


Abstract
Plasma protein–mediated uptake (PMU) and its effect on clearance (CL) prediction have been studied in various formats; however, a comprehensive analysis of the overall impact of PMU on CL parameters from hepatocyte assays (routinely used for IVIVE) has not previously been performed. The following work collated data reflecting the effect of PMU for 26 compounds with a wide variety of physicochemical, drug, and in vivo CL properties. PMU enhanced the unbound intrinsic clearance in vitro (CLint,u in vitro) beyond that conventionally calculated using fraction unbound and was correlated with the unbound fraction of drug in vitro and in plasma (fup) and absolute unbound intrinsic clearance in vivo (CLint,u in vivo) in both rat and human hepatocytes. PMU appeared to be more important for highly bound (fup < 0.1) and high CLint,u in vivo drugs. These trends were independent of species, assay conditions, ionization, and extended clearance classification system group, although the type of plasma protein used in in vitro assays may require further investigation. Such generalized trends (spanning fup 0.0008–0.99) may suggest a generic mechanism behind PMU; however, multiple drug-dependent mechanisms are also possible. Using the identified relationship between the impact of PMU on CLint,u in vitro and fup, PMU-enhanced predictions of CLint,u in vivo were calculated for both transporter substrates and metabolically cleared drugs. PMU was accurately predicted, and incorporation of predicted PMU improved the IVIVE of hepatic CL, with an average fold error of 1.17 and >50% of compounds predicted within a 2-fold error for both rat and human data sets (n ≥ 100).
SIGNIFICANCE STATEMENT Current strategies for prediction of hepatic clearance from in vitro data are recognized to be inaccurate, but they do not account for PMU. The impact of PMU on CLint,u in vitro is wide ranging and can be predicted based on fraction unbound in plasma and applied to CLint,u in vitro values obtained by standard procedures in the absence of plasma protein. Such PMU-enhanced predictions improved IVIVE, and future studies may easily incorporate this PMU relationship to provide more accurate IVIVE.
Introduction
The quantitative prediction of hepatic clearance (CL) of drugs from in vitro systems remains challenging and is currently inadequate for reliable estimation of first-in-human dosing (Bowman and Benet, 2016; Wood et al., 2017). The characteristic underprediction of high-CL compounds when using physiologically mechanistic scaling is deemed to be multifactorial in origin (Wood et al., 2017). Confounding in vitro phenomena such as the unstirred water layer (UWL) barrier, cofactor depletion, and loss of enzyme activity in vitro have been explored (Wood et al., 2018), but a consistent improvement in methodological accuracy and precision has yet to be achieved across a wide range of drugs. Conventionally, plasma protein binding has only been a consideration when extrapolating in vitro intrinsic clearance (CLint in vitro) to in vivo CL (and vice versa) using liver models [e.g., the well stirred liver model (WSLM)]. This methodology follows the free drug hypothesis (FDH), in which drug association to, and dissociation from, plasma protein is rapid (not rate limiting), only unbound drug penetrates hepatocytes (reversibly), and unbound drug (at equilibrium with plasma concentration) provides the driving concentration for metabolism.
However, numerous cases over several decades of both endogenous and exogenous compounds have shown hepatic uptake kinetics that appeared to be driven by bound rather than unbound compound—apparently challenging the FDH. Use of isolated perfused livers (IPL) during the 1980s provided evidence of “albumin-mediated uptake” for taurocholate, oleate, and warfarin (Forker and Luxon, 1981, 1983; Weisiger and Ma, 1987; Tsao et al., 1988). This plasma protein–mediated uptake (PMU) was initially hypothesized to involve either a specific albumin receptor or rate-limiting albumin-ligand dissociation (Weisiger et al., 1981), but alternative mechanistic explanations followed. IPL studies with warfarin led Tsao et al. (1988) to suggest a facilitated dissociation model involving a less specific interaction with the plasma membrane. Ichikawa et al. (1992) suggested that albumin may enhance drug diffusion through the UWL in hepatic sinusoids, as rate-limited diffusion was observed in the absence, but not presence, of albumin. More recently, Bowman et al. (2019) observed a reduction in unbound uptake affinity (Km,u) for several organic anion transporting polypeptide (OATP) substrates, suggesting PMU is driven by high-affinity drug transporters, which may be able to “strip” the drug directly from the albumin. Miyauchi et al. (2018) have recently applied the facilitated dissociation model to kinetics from hepatocyte assays, giving a mechanistic solution for in vitro observations of PMU. Although this type of analysis provides useful insight, it is retrospective and requires detailed uptake kinetic measurements from hepatocyte assays.
Although the mechanistic details behind PMU remain inconclusive, and potentially multiple, the importance of considering PMU in in vivo CL predictions has become compelling. In addition to the cases above, the use of plasma/isolated plasma protein in assay medium for the in vitro prediction of CL has been explored (prompted by raised unbound CLint in vitro relative to the conventional, protein-free approach), and improvements in in vitro-in vivo extrapolations (IVIVE) have been reported (Shibata et al., 2000; Blanchard et al., 2004, 2005, 2006; Li et al., 2020). Considering the overall number of cases that challenge conventional prediction strategies (and appear to challenge the FDH), progression of PMU to a mainstream component of the prediction of CL might now be appropriate. As such, there is a need for an integrated assessment of the experimental evidence from various sources, focusing on hepatocyte-based in vitro systems, as relied upon for CL prediction. Therefore, the main aim of the current study was to collate published data in which PMU has been quantified and assess trends common across all studies. A total of 74 individual observations (from 26 drugs in 11 studies) were identified involving rat and human hepatocytes. Correlations between the extent of protein binding and the impact on CL parameters were examined in relation to species, protein type, or other experimental approaches, as well as fundamental drug properties. Subsequently, a generic relationship was identified and incorporated into prediction methodology to assess the utility of this approach and its ability to improve IVIVE of CL.
Methods
DataBase Construction
The literature was searched to obtain studies that measured the impact of plasma protein (PP) on CLint in vitro. The CLint in vitro was obtained only from hepatocyte experiments, measured in both the presence and absence of PP, allowing for the assessment of the impact of PP (and thus PMU) under identical experimental conditions. Although other studies investigating the impact of PP using microsomes have been performed, these data sets were excluded from the current work. Microsomal studies only account for the impact of PP on cytochrome P450 metabolism (and not total clearance) and are likely to reflect a different mechanism [albumin has an anti-inhibitory effect on microsomes as a result of albumin binding avidly to long-chain unsaturated fatty acids, which inhibit many cytochrome P450 enzymes (Rowland et al., 2008; Fujino et al., 2018)] and thus is not representative of the PMU phenomenon under investigation here.
Data from all cellular species, sources of PP (isolated albumin, plasma, and serum), and compounds (drugs, in-house and nontherapeutic compounds) were considered. Only in vitro clearance parameters were included in the database (and back-calculated from scaled in vivo values when appropriate) to avoid potential bias of different scaling factors used across different studies. The in vitro fraction unbound (fu, under the exact same experimental conditions as the CLint in vitro studies were performed), was also recorded when possible. This includes nonspecific binding of the experimental conditions performed in the absence of PP as well as in the presence of PP. For a few studies, the nonspecific binding in the absence of PP was not measured, and in vitro fu was assumed to be 1. For one study (Bachmann et al., 2003), the in vitro fu in the presence of PP (in this case pure human serum) was not measured, so these in vitro fu values were sourced from independent studies (also obtained with pure human serum) for the five drugs examined in this study. All such cases are indicated in the footnotes of Supplemental Table 1.
The unbound CLint in vitro (CLint,u in vitro) was also included and was obtained directly from the original papers or calculated by eq. 1: (1)
(1)
When only the CLint,u in vitro was recorded by the authors, CLint in vitro could also be back-calculated based on eq. 1. When available, maximum uptake velocity (Vmax), Michaelis constant (Km), and Km,u values were also obtained directly as stated in the literature (which included both transporter and nontransporter substrates). Based on eqs. 2 and 3, these values were also used to calculate CLint in vitro or CLint,u in vitro, respectively, as required (see footnotes of Supplemental Table 1 for specific details on how individual values were calculated). As with eq. 1, back-calculations of eqs. 2 and 3 were also performed as required. (2)
(2) (3)
(3)
To assess the impact of PP on all in vitro clearance terms (Vmax, Km, CLint in vitro, Km,u, and CLint,u in vitro), the fold-change caused by the experimental addition of PP was calculated. The fold-change was calculated for each drug, in each study, comparing the relative change in the presence of PP to its experimentally matched no-PP controls [CLint,u in vitro (+PP)/CLint,u in vitro (no PP), with both values obtained under the same experimental conditions]. By using this approach to assess the impact of PP, comparisons across different species (e.g., rat and human) could be made despite clear species differences in the absolute CLint in vitro and CLint,u in vitro values. Because of the diverse nature of experimental conditions, the specific assay conditions (hepatocyte species, in vitro test system, cell source, type of albumin, cell density, plate format, shaking speed, drug concentration, and duration of experiments) were also recorded for comparison.
The physicochemical properties (molecular weight, water solubility, number of hydrogen donors and acceptors, polar surface area, log P, log D7.4, acidic pKa, basic pKa, and ionization) of each drug obtained in the database were also collected (Supplemental Table 1). The ionization of each drug was defined based on their charge and percent ionization at physiologic pH (7.4) as follows: neutrals, <3% ionized; acids, >10% negatively charged; weak acids, >3 <10% negatively charged; bases, >10% positively charged; weak bases, >3 <10% positively charged; zwitterions, >10% positively and negatively charged. The extended clearance classification system (ECCS), permeability, and clearance mechanism as reported by Varma et al. (2015) were also noted. For drugs not included in the Varma et al. (2015) database, the ECCS group was manually assigned based on its ionization and molecular weight (see footnotes, Table 1).
Overview of the physicochemical properties and in vivo values from rat and human of the compounds represented in the PMU database
References: [1] Varma et al. (2015), [2] Chung et al. (1990), [3] Miyauchi et al. (2018), [4] Avdeef, 2012, [5] Wood et al. (2017), [6] Li et al. (2020), [7] Yabe et al. (2011), [8] Brown et al. (2007), [9] Kim et al. (2019), [10] De Bruyn et al. (2018), [11] Watanabe et al. (2010), [12] Lave et al. (1997), [13] Blanchard et al. (2006), [14] Blanchard et al. (2004), [15] Brunton et al., 2011, [16] Bachmann et al. (2003).
Additional in vitro drug uptake parameters, passive permeability, uptake clearance, and active uptake (Pdiff, CLuptake, and CLactive, respectively) in the absence of PP were sourced for all drugs (mean values of multiple studies) when available from both rat and human hepatocyte assays. Finally, the fraction unbound in plasma and blood (fup and fub, respectively), unbound intrinsic CL in vivo (CLint,u in vivo) and total hepatic CL (CLH) values for rat and human were also collated, primarily based on the Wood et al. (2017) database, or independently sourced and mean values calculated. When necessary, some CLint,u in vivo values were calculated based on CLH and fub; see footnotes of Table 1 for details.
IVIVE Analysis Part 1: PMU DataBase.
Linear regression analysis was performed to quantify the identified relationship between log10-transformed fup and log10-transformed fold-change in CLint,u in vitro values caused by the addition of PP. No significant difference was observed between human and rat data sets, and thus a single equation was used to assess whether incorporation of predicted PMU could improve IVIVE for both species of this discrete set of drugs (see Results section for analysis). CLint,u in vitro (microliters per minute per 106 cells) obtained in the absence of PP and fup values from the PMU database was applied to eq. 4, predicting the fold-change in CLint,u in vitro caused by the addition of PP. (4)
(4)
This fold-change was then applied to the absolute CLint,u in vitro values obtained in the absence of PP to predict the CLint,u in vitro values in the presence of PP (microliters per minute per 106 cells). These in vitro values (CLint,u in vitro in the absence of PP, CLint,u in vitro in the presence of PP, and predicted CLint,u in vitro based on the observed PMU relationship) were subsequently scaled up to in vivo values (CLint,u in vivo, milliliters per minute per kilogram) after application of physiologically based scaling factors [hepatocellularity of 120 × 106 cells/g liver in both rat and human, and 40 g liver/kg bodyweight and 21.4 g liver/kg bodyweight in rat and human, respectively (Wood et al., 2017)] and compared with observed CLint,u in vivo values (Table 1). To assess accuracy and precision, the average fold error (AFE) and root-mean square error (RMSE) were calculated using eqs. 5 and 6, respectively: (5)
(5) (6)where n represents the number of predictions. The percentage within 2-, 3-, >3-fold, was also calculated to evaluate accuracy of the data set. CLint,u in vivo values were then applied to the WSLM (eq. 7) to assess IVIVE of CLH.
(6)where n represents the number of predictions. The percentage within 2-, 3-, >3-fold, was also calculated to evaluate accuracy of the data set. CLint,u in vivo values were then applied to the WSLM (eq. 7) to assess IVIVE of CLH. (7)where QH represents hepatic blood flow [100 ml/min per kg for rat and 20.7 ml/min per kg for human, Wood et al. (2017)]. These predicted CLH values were compared with observed CLH values (Table 1), and accuracy and precision were assessed as above.
(7)where QH represents hepatic blood flow [100 ml/min per kg for rat and 20.7 ml/min per kg for human, Wood et al. (2017)]. These predicted CLH values were compared with observed CLH values (Table 1), and accuracy and precision were assessed as above.
IVIVE Analysis Part 2: Wood et al. (2017) DataBase.
Because of the relatively small number of drugs within the PMU database, IVIVE analysis was also performed using the previously published database by Wood et al. (2017). This database contains 148 compounds for rat and 117 for human, all of which are mean values reported from the literature, and this was deemed a reliable and comprehensive source for such IVIVE analysis. The fup and CLint,u in vitro values (milliliters per minute per kilogram) measured in the absence of PP from hepatocytes, from both rat and human were used to calculate the predicted fold-change in CLint,u in vitro values caused by the addition of PP. This was applied to the absolute CLint,u in vitro values, as above, to predict CLint,u in vivo values in the presence of PP. Considering the uncertainty regarding PMU effect in vivo, CLH was also predicted from CLint,u in vivo values using the WSLM (eq. 7), thus avoiding the assumption of equivalent PMU effect in vivo. Full calculation of these predictions is shown in (Supplemental Table 2). IVIVE analysis (AFE, RMSE, and percent fold-within) was performed on CLint,u in vivo and CLH values, as described previously. These predictions were repeated using alternative parallel tube and dispersion liver models (Ito and Houston, 2004) to verify the choice of WSLM (Supplemental Fig. 1).
Statistical Analysis.
All data were analyzed using GraphPad Prism version 8.0. To assess the impact of PP on absolute CLint,u in vitro values, a paired two-tailed t test was performed (Supplemental Fig. 2). To assess the PMU phenomenon, log-log least-squares regression analysis with no weighting was performed on the fold-change in CLint,u in vitro and in vitro fu, with all data, and compared against a hypothetical slope of zero. This zero baseline would denote no effect of PP on CLint,u in vitro values as would be expected according to conventional normalization to unbound concentrations using fu. To identify whether in vitro assay conditions influenced the observed relationship between in vitro fu and fold-change in clearance parameters, log-log least-squares regression analysis with no weighting was performed, and an F-test was used to assess significant differences between the subsets (α = 0.05). When statistical differences were observed (e.g., Supplemental Fig. 3), linear regression analysis was repeated on the log10-transformed data, followed by an F-test to assess for significant differences between the subsets. To quantify the observed relationship between fup and the fold-change in CLint,u in vitro caused by the addition of physiologically equivalent concentrations of PP, linear least-squares regression analysis with no weighting was performed on the log10-transformed fup and log10-transformed fold-change in CLint,u in vitro values, followed by an F-test to assess whether there was a significant differences between the rat and human data sets (α = 0.05).
Results
Scope of PMU Database.
The PMU database consists of data from 11 studies on 26 different compounds with (cumulatively) 74 different reported values for the influence of PP on in vitro clearance parameters. Multiple studies on the same compound were considered independently (18 of 26 compounds contained multiple data points). These data were obtained from various experimental conditions, but all studies included direct comparisons to PP-free conditions, allowing for fold-change between the presence and absence of PP to be calculated (Supplemental Table 1). Although initially all cellular species were considered, only data from rat (n = 36) and human (n = 38) hepatocytes could be sourced and, thus, are evaluated here. The data set represents a broad range of compounds based on both their physicochemical properties and in vivo values, as summarized in Table 1. The majority of recent studies on the PMU phenomenon have focused on OATP transporter substrates (Miyauchi et al., 2018; Bowman et al., 2019; Li et al., 2020); however, this data set encompasses a wide range of drugs, including drugs cleared by passive hepatic uptake and metabolism, such as basic and neutral ECCS class 2 compounds (Fig. 1).

Distribution of physicochemical properties of the 26 compounds represented in the PMU database. (A) Ionization and (B) ECCS group. Legend as indicated, n numbers given in Table 1.
Impact of PMU on Clearance.
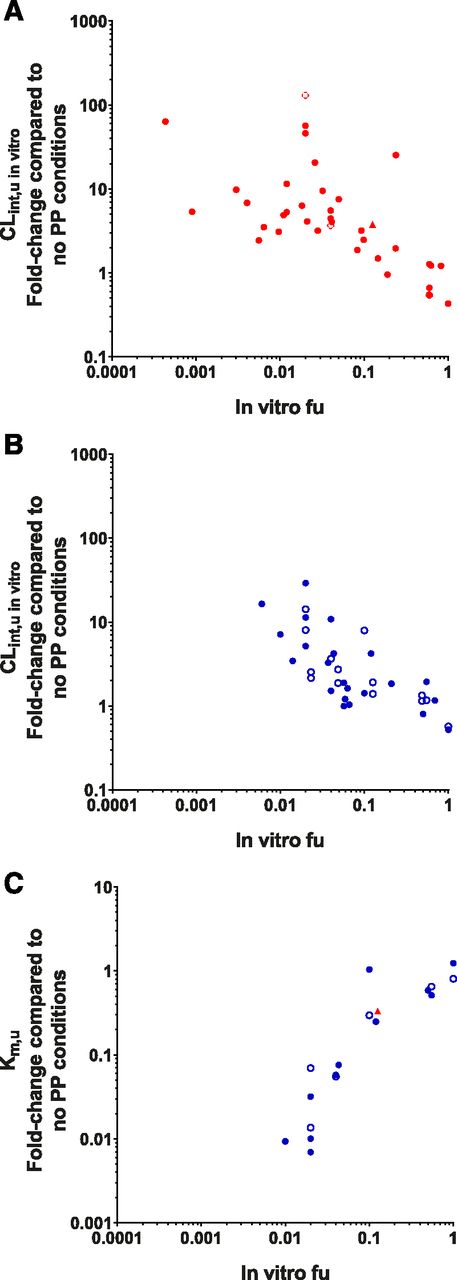
Initial analysis examined trends at the in vitro level and potential discrepancies caused by experimental conditions. Absolute CLint,u in vitro values were significantly higher in both rat and human data sets in the presence of PP compared with the absence (P = 0.0004 and P = 0.0025, respectively, see Supplemental Fig. 2 for details). Trends between fold-change in clearance parameters measured in in vitro hepatocyte systems (namely, the CLint in vitro, CLint,u in vitro, Km, Km,u, and Vmax) and the in vitro fu were investigated. A clear positive relationship between the fold-change in CLint in vitro values and in vitro fu values was observed in both human and rat hepatocyte systems (Fig. 2A). This trend is unsurprising, as lower in vitro fu values conventionally correspond to lower drug availability in vitro and ultimately lead to lower CLint, in vitro values measured. However, when the CLint in vitro was normalized to the fraction of unbound drug available at equilibrium (CLint,u in vitro), a clear negative trend was observed for both human and rat hepatocytes, with greater fold-change in the CLint,u in vitro observed for drugs that possess lower in vitro fu values (Fig. 2B). As expected for these data, this trend appears to contradict the assumption of the FDH; if only free drug could be cleared by hepatocytes, then CLint,u in vitro values should be unaffected by the presence of PP, and no fold-change in this parameter would be observed. This null hypothesis (in which CLint,u in vitro is unaffected by the presence of protein, and the slope would equal zero) was rejected with the presented (rat and human) data set [F(1,72) = 10.2, P = 0.0021]. In addition to confirming the PMU phenomenon, this finding highlights that highly bound drugs may be more significantly affected by PMU than drugs with low binding. No significant difference between the rat and human data sets was observed for this trend [F(2,70) = 0.8858, P = 0.417], demonstrating that the relationship between the fold-change in CLint,u in vitro and in vitro fu values is species-independent. However, higher variability in this trend was evident in human hepatocytes compared with rat and increased with lower in vitro fu values. This is likely to be due to inherent variability observed between human donors (Wood et al., 2017) in addition to sensitivity limitations in the quantification of low fu values.

General trends of the PMU database. Examining trends between in vitro fu values and (A) fold-change in CLint in vitro, (B) fold-change in CLint,u in vitro, (C) fold-change in Km, and (D) fold-change in Km,u. Red and blue indicate values obtained from human and rat hepatocytes, respectively.
When data were available, the fold-change in Km (n = 30), Km,u (n = 18), and Vmax (n = 30) was also investigated. No clear trends between the fold-change in the Km were observed in relation to the in vitro fu; however, a clear positive trend between the in vitro fu value and fold-change in Km,u was observed (Fig. 2, C and D, respectively). This observation is based entirely on rat data, so this relationship cannot currently be confirmed for human hepatocytes. There was some evidence of a positive relationship between the fold-change in Vmax and in vitro fu values in rat hepatocytes, but this trend was not observed within the human data set with a different subset of drugs (Supplemental Fig. 4).
Impact of Experimental Conditions Employed In Vitro.
Because of the diverse nature of experimental conditions used within this database, it was investigated whether any of the in vitro conditions themselves could influence the fold-change observed and, thus, whether this would lead to any bias in the interpretation of the effect of PP. Firstly, the hepatocyte assay format (suspension, monolayer, or liver slice) was investigated (Fig. 3). The majority of data using human hepatocytes was obtained using the suspension assay format, so no statistical comparisons could be made within the human data set (Fig. 3A). In contrast, both suspension and monolayer assays were used throughout the rat data set, but no statistical differences were observed in either the fold-change in CLint,u in vitro or Km,u values (Fig. 3, B and C, respectively). This suggests that the observed trends between in vitro fu and fold-change in CLint,u in vitro and Km,u values are independent of assay format.

Comparison of assay format on the CLint,u in vitro fold-change in (A) human (red) and (B) rat (blue) and (C) the Km,u fold-change in rat and human observed. Closed circles, suspension; open circles, monolayer; triangle, liver slice. (A) Human suspension, n = 34; human monolayer, n = 3; human liver slice, n = 1. (B) Rat suspension, n = 22; rat monolayer, n = 14. (C) Rat suspension, n = 11; rat monolayer, n = 6; human liver slice, n = 1.
It has previously been suggested that the UWL adjacent to hepatocytes is a key limitation to the clearance of high-permeability compounds and that albumin may enhance clearance of high-permeability compounds by promoting drug diffusion through the UWL (Ichikawa et al., 1992; Wood et al., 2018). Therefore, it was subsequently investigated whether performing the suspension assay under static or shaken conditions would lead to differences in the fold-change observed in clearance parameters. The experimental details on the use of shaking (and shaking speed) were included in the PMU database (Supplemental Table 1). For a number of studies, the use of shaking during drug incubation was not documented. This might imply that these assays were performed under static conditions, but given the indeterminate conditions, these data should be interpreted with caution.
Comparison of the confirmed or unknown shaking conditions in suspension assay formats on fold-change in CLint,u in vitro values is shown in Fig. 4 (fold-change in Km,u values was not included due to insufficient data). No statistical differences between confirmed or unknown shaking conditions were observed in the rat or human data sets (Fig. 4, A and B). The higher variability in the human data set previously observed appears to be attributed to the unknown shaking conditions (possibly reflecting a mixture of shaking conditions). This high variability likely limits the power of such testing, as otherwise the unknown shaking conditions appear to be associated with generally lower PMU effect. Omission of this unknown shaking condition resulted in CLint,u in vitro fold-change that was highly similar across both species (Fig. 4C).

Comparison of shaking conditions in the suspension assay on the CLint,u in vitro fold-change observed. (A) Human (red), (B) rat (blue), and (C) rat and human data from only assays with confirmed shaking. Closed circles represent experiments performed with confirmed shaking conditions; open circles represent experiments performed under unknown/unconfirmed shaking conditions. Rat shaking, n = 10; rat unknown shaking, n = 12; human shaking, n = 17; human unknown shaking, n = 17.
The final experimental variable investigated was the source of PP used in the in vitro studies. It is plausible that for drugs that bind to plasma proteins or lipoproteins other than albumin [e.g., α1-acid-glycoprotein 1 (AAG)], the specificity of protein used (e.g., whole plasma or isolated albumin) may lead to significant differences in the observed fold-change of clearance parameters. Furthermore, if albumin enhances the uptake of drugs via an albumin receptor mechanism (Weisiger et al., 1981) or albumin-specific interaction with the hepatocyte plasma membrane, it is possible that albumin from different species [human serum albumin (HSA) or bovine serum albumin (BSA), respectively] may also lead to differences in the fold-change observed. Within the database, five different types of PP were used—namely, BSA, HSA, human plasma, human serum, and rat serum. In human hepatocytes (Fig. 5A), no significant difference in the relationship of the fold-change in CLint,u in vitro values and in vitro fu values was observed between the various types of PP used in in vitro studies. It is noted, however, that this lack of significant difference is largely attributed to the high variability in the human data set. In rat hepatocytes, the type of albumin used did have a significant effect [F(4,28) = 6.957, P = 0.0005] on the relationship between the fold-change in the CLint,u in vitro values and in vitro fu values observed (Fig. 5B; full statistical details are displayed in Supplemental Fig. 3). Studies performed with rat serum appeared to show the greatest increases in fold-change in CLint,u in vitro values, whereas studies using BSA appeared to show the smallest increases in fold-change in CLint,u in vitro values. This highlights that protein type may play in important role when studying PMU in vitro; however, it should be noted that each protein subset (BSA, human plasma, and rat serum) only contains data from one or two studies per subset (albeit with multiple drugs), so it is also possible that the differences observed here may be driven by interlaboratory variation. Further studies would be required to clarify whether this observation is truly a result of differences in protein type or simply interlaboratory variation. In contrast, no difference was observed between the use of human plasma and rat serum in rat hepatocyte experiments on the relationship between the fold-change in Km,u values and in vitro fu values (Fig. 5C).

Comparison of the different types of PP used in experimental conditions. Fold-change in CLint,u in vitro compared with in vitro fu in (A) human (red) and (B) rat (blue). (C) Fold-change in Km,u compared with in vitro fu in rat. Open circles, BSA; open squares, HSA; X, human plasma; closed circles, human serum; closed squares, rat serum. (A) BSA, n = 1; HSA, n = 14; human plasma, n = 3; human serum, n = 20. (B) BSA, n = 19; human plasma, n = 4; rat serum, n = 13. (C) Human plasma, n = 4; rat serum, n = 13.
Properties of Drugs Potentially Important in PMU.
The in vitro fu value shows a clear correlation with the impact of PP on CL parameters measured in vitro. Although this strongly suggests that PP enhances unbound drug uptake beyond that determined using fu (when conventionally applying the FDH), it is important to understand the utility and implications of this in vivo. Therefore, the relationship between the fold-change in clearance parameters measured under physiologically relevant protein conditions in vitro and their fup and CLint,u in vivo values (Table 1) were examined. As this analysis focused on the implications at the in vivo level, only entries in the database that were performed under physiologically relevant albumin conditions were included [e.g., pitavastatin studies by Kim et al. (2019) that were conducted using 5% HSA were included; however, pitavastatin studies by Miyauchi et al. (2018) that were conducted at ≤1% HSA were excluded]. Such entries are annotated in Supplemental Table 1. Compounds for which no in vivo data were available were also excluded from this analysis.
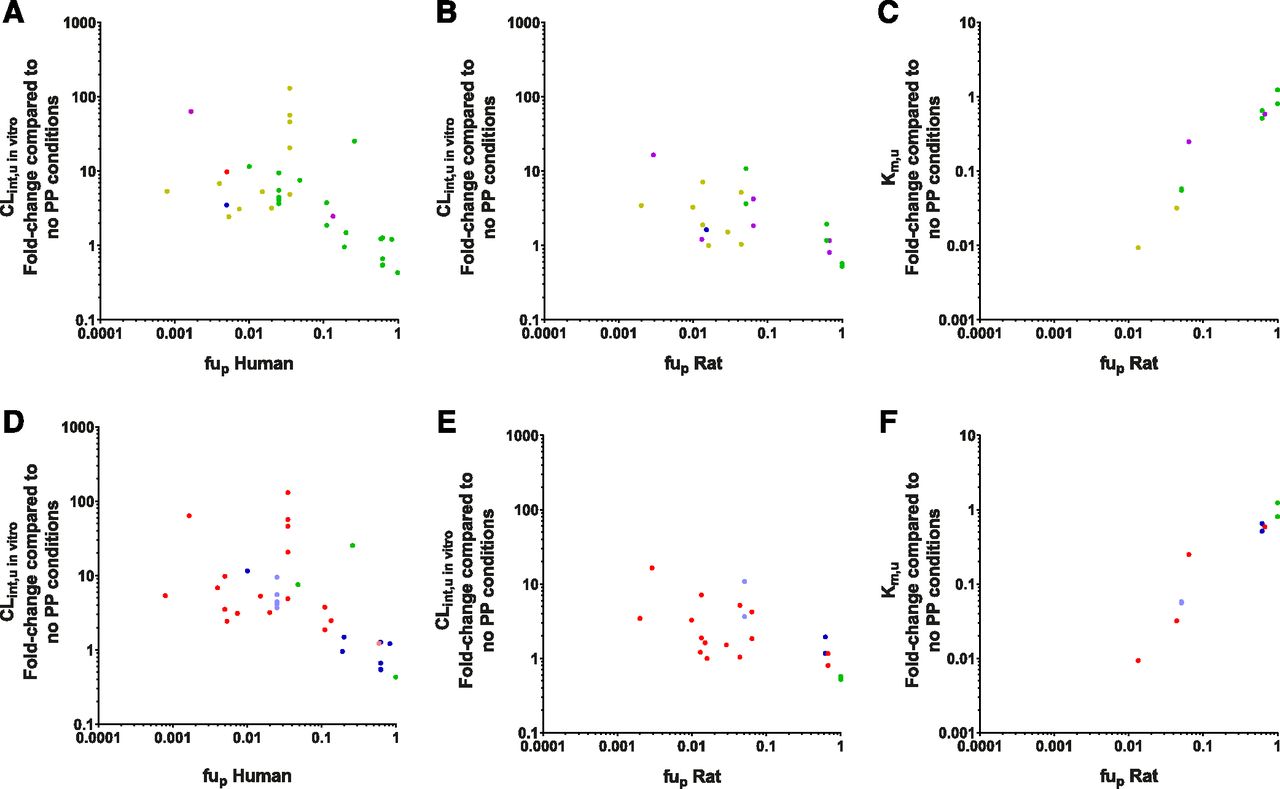
The fold-change in Km,u and CLint,u in vitro relative to their fup were investigated and segregated by species, ECCS classification, and ionization (Fig. 6). Overall, trends between fold-change in CLint,u in vitro and Km,u values were observed with fup that were similar to those observed with in vitro fu for both human and rat data. Drugs with lower fup values showed the greatest decrease in Km,u values and the greatest increase in CLint,u in vitro values. No clear differences were observed in the fold-change in CLint,u in vitro values between the rat and human data sets, although fewer data are present in the rat data set (mainly as a result of a lack of rat fup values), and greater variability was observed in the human data set. No cross-species comparison could be made for the fold-change in Km,u as a result of the negligible number of human studies.

Fold-change in in vitro clearance parameters in relation to their fup segregated according to their species and ECCS group [(A–C) 1a, red; 1b, yellow; 2, green; 3a, blue; 3b, purple) or ionization [(D–F) acid, red; weak acid, pink; neutral, green; base, blue; weak base, light blue). (A and D) Fold-change in CLint,u in vitro values from human data (ECCS: 1a = 1, 1b = 11, 2 = 19, 3a = 1, 3b = 2; ionization: acid = 17, weak acid = 1, neutral = 3, weak base = 5, base = 8). (B and E) Fold-change in CLint,u in vitro values from rat data (ECCS: 1b = 8, 2 = 10, 3a = 1, 3b = 6; ionization: acid = 15, neutral = 4, weak base = 2, base = 4). (C and F) Fold-change in Km,u values from rat data (ECCS: 1b = 2, 2 = 10, 3b = 2; ionization: acid = 4, neutral = 4, weak base = 2, base = 4).
The observed relationship between the fold-change in CLint,u in vitro and fup appears to be independent of ECCS classification in both human and rat (Fig. 6, A and B, respectively). The ECCS class 2 compounds (representing basic/neutral compounds with high permeability and predominantly cleared via metabolism), showed the clearest trend within the data, spanning fup values 0.01–1 (in human). All other ECCS classes appeared to conform to this trend, although fewer data were available for the other ECCS classes, and thus direct comparisons were difficult to assess. For high fup values (approaching 1), the fold-change in CLint,u in vitro ranged around 1, with the apparent negative effect in some cases probably reflecting experimental uncertainty (data within a 2-fold error margin). A few clear outliers of the ECCS class 1b were also noted in the human data set; in particular, the vertical spread of points at a fup of 0.035 represent different data for just one drug, bosentan, measured across multiple studies. On further investigation of these bosentan data, it was noted that the large variation in CLint,u in vitro fold-change (ranging from 5 to 130) could be accounted for by their differences in (absolute) CLint,u in vitro values measured without PP (ranging from 1.5 to 23.2 μl/min per 106 cells, Supplemental Table 1); thus, the >10-fold difference observed under the control conditions were reflected in the calculated fold-change here. In this particular case, it was difficult to ascertain whether the differences were due to interlaboratory or human donor variation, but it is nevertheless a reality for some drug cases in vitro and serves to illustrate the utility of database analysis in finding trends across multiple sources. For this reason, all bosentan (and other) data were used in the present database analysis without discrimination.
No clear differences based on ionization of the compounds were observed for either the fold-change in CLint,u in vitro or Km,u values in both rat and human (Fig. 6, D–F). Although it is clear that acidic and basic compounds are well represented in the human data set, the rat data set is marginally over-represented by acidic compounds (representing 60% of the data available), likely representing recent studies focused on the OATP transporter substrates. Within the human data set, the acidic drug bosentan is again a clear outlier due to its high variability.
Higher CLint,u in vivo compounds were shown to have the greatest fold-change in CLint,u in vitro values, implying that high metabolic turnover compounds may be more influenced by PMU. In rat, clear trends for both fold-change in Km,u (data not shown) and CLint,u in vitro values were observed in relation to their CLint,u in vivo values, both of which were independent of ECCS classification and ionization (Fig. 7, A and B). Specifically, the ECCS class 2 compounds showed this trend the clearest (across rat CLint,u in vivo values 5.4–78,300 ml/min per kg), and the ECCS class 3b agreed with this trend. The ECCS 1b compounds in this data set also appeared to agree with this trend, but because of their relatively narrow range of rat CLint,u in vivo values (1500–4000), this is stated with less certainty. This may insinuate that the PMU effect is associated with drug permeability, and thus correlations between drug-specific uptake parameters from PP-free studies (i.e., Pdiff, CLuptake, or CLactive) and fold-change in CLint,u in vitro values were also explored. No clear trends between any of the in vitro uptake parameters were observed (Supplemental Fig. 5), and thus the effect of permeability on PMU remains inconclusive.
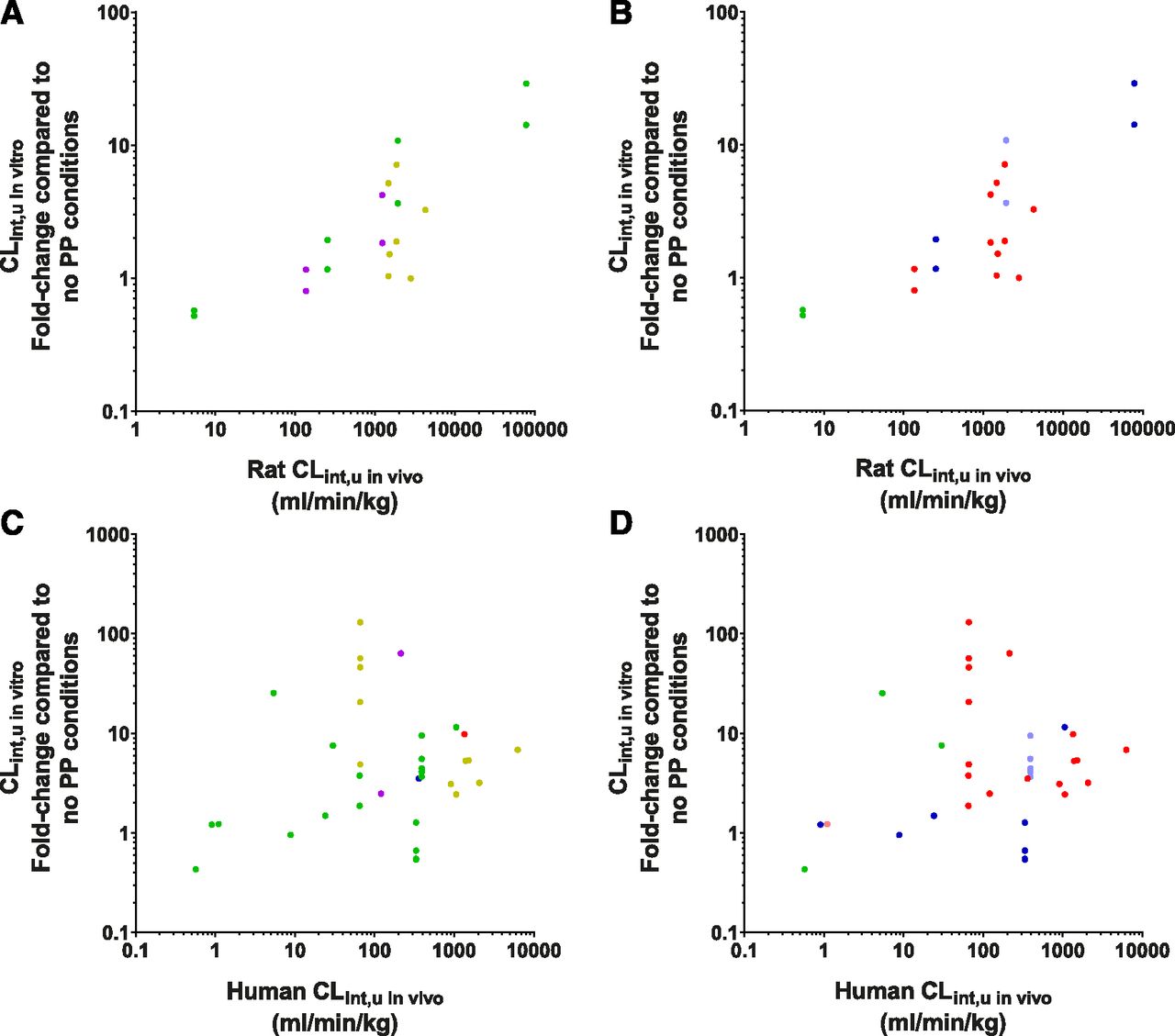
Fold-change in CLint,u in vitro in comparison with their total CLint,u in vivo, segregated by their species and ECCS group [(A and C) 1a, red; 1b, yellow; 2, green; 3a, blue; 3b, purple) or ionization [(B and D) acid, red; weak acid, pink; neutral, green; base, blue; weak base, light blue). Fold-change in CLint,u in vitro in rat [(A and B) ECCS: 1b = 8, 2 = 10, 3a = 1, 3b = 6; ionization: acid = 15, neutral = 4, weak base = 2, base = 4). Fold-change in CLint,u in vitro in human [(C and D) ECCS: 1a = 1, 1b = 11, 2 = 19, 3a = 1, 3b = 2; ionization: acid = 17, weak acid = 1, neutral = 3, weak base = 5, base = 8).
Similar trends were observed within the human data set (Fig. 7, C and D), although much higher variability was noted with a few drugs (e.g., bosentan, naloxone, and carbamazepine), noticeable as outliers to this trend.
Application of PMU to Improve IVIVE 1: PMU DataBase.
The current work has shown that the extent of PMU on CLint,u in vitro was correlated to the fu, both in vitro and in vivo (fup). This relationship appears to be independent of species, assay conditions, and drug properties, although human donor variability persists. It is important to note that this does not necessarily exclude these variables from influencing the PMU mechanism, but that the fu parameter itself may be capable of capturing, and thus accounting for, the majority of these potential effects.
Linear regression analysis was performed to quantify the identified relationship between log10-transformed fup and log10-transformed fold-change in CLint,u in vitro values caused by the addition of PP (Fig. 8). No significant difference was observed between human and rat data sets [F(2,51) = 2.996, P = 0.0589], and thus a single equation (eq. 4) was used to assess whether incorporation of predicted PMU could improve IVIVE for both species of this discrete set of drugs. Because of the relatively weak correlation (r2 for all data are 0.3), reanalysis without bosentan was performed, which yielded a stronger relationship while demonstrating no significant difference between rat and human data sets (r2 −0.46, P = 0.36, full details and statistical analysis are displayed in Supplemental Fig. 6). Therefore, despite the variability in the data, the relationship identified in Fig. 8 was used to assess whether accounting for the effect of PMU on CLint,u in vitro could improve IVIVE of hepatic CL. If the PMU phenomenon only occurs in vitro, it is still plausible that the incorporation of predicted PMU will improve prediction of CL by optimization of the CLint,u in vitro value used. In contrast, if PMU represents a true physiologic process occurring in vivo, then improvements in IVIVE would be anticipated as a result of the complete incorporation of all key processes.
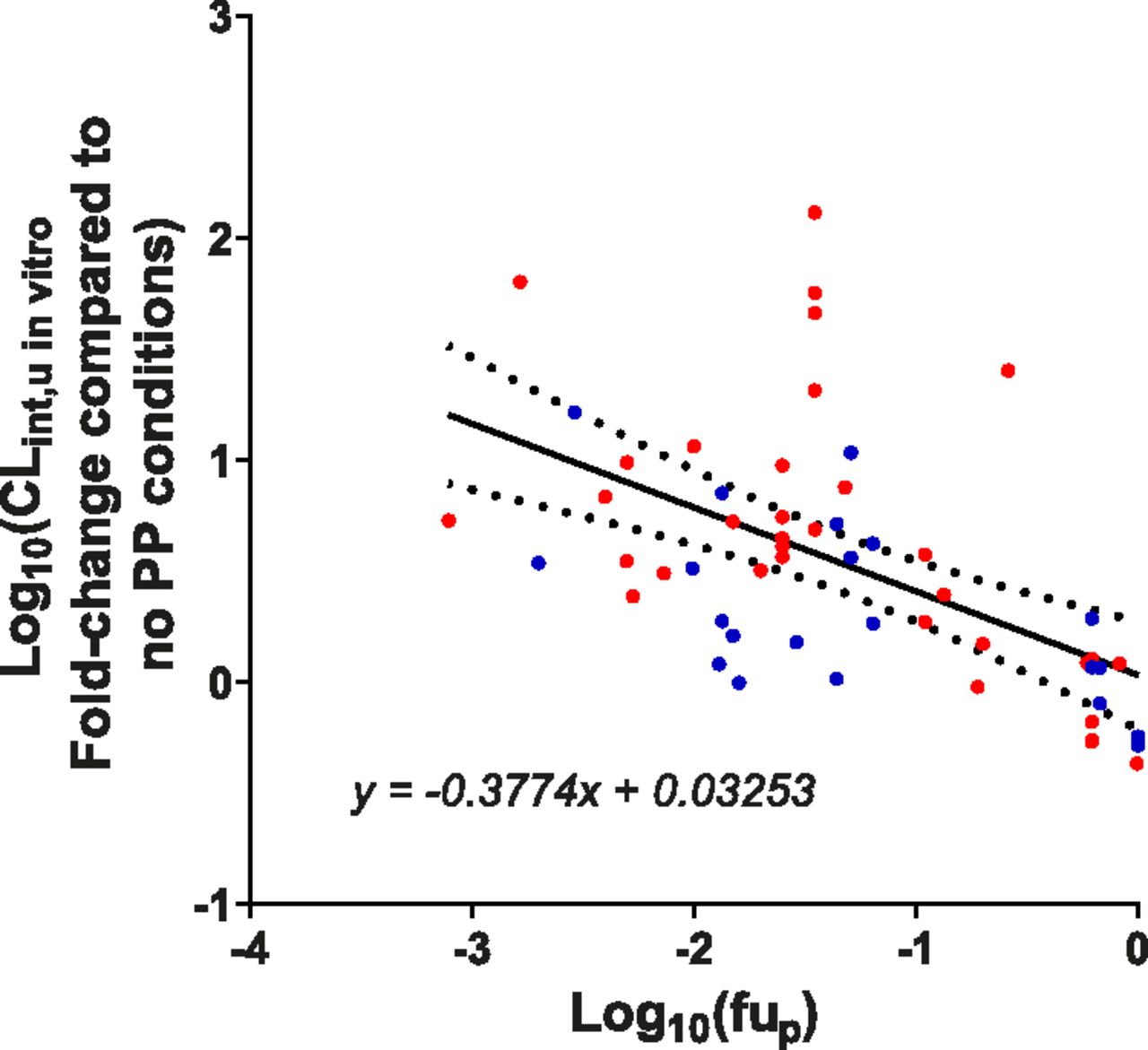
Relationship between fup and the fold-change in CLint,u in vitro caused by the addition of physiologic relevant concentrations of PP compared with no-PP conditions. Linear regression analysis was performed on the log10-transformed fup and fold-change in CLint,u in vitro data. All data (human and rat) were shown to have a significant slope [F(1,53) = 22.95, P < 0.0001]. No significant difference was observed between the human (red) and rat (blue) data sets [F(1,51) = 2.996, P = 0.0589], and thus the same equation (displayed) can be used for both species. The 95% confidence bands (dotted lines) are also displayed. r2 values for rat, human, and total data for this line are 0.1099, 0.2782, and 0.3022, respectively.
Firstly, the PMU database was used to assess the prediction of CLint,u in vivo and CLH in both rat and human based on experimental values obtained in the absence of PP, experimental values obtained in the presence of PP, and the predicted values incorporating PMU (based on eq. 4). Overall, clear improvements for rat and human were observed for both CLint,u in vivo and CLH values by either performing in vitro experiments with PP or by accounting for predicted PMU effect in comparison with data obtained in the absence of PP (Table 2). Graphical outputs of these results are shown in Supplemental Fig. 7. In the absence of PP, AFE for rat and human CLint,u in vivo were 3.02 and 3.55, respectively, and this reduced to <2 by either incorporation of predicted PMU enhancement or experimental addition of PP. For both rat and human CLint,u in vivo values, addition of PP showed no improvements to the precision of the in vivo predictions (compared with the absence of PP). However, use of the predicted PMU appeared to increase precision in rat (RMSE of 3200 compared with 25,000 in the absence of PP), although the high-CL drug mibefradil was not included in the rat PMU data set, as a rat fup could not be sourced, which may have skewed the analysis. The human CLH predictions showed similar trends, with the predictions made in the experimental presence of PP or incorporation of predicted PMU enhancement showing improvements (by AFE values) compared with the predictions made in the absence of PP. The rat CLH predictions were less conclusive, as reasonably accurate predictions were observed in the absence of PP (AFE = 1.39) for the small number of drugs analyzed within this data set (n = 13). Overall, predictions in the PMU database made with either addition of PP to experimental conditions or predicted PMU enhancement based on the identified trend in this work, showed equivalent improvements compared with predictions made in the absence of PP. This is despite the expected limitations in judging prediction based on a small data set comprising drugs with diverse CL routes [transport, metabolic, and potentially biliary (the latter, ECCS class 3b)].
IVIVE Analysis of CLint,u in vivo and CLH from the PMU database
Application of PMU to Improve IVIVE 2: Wood et al. (2017) DataBase.
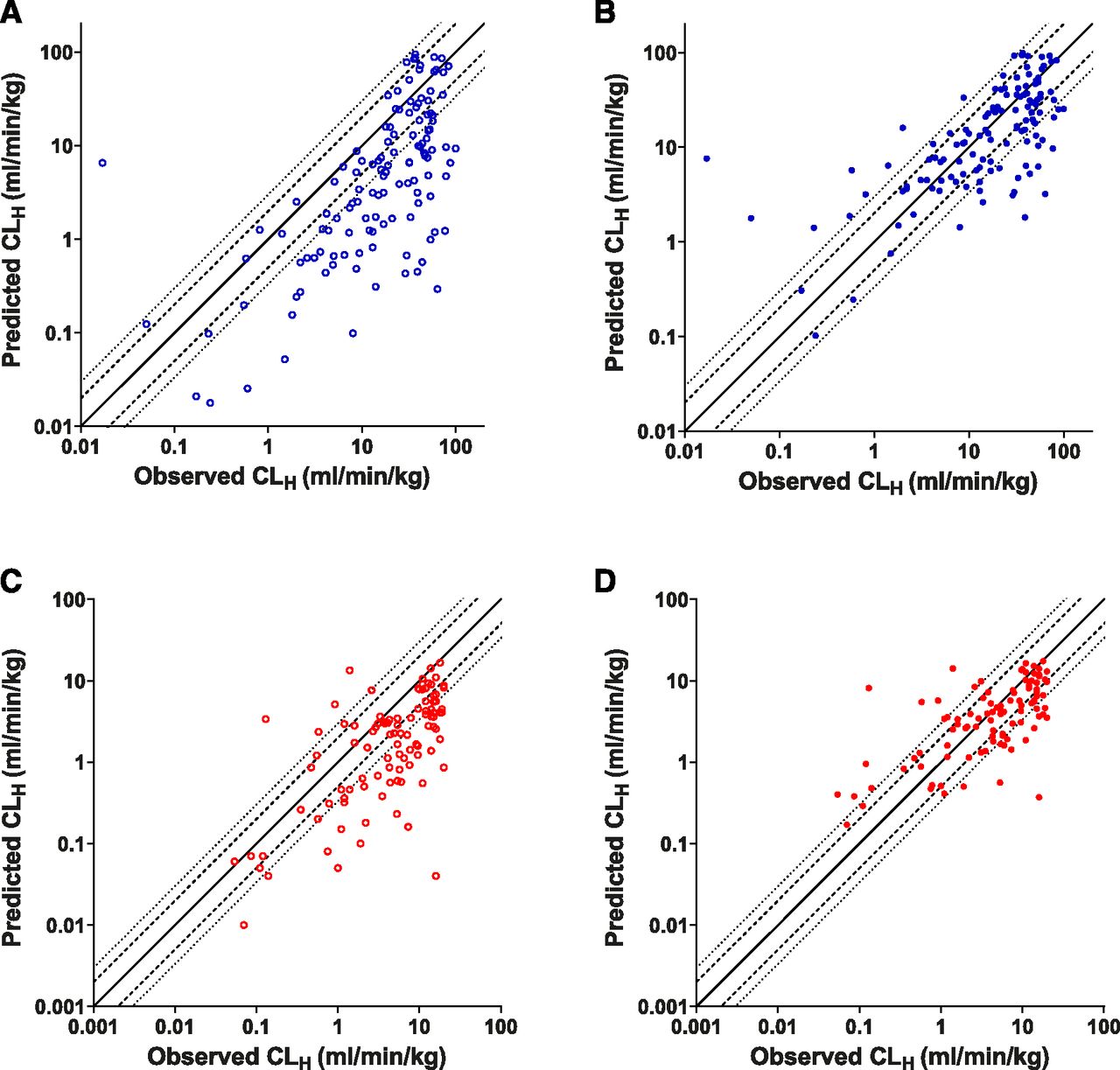
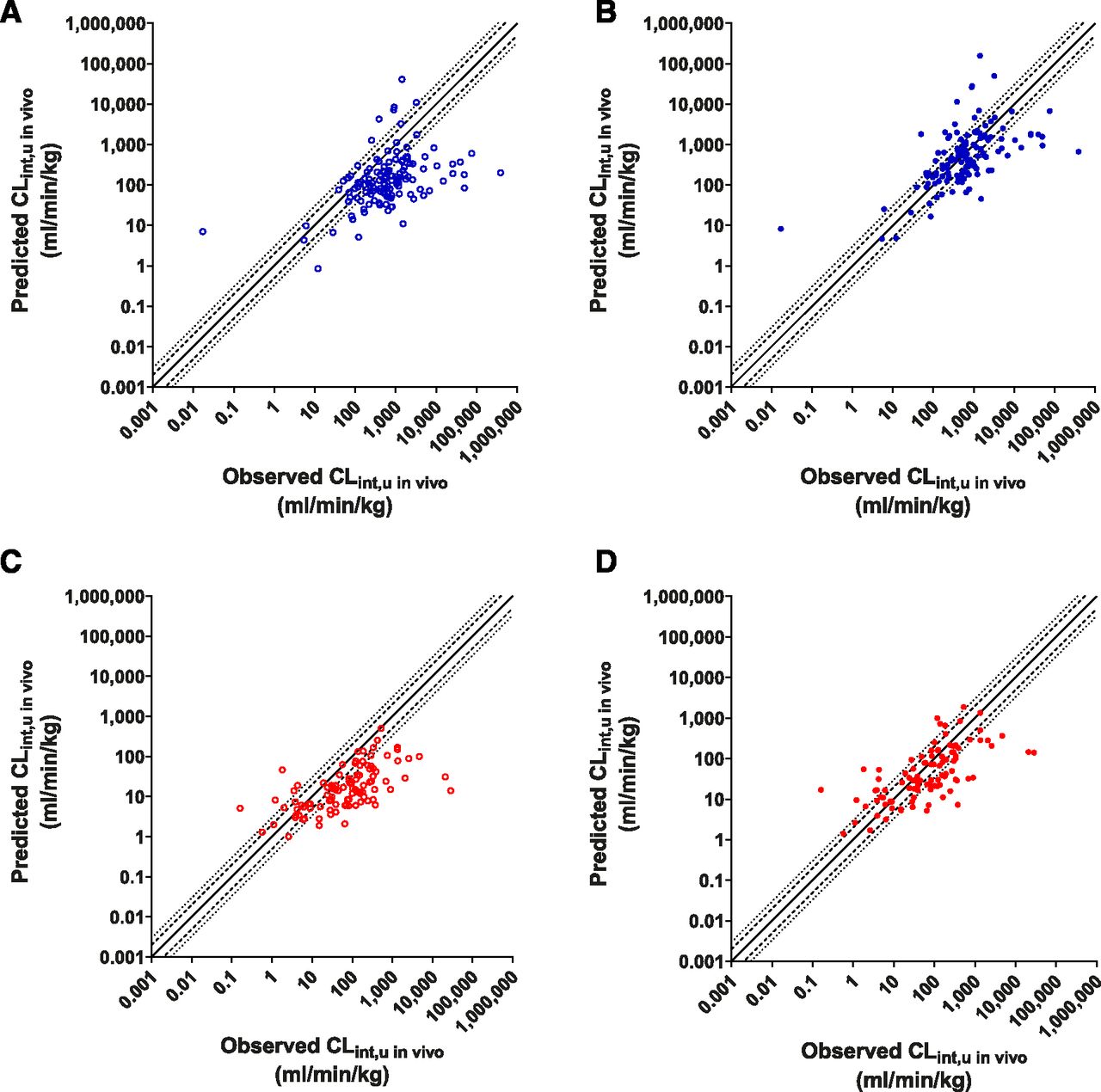
To further assess the utility of the PMU in IVIVE predictions, a broader data set, the Wood et al. (2017) database (which contains rat and human data, both n ≥ 100), was used. This data set predominantly comprised metabolically cleared drugs (ECCS class 1a and 2). Incorporation of PMU effect in both rat and human data sets showed clear improvements in CLint,u in vivo predictions (Fig. 9). Predictions incorporating PMU effect were more accurate, as demonstrated by improved AFE values (from 4.67 to 1.21, and 4.20 to 1.56 in rat and human, respectively), and greater percentage of values within the 2- and 3-fold limits (Table 3). However, no improvements in precision (similar RMSE values) were observed.

IVIVE analysis of CLint,u in vivo for compounds obtained from the Wood et al. (2017) database. Observed vs. predicted values without PMU-enhanced prediction [open circles, (A and C)] or with PMU-enhanced prediction [closed circles, (B and D)] in rat [blue, (A and B)] and human [red, (C and D)]. Solid line, dashed line, and dotted line represent unity, 2-fold, and 3-fold error.
IVIVE analysis of PMU effect in rat and human on CLint,u in vivo and CLH values using the Wood et al. (2017) database
Examining predictions for CLH, it was again observed that incorporation of PMU improved the accuracy of IVIVE predictions (Fig. 10), reducing AFE values from 3.81 to 1.17 and from 2.73 to 1.17 in rat and human, respectively. Over 50% of compounds were predicted within 2-fold, and almost 75% were predicted within 3-fold of the observed values for both species after application of PMU enhancement (Table 3). Without incorporation of PMU enhancement, <50% of CLH values were predicted within 3-fold (both species). Precision is also improved within the rat data set (RMSE decreased from 42.3 to 24.1), but only a minor improvement in precision was observed for the human data set (6.65–5.51). Some overprediction of low-clearance compounds (<1 ml/min per kg) was observed.
IVIVE analysis of CLH for compounds obtained from the Wood et al. (2017) database. Observed vs. predicted values without PMU-enhanced prediction [open circles, (A and C)] or with PMU-enhanced prediction [closed circles, (B and D)] in rat [blue, (A and B)] and human [red, (C and D)]. Solid line, dashed line, and dotted line represent unity, 2-fold, and 3-fold error.
Discussion
There is a vital need to address the shortcomings of existing IVIVE methodology for prediction of drug CL. Currently, a major yet unresolved issue is the role of PP in vitro and its apparent challenge to the conventional FDH. Although considerable compelling evidence of drug uptake into hepatocytes being dependent on bound rather than unbound drugs has accumulated, cases remain unresolved with mechanistic hypotheses and solutions that vary. The basis of the FDH is a rapidly maintained equilibrium of unbound drug in either side of a membrane in the absence of (asymmetric) energy-requiring transport processes. Although this may appear to be violated in the presence of PP (for which higher CLint,u in vitro than expected is reported), it is possible that the concentration of unbound drug at the hepatocyte surface is enhanced because of PMU, and thus the FDH would remain valid at a biochemical level while appearing violated as a result of discrepancy in fu between the hepatocyte surface and the bulk plasma. On this basis, a drug’s hepatic disposition may be affected by PMU regardless of involvement of hepatic uptake transport.
In the present study, we have unified the reported quantitative effects of PP (or isolated albumin) in hepatocyte assays of CL (reported between 1997 and 2020) into a database to facilitate global trend analysis and an improved understanding of the impact of PP across the widest range of drugs (including drugs cleared predominantly by either transport or metabolism), potentially leading to a more generic prediction approach, was sought.
Initial analysis focused on the impact of PP at the in vitro level, with the impact of PP calculated as the fold-change in CL parameters caused by the addition of PP. A distinct, inverse linear trend (spanning several orders of magnitude) was observed between the fold-change in CLint,u in vitro and in vitro fu, demonstrating a clear effect of PMU beyond that expected from conventionally determined unbound drug concentrations. This is in agreement with previous studies that also indicated that the enhancement of CLint,u in vitro by PP might be dependent upon the extent of binding (Miyauchi et al., 2018; Bowman et al., 2019). Of extra significance, there was essentially no difference in this trend between human and rat hepatocytes, supporting potentially useful cross-species equivalence. No clear trends were observed for the fold-change in Km in relation to the in vitro fu; however, Km,u showed a clear positive relationship [in agreement with Bowman et al. (2019)], suggesting that this parameter may be underestimated during conventional adjustment for unbound drug (based on equilibrium fu values).
The impact of experimental conditions (assay format, shaking, and PP type) on the observed impact of PMU in relation to in vitro fu was also investigated to assess possible experimental bias and reveal potential mechanistic insight into PMU. No clear difference was observed between monolayer and suspension assays. However, for suspension assays with confirmed shaking, there appeared to be a marginally greater effect on fold-change in CLint,u in vitro compared with shaking assays performed under indeterminate shaking conditions. This suggests a potential (at least minor) role for the UWL barrier in PMU. The type of PP used was also investigated. To date, it has not been clarified whether PMU is specifically limited to albumin or if other PPs (i.e., AAG and lipoproteins) contribute, as only albumin has been tested repeatedly, in isolation. Although no distinction of effect between PP was observed in the human studies (confounded by high variability), a possible effect of PP type was observed in the rat data set; however, further studies are required to resolve this from potential interlaboratory variability.
The impact of PP on clearance parameters in vitro was also investigated at in vivo levels of PP binding, as well as whether drug properties (ionization and ECCS group) could affect these trends. Similar to the in vitro fu relationship, trends were observed between the fold-change in CLint,u in vitro and the fup, with the greatest increase in CLint,u in vitro values correlating with lower fup. This relationship is potentially useful considering the utility of readily obtained fup in prediction methodology. The fold-change also demonstrated a positive correlation with CLint,u in vivo values, indicating that highly bound (fup < 0.1) and higher-metabolic-turnover drugs tend to be more affected by PMU (particularly evident in rat). Again, no statistical difference between human and rat data sets was observed for either of these trends, further highlighting the usefulness of such cross-species comparison (Hallifax and Houston, 2019). The clear trends in the rat data helped provide confidence in the human data, despite the higher variability of the latter [due to multiple drug-dependent factors and human donor variability, Wood et al. (2017)]. Although the correlation between CLint,u in vivo and fup could be interpreted as a coincidental consequence of lipophilicity, it might reflect modulation of CL by plasma protein. For rapidly metabolized high-permeability drugs, the rate-limiting step in the CL could be drug diffusion through the UWL (Wood et al., 2018). If the presence of PP enhances drug diffusion through the UWL, as proposed by Ichikawa et al. (1992), then the extent of binding may be a critical rate determinant for such drugs, and the ability of PP to overcome such diffusional/permeation barriers may be reflected in the enhancement of CLint,u in vitro.. Unfortunately, the trend of protein effect with absolute CLint,u in vivo could not be resolved further into uptake transport or metabolic CL because of the lack of specific data.
The relationship between fold-change in CLint,u in vitro and fu (in vitro and in vivo) appeared to be independent of ionization or ECCS group, suggesting that PMU involves a more general drug uptake process applicable across both passive and active uptake mechanisms. The majority of drugs studied were either ECCS class 1b or 2; the acidic class 1b drugs were generally more highly bound, and the basic or neutral class 2 drugs were less so, overlapping at about fup = 0.2 (in human). This continuum of PMU effect therefore clearly spanned drug type and, potentially, different responsible proteins. Basic drugs bind to albumin to a much lesser extent than acids, which, combined with their more avid binding to AAG, suggests that basic drugs would not share the same effect with acids, proportionally, if the mechanism relied on dissociation facilitated by an interaction of drug-protein complex with the hepatocyte plasma membrane, as has been explored with respect to albumin and acidic compounds (Stremmel et al., 1983; Miyauchi et al., 2018). This further highlights that future in vitro studies with various types of PP are required to improve our mechanistic understanding of PMU for acids and bases. Despite the mechanistic uncertainty, the fup parameter may adequately account for differences in the binding of acidic and basic drugs to PP in prospective prediction of PMU enhancement of CL.
Previous IVIVE assessment has suggested that the underprediction of CL is related to the extent of binding to PP (Ring et al., 2011; Bowman and Benet, 2016)—a possibility that might now be interpreted as a consequence of the PMU effect. To improve IVIVE, addition of PP into in vitro CL assays (Shibata et al., 2000; Blanchard et al., 2004, 2005, 2006; Li et al., 2020), determination of unbound drug Kp in hepatocytes in the presence of PP (Li et al., 2020; Riccardi et al., 2020), or semiempirically predicting the PMU effect based on an apparent shift in fu between plasma and interstitial fluid (fup-adjusted) has previously been suggested (Poulin and Haddad, 2015). Although all methods reported improved IVIVE, determination of unbound Kp in hepatocytes requires additional experimentation, incorporation of fup-adjusted has presently only been justified for acidic compounds, and the addition of PP in vitro has the disadvantage of reducing the total CLint in vitro measured, making reliable quantification of highly bound and/or low-CL compounds challenging. The current work has shown that the PMU effect may be predicted using fup (independent of drug type), specific PP, and hepatocyte species; hence, CLint,u in vitro measured in the absence of PP can be converted to a PMU-corrected value by applying the formula in eq. 4. There is, however, considerable variability in the relationship between PMU effect and fu, which indicates that some procedural (e.g., protein type) or drug-specific factors are influential if as yet unresolved among available data. Nevertheless, the application of predicted PMU effect within the PMU database (n = 26) appears to improve IVIVE predictions to a similar extent to those predictions performed directly in the presence of PP, suggesting that the identified relationship accurately captures the observed PMU enhancement and that it is an appropriate tool to predict PMU for CL assays performed in the absence of PP. This proposition was subsequently tested with an unrelated data set [Wood et al. (2017), n ≥ 100]. Clear improvements were observed in the IVIVE for both rat and human subsets (regardless of hepatic model used, Supplemental Fig. 1), achieving an AFE <1.2, and over 50% of compounds predicted within 2-fold of their observed values, demonstrating the utility of the relationship identified in this study. Although incorporation of this PMU prediction step effectively eliminates bias, there remains considerable imprecision in prediction, and further experimental effort to understand the variability between, for example, protein type or the impact of shaking in vitro is warranted. Beyond this, drug-specific factors such as rate-limiting elimination pathways may need more detailed assessment. Nevertheless, considering the lack of bias using this relationship for IVIVE and the occurrence of PMU in numerous IPL studies, PMU might be considered an in vivo as well as in vitro phenomenon.
In this study, we have demonstrated a generic relationship between fu and fold-change in CLint,u in vitro (for both uptake transport and metabolically cleared drugs) caused by the addition of PP, despite numerous potential underlying mechanisms. This offers a practical benefit for prospective prediction of CL across a broad range of drug uptake and clearance mechanisms. A simple empirical correction for PMU based on independently determined equilibrium fup can be applied to a conventional hepatocyte assay (rat or human) without the need to add PP, thereby avoiding compromising CL assay sensitivity.
Acknowledgments
The authors would like to thank Leon Aarons for useful discussion on the data analysis.
Authorship Contributions
Participated in research design: Francis, Houston, Hallifax.
Performed data analysis: Francis, Hallifax.
Wrote or contributed to the writing of the manuscript: Francis, Houston, Hallifax.
Footnotes
- Received October 23, 2020.
- Accepted December 7, 2020.
This work was supported by the Centre for Applied Pharmacokinetic Research consortium membership, which included Certara, Genentech, Johnson & Johnson, Lilly, Merck, and Takeda.
The authors declare no conflict of interest.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AAG
- α1-acid-glycoporotein 1
- AFE
- average fold error
- ANS
- 1-anilino-8-naphthalene sulfonate
- BSA
- bovine serum albumin
- CL
- clearance
- CLH
- hepatic clearance
- CLint in vitro
- intrinsic clearance in vitro
- CLint,u in vitro
- unbound intrinsic clearance in vitro
- CLint,u in vivo
- unbound intrinsic clearance in vivo
- ECCS
- extended clearance classification system
- FDH
- free drug hypothesis
- fu
- fraction unbound
- fub
- fraction unbound in blood
- fup
- fraction unbound in plasma
- HSA
- human serum albumin
- IPL
- isolated perfused liver
- IVIVE
- in vitro-in vivo extrapolation
- OATP
- organic anion transporting polypeptide
- PMU
- plasma protein–mediated uptake
- PP
- plasma protein
- RMSE
- root-mean square error
- UWL
- unstirred water layer
- WSLM
- well stirred liver model
- Copyright © 2021 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}