepatic blood clearance (Clh,iv,blood) was determined by adjusting the hepatic plasma clearance by the linerixibat blood-to-plasma ratio of 0.678 determined in vitro (unpublished data). The hepatic extraction ratio (Eh) was then determined by dividing Clh,iv,blood by hepatic blood flow utilizing the literature reference value of 1660 ml/min (Edginton et al., 2006). The fraction of linerixibat that escapes first-pass liver extraction (Fh) was determined as one minus the Eh. The fraction of linerixibat absorbed (fa), including the fraction escaping first-pass gut metabolism (fg ∼100%), was determined by dividing the absolute oral bioavailability (F) by the Fh.
Results
A total of six participants were enrolled in this study, and all participants completed the study. Mean age was 41.2 (S.D. 8.1; range 33–53) years, and mean body mass index was 24.0 (S.D. 1.1; range 22.9–26.0) kg/m2.
Linerixibat and total drug-related radioactivity in the systemic circulation rapidly attained steady state during the 3-hour intravenous infusion and declined rapidly at the end of the infusion with detectable concentrations at up to 6 hours (Fig. 2). The short elimination half-life (0.828 hours) after intravenous administration was consistent with the high systemic clearance (61.8 l/h) and low volume of distribution (16.3 l) of linerixibat (Table 1). The exposure ratio of plasma linerixibat to plasma total radioactivity as well as the metabolite profiling showed that linerixibat was the predominant contributor to total radioactivity in the circulation (86% of total radioactivity in plasma), demonstrating linerixibat was minimally metabolized. Elimination of intravenous radioactivity was 80% fecal and 20% renal, predominantly as unchanged parent drug (90%–97% of fecal, bile-string, and urinary radioactivity) (Fig. 3A; Tables 1 and 2).
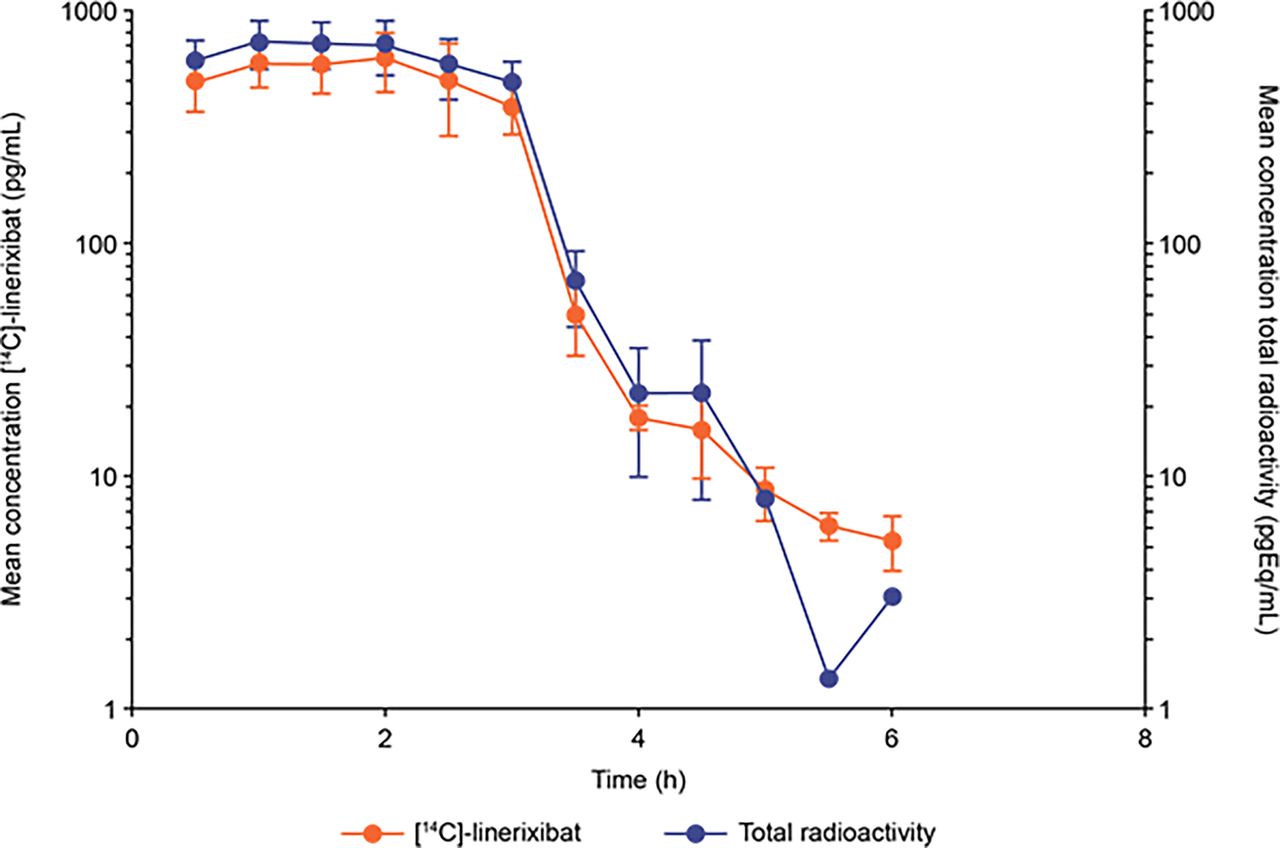
Parent [14C]-linerixibat and total drug-related radioactivity concentration-time profiles after 100 μg i.v. microdose of [14C]-linerixibat (n = 6). The intravenous microdose of [14C]-linerixibat (infused over 3 hours) was administered concomitantly with a nonradiolabeled 90-mg tablet oral dose.

Fecal, urinary, and total recovery of radioactivity after administration of (A) an intravenous [14C]-linerixibat microtracer over 3 hours (n = 6, mean ± S.D.) and (B) a 90-mg [14C]-linerixibat oral solution (n = 5a, mean ± S.D.). n = 5 for all time points, as one participant’s recovery was only 20.8% of the dose and considered anomalous compared with the 97% recovery in the other five participants (outlier by Q test; no fecal samples on study days 2 and 6 when the other five participants produced daily fecal samples; prune juice given from day 6 to elicit bowel movements, resulting in feces with no detectable radioactivity from day 7 onward).
Summary of pharmacokinetic parameters for linerixibat (n = 6, unless stated otherwise)
[14C]-Linerixibat–related radioactivity after intravenous infusion of radiolabeled microdose
Standard in vitro studies identified oxidative metabolism in human liver microsomes as the most likely route of linerixibat clearance, which was mediated by cytochrome P450 (CYP) 3A4 but not the other major CYP enzymes (unpublished data). Scaled unbound intrinsic clearance of linerixibat in human liver microsomes was high (241 l/h) based on an in vitro incubation half-life of 39.4 minutes at 1 mg/ml microsomal protein concentration [microsome fraction unbound = 0.299; 39.7 mg microsomal protein/g of liver and 24.5 g of liver/kg body weight (Barter et al., 2007, 2008), assuming 70 kg human body weight, as described previously (Obach et al., 1997, 1999)]. This microsomal intrinsic clearance was extrapolated to human using IVIVE based on the well stirred liver model, and the human clearance was predicted to be 31 l/h [plasma fraction unbound = 0.245; blood-to-plasma ratio = 0.678; human hepatic blood flow = 96.6 l/h (Yang et al., 2007)]. This prediction was within 1.5-fold of the high hepatic clearance observed in this study (80% of human systemic elimination of intravenous dose).
The geometric mean half-life of linerixibat was approximately an order of magnitude longer after oral versus intravenous administration (6–7 hours vs. 0.8 hours). Based on the observed shorter half-life after intravenous administration compared with oral administration, linerixibat displays absorption-limited, flip-flop kinetics; therefore, the terminal slope of the oral concentration-time profile (Table 1, λ z) reflects the oral absorption rate constant, which was estimated to be 0.1 hour−1. Linerixibat exhibited minimal oral absorption (absolute oral bioavailability of 0.0517%) primarily due to the very low fraction absorbed (0.167%; fg ∼100%, see Table 3 results below). High hepatic extraction (77.0%) acted as a secondary barrier to systemic exposure, accounting for the approximately 4-fold lower oral bioavailability versus fraction absorbed. Time of Cmax was highly variable after oral administration of both tablet and solution formulations with the large range overlapping across the two (Table 1).
As expected for a minimally absorbed drug, the oral [14C]-linerixibat dose was almost entirely excreted (>99% of the recovered radioactivity) in feces as unchanged and unabsorbed linerixibat. Three very minor oxidative metabolites, M8, M9, and M10, were detected, but they accounted for negligible radioactivity after correcting for coeluting radiochemical impurity E and degradant K (Table 3), thus supporting fg being approximately 100%. Consistent with low oral fraction absorbed and approximately 80%/20% fecal/renal excretion of intravenous [14C]-linerixibat, urinary elimination of orally administered radioactivity was negligible (<0.04% of dose).
After administration of oral [14C]-linerixibat solution, the exposure to total radioactivity was far higher and longer than to parent drug (Fig. 4; individual participant data in Supplemental Figure S1). The geometric mean Cmax and exposure ratios of plasma linerixibat to plasma total radioactivity showed that linerixibat represented only 2%–6% of the total radioactivity in plasma. This low fraction of parent linerixibat accounting for oral plasma total radioactivity concentrations was conceptually inconsistent with parent linerixibat accounting for the majority of radioactivity administered via intravenous infusion. It also differs from previous profiling of human plasma obtained after 14 days of oral 90-mg twice-daily nonradiolabeled linerixibat dosing, wherein only the parent drug but no linerixibat-related metabolites were observed in circulation (Nunez et al., 2016). The low fraction of parent linerixibat accounting for plasma total radioactivity after oral dosing in treatment period 2 is likely an artifact of 2.4% radiochemical impurities for a drug with <0.2% fraction absorbed. Furthermore, these radiochemical impurities had a 70-fold–higher specific activity than isotopically diluted [14C]-linerixibat in the oral dose formulation and were profiled using AMS, which provides a measure of total radiocarbon (see supplement for discussion of results presented in Fig. 5).

Parent linerixibat and total drug-related radioactivity concentration-time profiles after 90-mg oral solution of [14C]-linerixibat (n = 6). Individual participant data shown in Supplemental Figure S1.
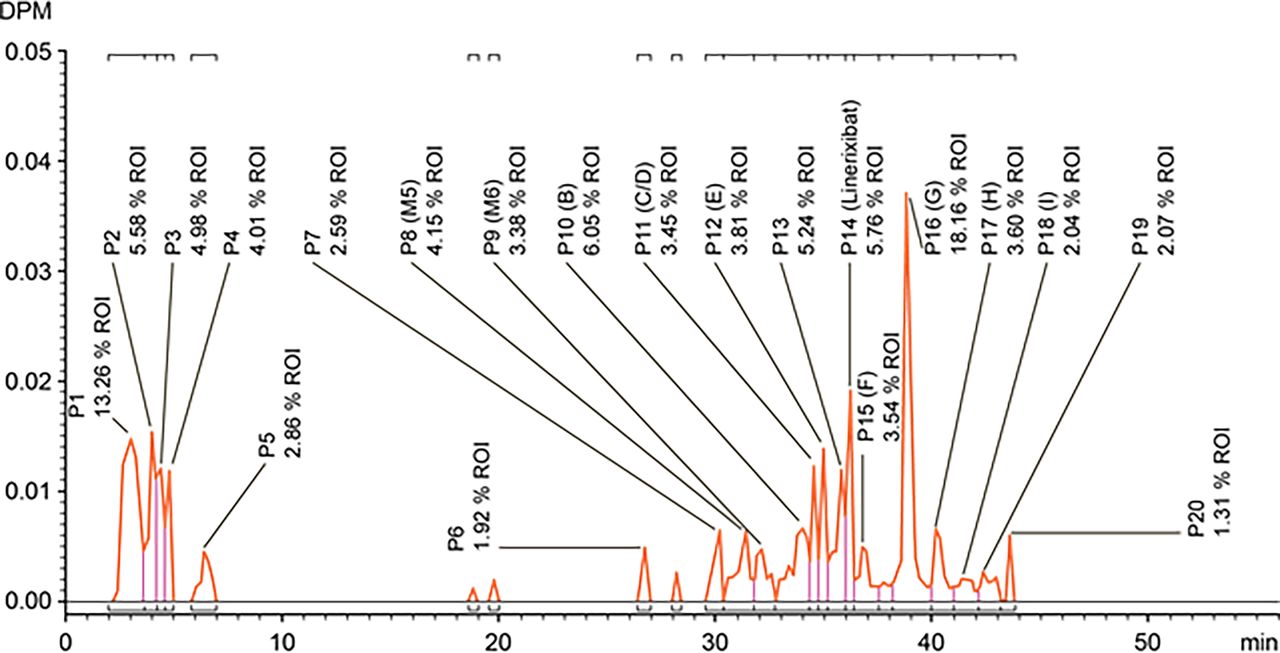
AMS radiochromatogram of cross-participant plasma pool extract (AUC0-12 h) after oral administration of 90-mg oral solution of [14C]-linerixibat. DPM, disintegrations per minute; ROI, region of interest.
Discussion
Characterization of the human PK and metabolism of [14C]-linerixibat after both intravenous and oral administration is important in the context of a drug that has minimal systemic exposure and in which the GI lumen is the site of pharmacology. Only by assessing both modes of administration can we quantify the contribution of fraction absorbed from the GI tract and of first-pass hepatic extraction to the minimal systemic exposure. The current study established that linerixibat exhibits minimal systemic exposure due to low intestinal absorption. Furthermore, intestinal absorption was slow, resulting in flip-flop kinetics that were limited by absorption rate, as evidenced by a longer oral versus intravenous half-life (6–7 hours vs. 0.8 hours). Technically, fraction absorbed is fa × fg and, typically, direct calculation of fg would have been possible based on oral and intravenous metabolite load data given that oral metabolite load = intravenous metabolite load + metabolites generated on first pass through the GI and the liver (Harrell et al., 2019). Unfortunately, the oral metabolite load calculation for linerixibat is not feasible since the major contributors to oral radioactivity exposure were radiochemical impurities and their metabolites. However, minimal first-pass gut-wall metabolism (fg ∼100%) is substantiated by negligible recovery of metabolites in excreta after oral administration of [14C]-linerixibat (Table 3). Finally, there is no evidence that linerixibat accesses enterocytes and their metabolic enzymes because of exceedingly low passive permeability, and although linerixibat is taken up into hepatocytes by OATP 1B, it is not transported by intestinal OATP2B1 (unpublished data).
These results have a direct impact on late-stage clinical development of linerixibat. In vitro screening of both perpetrator and victim DDI risks flagged linerixibat as both a substrate and inhibitor of OATP1B1 (IC50 = 2.69 μM), OATP1B3 (IC50 = 0.265 μM) and CYP3A4 (KI = 1.3 mM, kinact = 9.6 hour−1, fu,inc = 0.3) (unpublished data). Based on PK results from this study and current regulatory guidelines, linerixibat’s potential as a perpetrator or victim of DDIs via CYP3A4 and OATP1B does not meet the criteria for clinical evaluation (US FDA, 2020a).
Although linerixibat is an OATP1B1 and OATP1B3 substrate in vitro, the present study clearly established minimal intestinal absorption as the basis of minimal systemic exposure of linerixibat, with the liver acting as a secondary barrier to systemic exposure with a hepatic extraction of 77.0%. Assuming complete inhibition of hepatic OATP1B, oral bioavailability would increase at the most from 0.05% to the fraction absorbed of <0.2%. Consequently, systemic exposure would increase at most only by approximately 4-fold, which would be well within systemic safety margins. Because of linerixibat exhibiting flip-flop kinetics limited by absorption rate, potential inhibition of hepatic clearance would not increase exposure after oral dosing because absorption is approximately 10-fold–slower than elimination. As such, only the hepatic first-pass effect is relevant to victim DDI potential.
Based on the results of the present study, further clinical studies to assess the effect of renal impairment are not scientifically justified. The systemic exposure of linerixibat after oral administration is not expected to be impacted by renal impairment or by a reduction in hepatic clearance due to circulation of elevated levels of uremic toxins due to its minimal oral absorption, negligible dose recovery in urine after oral administration, and flip-flop kinetics.
Although linerixibat is a CYP3A4 substrate in vitro, inhibition of metabolic clearance by a coadministered CYP3A4 inhibitor is not expected to impact linerixibat disposition or systemic PK. This is because orally administered linerixibat was eliminated almost entirely as unabsorbed and unchanged parent drug in feces. About 1 in 2000 orally administered linerixibat molecules that do enter the systemic circulation is eliminated as unchanged drug in bile/feces (approximately 80%) and urine (approximately 20%).
Intrinsic clearance of linerixibat in human liver microsomes was high (241 l/h) with metabolism exclusively via CYP3A4 and no other major CYP isoforms (unpublished observations). IVIVE based on the well stirred liver model predicted a human hepatic clearance of 31 l/h, which was within 1.5-fold of the observed human hepatic clearance (80% of systemic clearance). Therefore, it was surprising that human clearance occurred primarily by direct excretion of parent drug with negligible metabolism. Even after a thorough literature review, it appears that linerixibat is the only reported example of this phenomenon, which may be related to the unusual physicochemical properties needed to achieve minimal absorption (Wu et al., 2013). An interesting future direction may be to determine whether a more complex in vitro hepatocyte system that re-establishes bile canaliculi while maintaining CYP metabolic activity is capable of describing in vivo hepatobiliary disposition of linerixibat.
Biliary excretion was studied in bile duct–cannulated male Han Wistar rats and male Beagle dogs after oral administration of [14C]-linerixibat (unpublished data). The original conclusion of these studies was that fecal excretion was the major elimination pathway for oral [14C]-linerixibat (rat = 93.9% and dog = 82.0% of dosed radioactivity), whereas biliary and urinary excretion represented minor elimination routes (rat bile = 3.0% and urine = 0.1%; dog bile = 6.7% and urine = 2.7% of dosed radioactivity). The outcome of the present clinical study resulted in re-examination of preclinical oral [14C]-linerixibat mass balance studies, assuming radioactivity recovered in bile and urine represents absorbed radioactivity. Upon re-examination of these preclinical results in terms of elimination of absorbed radioactivity, it becomes evident that biliary excretion of unchanged linerixibat is also the major route of elimination in rats and dogs. Biliary/urinary recovery of absorbed radioactivity in rats and dogs was 97%/3% and 71%/29%, respectively, which is consistent with ∼80%/20% linerixibat-related material excretion in humans. Rat and dog biliary radioactivity consisted primarily of parent linerixibat 73% and 75%, respectively; mono-oxygenated metabolites ≤6.5% and 19.5%, respectively; and radiochemical impurity-related radioactivity ≥13% and 3.4%, respectively. Mechanistically, the first step in linerixibat biliary excretion is mediated by OATP1B1 and OATP1B3 hepatic uptake (unpublished data); subsequent canalicular secretion is not mediated by P-glycoprotein or breast cancer resistance protein (unpublished data), leaving multidrug resistance-associated protein 2 as the most likely canalicular transporter (Patel et al., 2016) but one that is not studied during drug development because of lack of clinical evidence supporting involvement of multidrug resistance-associated protein 2 in clinical DDIs (US FDA, 2020a).
Typically, in the development of small-molecule drugs, the primary purpose of a radiolabeled study is the characterization of human in vivo metabolism to ensure that preclinical rodent and nonrodent toxicology species provide adequate coverage of the major human metabolites (US FDA, 2020b). After intravenous administration, unchanged linerixibat was the predominant form in human circulation and excreta (Fig. 2; Table 2). This was consistent with nonradiolabeled metabolite profiling of human plasma obtained after 90-mg oral linerixibat dosed twice-daily for 2 weeks (Nunez et al., 2016). However, the plasma radiochromatogram (derived by AMS) after oral administration was complex (Fig. 5). As described in Results, the observed complexity can be attributed to 2.4% of radiochemical impurities, with an estimated 70-fold–higher specific activity than the isotopically diluted parent drug, which exhibits low oral absorption (<0.2%). Despite this complex finding, the overall metabolite profile supports the absence of major human metabolites. AMS radiochromatogram of crossparticipant plasma pool extract (AUC0-12 hour) after oral administration of 90-mg oral solution of [14C]-linerixibat is described in detail in the Supplemental Results and depicted in Fig. 5.
In conclusion, the present two-period clinical study of [14C]-linerixibat intravenous and oral PK and disposition established linerixibat as a minimally absorbed drug that after oral administration is almost entirely recovered as unabsorbed and unchanged parent drug in feces. This study supports that, after oral administration, linerixibat is effectively restricted to the GI lumen, the site of pharmacology for IBAT inhibition. Additionally, this study demonstrated flip-flop oral absorption–rate-limited systemic PK. These results have considerable impact on late-stage clinical pharmacology studies as discussed above. Finally, linerixibat presents a fascinating novel case study in which human clearance by CYP3A4 metabolism was quantitatively predicted to be high, but in vivo metabolism was found to be minimal, with clearance predominantly by direct biliary/fecal excretion of the unchanged parent drug.
Acknowledgments
Editorial support (in the form of writing assistance, including preparation of the draft manuscript under the direction and guidance of the authors, collating and incorporating authors’ comments for each draft, assembling tables and figures, grammatical editing, and referencing) was provided by Mary E. Morgan and Hayley S. Butler of Fishawack Indicia Ltd., which is part of Fishawack Health, and funded by GlaxoSmithKline.
Authorship Contributions
Participated in research design: Zamek-Gliszczynski, Kenworthy, Mudunuru, O’Connor-Semmes, Young.
Conducted experiments: Bershas, Sanghvi, Pereira, Crossman, Thorpe, Dennison.
Performed data analysis: Zamek-Gliszczynski, Kenworthy, Bershas, Sangvhi, Pereira, Crossman, Pirhalla, Thorpe, McLaughlin, Allinder, Swift, O’Connor-Semmes, Young.
Wrote or contributed to the writing of the manuscript: Zamek-Gliszczynski, Kenworthy, Bershas, Sanghvi, Pereira, Mudunuru, Crossman, Pirhalla, Thorpe, Dennison, McLaughlin, Allinder, Swift, O’Connor-Semmes, Young.
Footnotes
- Received June 23, 2021.
- Accepted September 24, 2021.
This study was funded by GlaxoSmithKline (Study 205895).
M.J.Z.-G., D.K., D.A.B., A.I.P., J.M., J.L.P., K.M.T., M.M.M., M.A., B.S., and G.C.Y. are GlaxoSmithKline (GSK) employees and hold GSK shares. M.S. is an employee of Pharmaron US. L.C. is an employee of Covance Laboratories Limited, UK. R.L.O.-S. and J.D. do not have any conflicts of interest.
Information on GSK’s data sharing commitments and requesting access can be found at: https://www.clinicalstudydatarequest.com.
The data presented in this manuscript was presented in part at the American Association for the Study of Liver Diseases November 13–16, 2020 (Zamek-Gliszczynski et al. AASLD 2020; Poster 1257).
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AMS
- accelerator mass spectrometry
- AUC
- area under the plasma concentration-time curve
- Clh,iv,blood
- hepatic blood clearance
- CYP
- cytochrome P450
- DDI
- drug-drug interaction
- Eh
- hepatic extraction ratio
- F
- absolute oral bioavailability
- fa
- fraction absorbed
- fg
- fraction escaping first-pass gut metabolism
- Fh
- fraction escaping first-pass hepatic extraction
- GI
- gastrointestinal
- IBAT
- ileal bile acid transporter
- IVIVE
- in vitro–to–in vivo extrapolation
- LC
- liquid chromatography
- LC-MS/MS
- LC with tandem mass spectrometry
- LLQ
- lower limit of quantification
- LSC
- liquid scintillation counting
- OATP
- organic anion–transporting protein
- PBC
- primary biliary cholangitis
- PK
- pharmacokinetics
- QC
- quality control
- Copyright © 2021 by The Author(s)
This is an open access article distributed under the CC BY Attribution 4.0 International license.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}