


Abstract
Fevipiprant, an oral, nonsteroidal, highly selective, reversible, and competitive prostaglandin D2 receptor 2 antagonist, is eliminated by glucuronidation and by direct renal excretion predominantly via organic anion transporter (OAT) 3. This study aimed to assess the effect of simultaneous UDP-glucuronosyltransferase (UGT) and OAT3 inhibition by probenecid on the pharmacokinetics of fevipiprant and its acyl glucuronide (AG) metabolite to support the dosing recommendation of fevipiprant in the presence of drugs inhibiting these pathways; however, phase III clinical trial results did not support its submission. This was a single-center, open-label, single-sequence, two-period crossover study in healthy subjects. Liquid chromatography with tandem mass spectrometry was used to measure concentrations of fevipiprant and its AG metabolite in plasma and urine. In the presence of probenecid, the mean maximum concentrations of fevipiprant increased approximately 1.7-fold, and the area under the concentration-time curve in plasma increased approximately 2.5-fold, whereas the mean apparent volume of distribution and the AG metabolite:fevipiprant ratio decreased. The apparent systemic clearance decreased by approximately 60% and the renal clearance decreased by approximately 88% in the presence of probenecid. Using these data and those from previous studies, the relative contribution of OAT and UGT inhibition to the overall effect of probenecid was estimated. Furthermore, a general disposition scheme for fevipiprant was developed, showing how a perpetrator drug such as probenecid, which interferes with two key elimination pathways of fevipiprant, causes only a moderate increase in exposure and allows estimation of the drug-drug inhibition when only one of the two pathways is inhibited.
Significance Statement In this drug-drug interaction (DDI) study, probenecid was used as a tool to inhibit both glucuronidation and active renal secretion of fevipiprant. The combination of plasma and urine pharmacokinetic data from this study with available data allowed the development of a quantitative scheme to describe the fate of fevipiprant in the body, illustrating why the DDI effect on fevipiprant is weak-to-moderate even if a perpetrator drug inhibits several elimination pathways.
Introduction
Fevipiprant is an oral, nonsteroidal, highly selective, reversible, and competitive antagonist of the prostaglandin D2 receptor 2 (DP2) (Sykes et al., 2016). The DP2 receptor, a G-protein–coupled receptor, is an important regulator of the inflammatory cascade with a key role in the pathophysiology of asthma (Domingo et al., 2018). Fevipiprant selectively antagonizes the DP2 receptor, thereby targeting and reducing DP2 receptor–mediated inflammation in the airways of people with asthma (Erpenbeck et al., 2016a; Gonem et al., 2016; Bateman et al., 2017); however, phase III clinical trial results did not support submission in this indication (Brightling et al., 2021).
Elimination of fevipiprant occurs via glucuronidation as well as by direct renal and possible biliary excretion (Pearson et al., 2017). Fevipiprant is metabolized to an acyl glucuronide (AG) metabolite (1-O-β form, which can rearrange to isomers), representing the only relevant metabolite in systemic circulation and excreta that is not pharmacologically active. Data from clinical mass balance and drug-drug interaction (DDI) studies revealed that hepatic and renal clearance contribute to the total systemic elimination of fevipiprant and that renal clearance involves an active secretion process (Pearson et al., 2017; Weiss et al., 2020). Because the contributions of both glucuronidation and renal excretion exceed 25% of the clearance of fevipiprant, clinical studies are recommended by health authority guidelines to study the DDI risk in humans (Zhang et al., 2009; European Medicines Agency, 2013; Center for Drug Evaluation and Research (CDER), 2020).
Fevipiprant was tested in vitro as a substrate of the major human UDP-glucuronosyltransferase (UGT) enzymes and drug transporters (Pearson et al., 2017). Fevipiprant is a substrate of the organic anion transporter (OAT) 3, P-glycoprotein (P-gp), and organic anion–transporting polypeptide (OATP) 1B3, and its metabolism is mediated by the human UGT enzymes UGT1A3, UGT2B7, and UGT2B17 (Pearson et al., 2017).
A previous DDI study showed that coadministration of cyclosporine, which is an inhibitor of OATP1B3 and P-gp, increased oral fevipiprant 150 mg area under the curve (AUC) by 2.5-fold and Cmax by 3-fold (Weiss et al., 2020). Our study investigates the effect of inhibition of the other relevant clearance pathways of fevipiprant by probenecid (i.e., metabolism by UGTs and OAT3-mediated renal clearance). OAT3 is expressed in proximal kidney tubule cells and plays an important role in the active secretion of low-permeable anionic compounds (Wang and Sweet, 2013). UGT1A3 and UGT2B7 are expressed in the liver; UGT2B7 and UGT2B17 are reported to have an important role in the intestine (Strassburg et al., 2001; Kiang et al., 2005; Zhang et al., 2018).
Probenecid was used as a nonselective inhibitor of UGTs (Uchaipichat et al., 2004) and is recommended by the Food and Drug Administration to assess sensitivity to DDI for drugs that are metabolized by several UGTs, such as fevipiprant. It is also recommended as an index perpetrator of OAT1 and OAT3 (FDA Center for Drug Evaluation and Research, 2020). The purpose of this study was to assess the effect of concurrent UGT and OAT3 inhibition by probenecid on the pharmacokinetics (PK) of fevipiprant. The assessment of the systemic PK as well as urinary excretion of both fevipiprant and its pharmacologically inactive AG metabolite allows for some distinction of the metabolic and renal effects of probenecid. The results are discussed further in the context of existing in vitro data and clinical results from human mass balance and DDI studies to establish an overall quantitative understanding of the disposition of fevipiprant.
Materials and Methods
Regulatory and Ethical Compliance.
The study protocol was reviewed by the Institutional Review Board, and the study was conducted according to the ethical principles of the Declaration of Helsinki. Informed consent was obtained from each subject in writing before any study-specific procedures took place.
Study Objectives.
The primary objective of the study was to determine the effect of probenecid 1000 mg twice daily on the key PK parameters of fevipiprant 150 mg in healthy subjects. The secondary objectives were to assess the safety and tolerability of fevipiprant with and without coadministration of probenecid and to determine the effect of probenecid on the PK of the AG metabolite of fevipiprant.
Study Design.
This was a single-center, open-label, single-sequence, two-period crossover study in healthy subjects. The study consisted of a 28-day screening period with two baseline visits and two treatment periods, which were separated by a washout of 7–14 days, and an end-of-study assessment (Fig. 1). In treatment period 1, subjects received a single oral fevipiprant dose of 150 mg on day 1, and this was followed by collection of reference plasma PK samples over 96 hours and urine collection over 24 hours. In treatment period 2, subjects received oral doses of probenecid 1000 mg twice daily on days 16–21 and a single oral fevipiprant dose of 150 mg (which was followed by collection of plasma PK samples over 96 hours and urine over 24 hours) together with the probenecid morning dose on day 18.
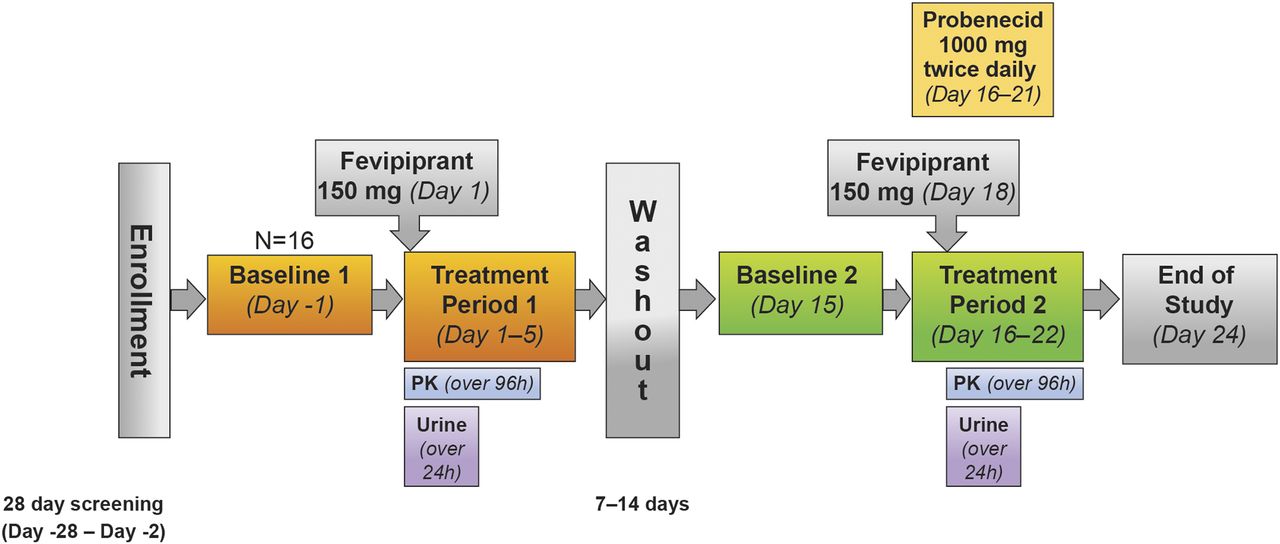
Study design.
Key Inclusion and Exclusion Criteria.
Healthy men and women aged 18–55 years weighing between 50 and 90 kg were eligible to participate in the study and were required to have a body mass index within the range of 18–30 kg/m2. Written informed consent was obtained before any assessment was carried out. Subjects using any prescription drugs (with the exception of oral or injectable contraceptives) were excluded from the study.
Pregnant or nursing women and those who smoked were excluded. Those with a history of clinically significant ECG abnormalities or history or presence of long QT syndrome were also excluded from the study. Full inclusion and exclusion criteria may be found in the online data supplement.
Pharmacokinetic Analyses.
Blood samples were collected in K3EDTA (anticoagulant) collection tubes. Because the AG metabolite is unstable at the physiologic pH of plasma and urine (pH > 6.8), both urine and plasma samples were transferred after collection to commercially available Vacuette tubes (FC) mixture (catalog number 454513; Greiner) for stabilization of the AG metabolite. Further details may be found in the supplement.
The concentrations of fevipiprant and its AG metabolite in plasma and urine were measured using a validated liquid chromatography with tandem mass spectrometry combo method (simultaneous quantification of both analytes with same injection), which consisted of a robotized supported liquid extraction using Isolute SLE+ 200-µl, 96-well plates for extraction plasma samples and liquid-liquid extraction for urine samples, respectively, which was followed by reverse-phase liquid chromatography with tandem mass spectrometry using ElectroSpray Ionization in the positive ion mode (Erpenbeck et al., 2016b). The plasma method was suitable for quantification of fevipiprant in the range of 1–400 ng/ml and in the range of 0.48–192 ng/ml for the AG metabolite using 50 μl plasma. The method for urine could quantify fevipiprant in the range of 0.2–80.0 µg/ml and its AG metabolite in the range of 0.096–38.4 µg/ml using 50 µl urine.
The bioanalytical method for probenecid consisted of protein precipitation followed by reverse-phase liquid chromatography with tandem mass spectrometric detection using ElectroSpray Ionization in the negative ion mode. The method was suitable for the determination of probenecid in human blood over the range of 1.00 (lower limit of quantitation) to 400 μg/ml using 50 μl of human blood (further details may be found in the supplement).
Fevipiprant PK parameters were determined using the actual recorded sampling times and noncompartmental method(s) with Phoenix WinNonlin (Version 6.4 or higher) from the plasma and urine concentration data.
Key Safety and Tolerability Assessments.
All adverse events (AEs) and serious adverse events with their severity and relationship to study drug were collected. Evaluations included safety assessments (hematology, clinical chemistry, and urinalysis), pregnancy and assessments of fertility, and regular assessments of vital signs, physical condition, and body weight as well as a standard 12-lead ECG.
Statistical Analyses.
To assess the effect of probenecid on the PK of fevipiprant, log-transformed primary plasma/urine PK parameters of fevipiprant were analyzed using a mixed-effects model with a fixed effect for treatment (fevipiprant plus probenecid vs. fevipiprant alone) and a random effect for subject. Least-squares mean differences between treatments and associated 90% confidence intervals (CIs) in the logarithmic scale were back-transformed to produce geometric mean ratio and associated 90% CIs for each PK parameter.
Results
Demographics.
In total, 16 subjects entered the study, all of whom completed treatment period 1 and were included in the safety and PK analysis sets. All subjects except one who discontinued because of an adverse event completed treatment period 2. Fourteen out of sixteen subjects (87.5%) were female, and most were white (68.8%; Supplemental Table 1 provides additional demographic information). The median age was 27.5 (range, 19–55) years; mean weight was 68.3 ± 10.4 kg; and mean body mass index was 24.2 ± 2.44 kg/m2 (Supplemental Table 1).
Pharmacokinetics of Fevipiprant Administered with and without Probenecid.
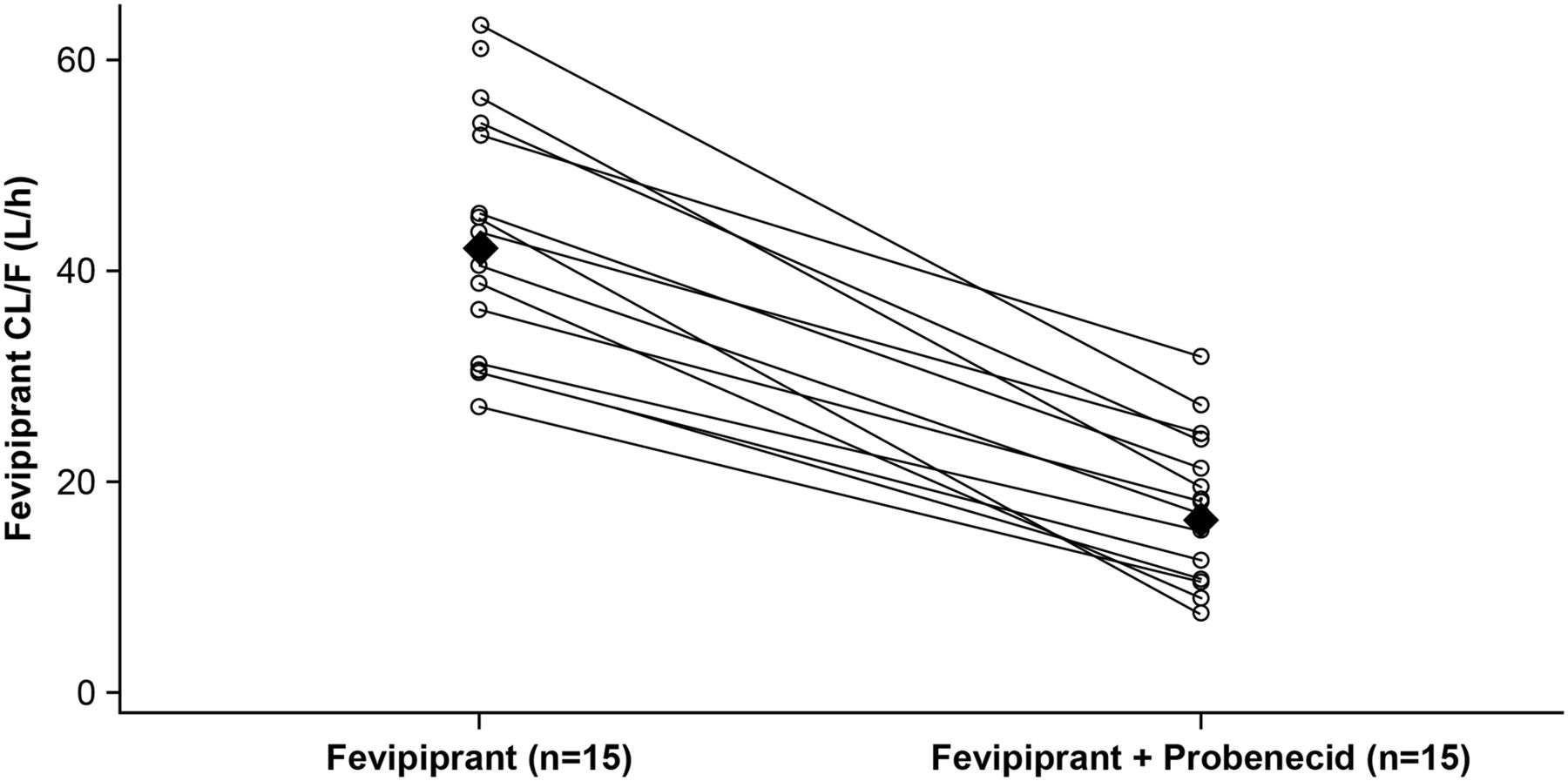
Coadministration of probenecid increased mean fevipiprant plasma concentrations (Fig. 2a); mean concentration-time data for fevipiprant and its AG metabolite with and without probenecid are provided in Supplemental Table 2. PK parameters for fevipiprant with and without probenecid are summarized in Table 1, and corresponding statistical analysis is in Table 2. Fevipiprant mean Cmax increased approximately 1.7-fold, and AUClast and AUCinf increased approximately 2.5-fold in the presence of probenecid (Fig. 2a; Table 2). The mean Vz/F decreased from 1470 to 600 l. Geometric mean ratios showed a decrease in CL/F of 25.8 l/h (a reduction of approximately 60% with reduction observed across all subjects, Fig. 3) and a decrease in renal clearance (CLr) of 8.5 l/h (a reduction of 88%) in the presence of probenecid (Fig. 2; Table 2). The mean amount of fevipiprant excreted in urine (Ae0–24 h) decreased from 27.7 ± 8.57 mg (approximately 19% of the dose) for fevipiprant treatment alone to 7.08 ± 2.15 mg (approximately 5% of the dose) for treatment with fevipiprant plus probenecid (Table 1).

(A) Arithmetic mean (± S.D.) plasma concentration of fevipiprant 150 mg once daily in the presence (black triangles) and absence (black circles) of probenecid over time (0–24 h) on a linear scale. Data presented on a semilogarithmic scale (0–96 h) are shown as an inset. (B) Arithmetic mean plasma (± S.D.) concentration of the AG metabolite of fevipiprant in the presence (black triangles) and absence (black circles) of probenecid over time (0–24 h) on linear scale. Data presented on a semilogarithmic scale (0–96 h) are shown as an inset.
Summary of pharmacokinetic parameters for fevipiprant and AG metabolite by treatment
Data are arithmetic mean ± S.D. (CV%) [n]. CV% = sqrt(exp(variance for log-transformed data)−1) × 100.
Geometric mean ratio (90% confidence intervals) for fevipiprant 150 mg pharmacokinetic parameters with and without probenecid 1000 mg twice daily (pharmacokinetic analysis set)
Mixed-effects model with a fixed effect for treatment and a random effect for subject. Subjects with missing PK parameters for any treatment but not all treatments are included in the analysis assuming “missing” as random. Geometric mean ratios and 90% CI are back-transformed from log scale.
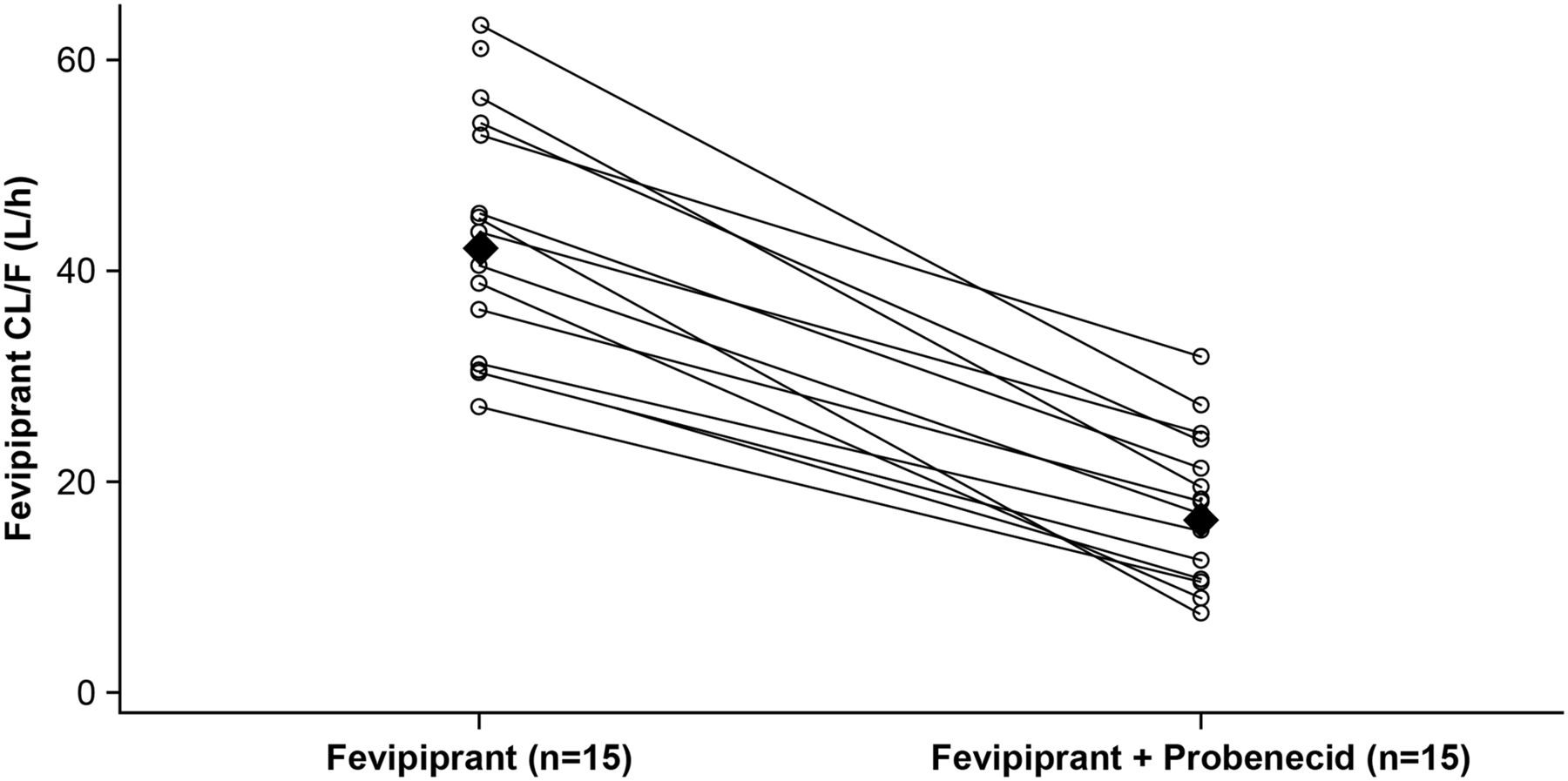
Fevipiprant individual and geometric mean CL/F by treatment. Individual subjects are shown as an open circle with a line connection for fevipiprant (day 1) and fevipiprant plus probenecid (day 18) values. Geometric mean values are represented by closed diamonds.
Pharmacokinetics of Fevipiprant AG Metabolite Administered with and without Probenecid.
Although coadministration of probenecid is expected to reduce glucuronidation of fevipiprant, it resulted in an increased plasma exposure to the metabolite (Table 1). However, the metabolite-to-parent drug ratios (MRs) (AG metabolite to fevipiprant) for maximum concentration (MR Cmax) and overall exposure (MR AUClast and MR AUCinf) decreased in the presence of probenecid (Table 1). The decrease in the MR for systemic exposure suggests a reduced rate of metabolite formation resulting from reduced UGT activity in presence of probenecid. The net increase in exposure to the metabolite results from the 4.3-fold decrease in CLr of the AG metabolite by probenecid (Fig. 2b; Table 1). The mean amount of the AG metabolite excreted in urine (Ae0–24 h) decreased from 36.5 ± 9.62 mg (approximately 17% of the dose considering difference in molecular weight) with fevipiprant alone to 13.0 ± 5.80 mg (approximately 6% of the dose) when probenecid was coadministered.
Adverse Events.
Headache was the most frequently reported AE (see Supplemental Table 3 for all adverse events). Two AEs (postural dizziness, headache) reported for one (6.3%) subject were considered to be related to both fevipiprant and probenecid. Eight AEs reported for four (25.0%) subjects were considered to be related to probenecid. All reported AEs were mild in severity (Supplemental Table 3). One subject discontinued from the study because of an AE. No deaths or serious AEs were reported.
Discussion
In this study we investigated the effect of simultaneous inhibition of UGTs and OAT3 by probenecid on the PK of fevipiprant. The inhibition of two important clearance pathways resulted in a weak (<2-fold) effect on Cmax and a moderate (2.5-fold) increase in AUC considering DDI categories also used by the health authorities (European Medicines Agency, 2013; Center for Drug Evaluation and Research (CDER), 2020). The exposure of healthy subjects to 150 mg fevipiprant either with or without coadministration of probenecid was found to be safe and generally well tolerated with no unexpected adverse events reported. This is consistent with previous safety and tolerability findings (Erpenbeck et al., 2016a,b; Gonem et al., 2016; Bateman et al., 2017).
The metabolic and renal effects of probenecid can be distinguished using the following information: plasma concentration data and the urinary excretion of both fevipiprant and its AG metabolite; complementary literature providing in vitro and absorption, distribution, metabolism, and excretion data (Pearson et al., 2017); and oral and intravenous DDI data with and without the OATP1B3 and P-gp inhibitor cyclosporine (Weiss et al., 2020). Based on this, the fractional contribution of OAT and UGT inhibition to the observed effect was estimated, and a general disposition scheme for fevipiprant was established. This information in combination with safety and efficacy data from patient trials would have provided the basis to assess the need for a dose adjustment or contraindication of fevipiprant in presence of comedications affecting its pharmacokinetics.
Renal elimination of fevipiprant involves glomerular filtration as well as OAT3-mediated active secretion. Fevipiprant has an unbound fraction (fu) in plasma of 0.118 (Pearson et al., 2017); therefore, the clearance by glomerular filtration can be estimated to be approximately 0.9 l/h (fu × GFR = 0.118 × 7.5 l/h ∼0.9 l/h), assuming a GFR of 7.5 l/h (Davies and Morris, 1993). This suggests a contribution of active secretion to the renal clearance of fevipiprant of approximately 91% (9.0 l/h, Table 3). In the presence of probenecid, the active secretion was reduced to approximately 0.3 l/h (1.2–0.9 l/h), indicating near-complete (97%) inhibition of active secretion by probenecid. The AG metabolite has an unbound fraction in plasma of 0.234, and therefore, glomerular filtration can be estimated to be approximately 1.8 l/h (fu × GFR = 0.234 × 7.5 l/h ∼1.8 l/h), indicating a contribution of active secretion to the renal clearance of approximately 75% (5.3 l/h, Table 3). In the presence of probenecid, urinary secretion decreased to a value close to the estimated glomerular filtration (1.7 vs. 1.8 l/h), indicating near-complete inhibition of active metabolite secretion by probenecid. This sensitivity to probenecid suggests that the metabolite is subject to active renal secretion likely involving transporters of the OAT family also.
Renal clearance of fevipiprant and its AG metabolite with and without probenecid
The systemic clearance (CL) of fevipiprant based on intravenous data is 19 l/h (Weiss et al., 2020), which allows an estimation of bioavailability (F) of 0.43 using the observed CL/F of 43.7 l/h in this study population. The measured CLr (9.87 l/h) accounts for half of CL (19 l/h) and is comparable to the observed CLr reported previously (9.49 l/h) (Weiss et al., 2020). Inhibition of OAT3-mediated active renal excretion by probenecid reduces CLr by 8.7 l/h (Table 1) and, consequently, CL to 10.3 l/h (19 –8.7 l/h) and CL/F to 24 l/h (10.3 l/h/0.43). Therefore, the expected exposure increase of only inhibiting renal excretion is ∼1.8-fold (43.7/24 l/h), which is lower than the observed ∼2.5-fold AUC change. Accordingly, inhibiting glucuronidation is expected to have a smaller impact (2.5/1.8 = 1.4-fold). The smaller impact of inhibiting glucuronidation is consistent with findings that the clinical effects of UGT inhibitors on the clearance of UGT substrates are generally weak (Williams et al., 2004). Taken together, inhibitors of a single pathway (OAT3 or UGTs) are expected to cause only a weak increase in AUC of fevipiprant, whereas simultaneous inhibition results in a weak-to-moderate exposure increase.
Although UGT1A3 and UGT2B7 are expressed in the liver, extrahepatic glucuronidation of fevipiprant cannot be excluded. Based on recent protein abundance data, UGT2B7 and UGT2B17 may contribute to the intestinal first pass, and both enzymes are also expressed in the kidney (Margaillan et al., 2015). Probenecid inhibits all three UGT isoforms in vitro (Uchaipichat et al., 2004), with highest potency for UGT2B7. Therefore, inhibition of the intestinal UGT2B7 and UGT2B17 could result in a reduced intestinal first-pass metabolism, which is consistent with the reduction in Vz/F seen in this study. In addition, reduced distribution of fevipiprant into the kidney in the presence of probenecid can also contribute to the reduced Vz/F.
Previous in vitro and clinical DDI data revealed that OATP1B3-mediated uptake into the liver is a key mechanism of fevipiprant systemic elimination (Weiss et al., 2020). The results from the present study further corroborate that hepatic and renal elimination both contribute approximately 50% to the total systemic clearance of fevipiprant (Fig. 4). When fevipiprant was administered together with cyclosporine or probenecid, hepatic clearance and active renal secretion, respectively, were nearly completely inhibited. Hence, the data indicate that transporter-mediated uptake processes (OATP1B3 and OAT3) are the rate-limiting clearance steps in both organs, which is in line with moderate passive permeability of fevipiprant (Pearson et al., 2017).
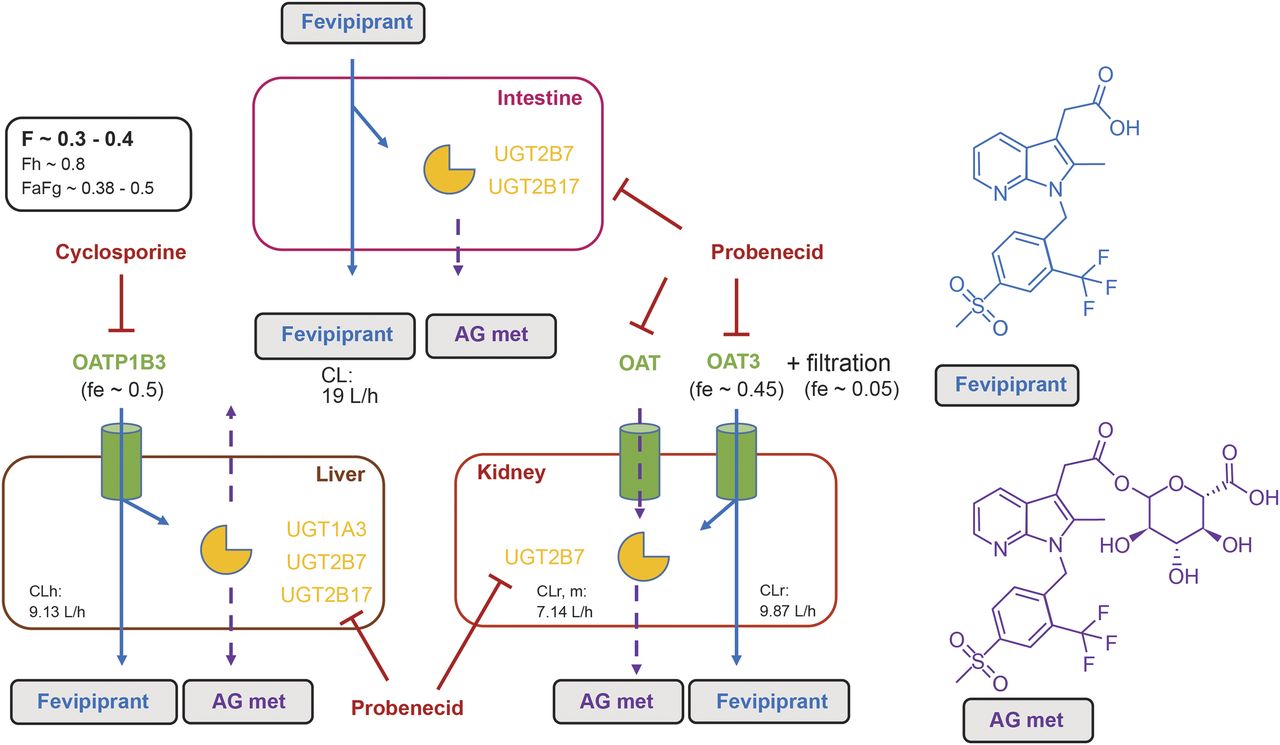
Drug disposition of fevipiprant (blue arrows) and the AG metabolite (AG met, dashed purple arrows) in humans based on in vitro phenotyping data (Pearson et al., 2017) and clinical study results. Oral F was determined in Weiss et al. (2020). Hepatic first pass (Fh) was calculated with Fh = 1−((CLh/Rb)/Qh) = 0.8, wherein CLh represents the hepatic plasma clearance (CLh = CL−CLr = 19−9.87 l/h = 9.13 l/h), Rb is the blood-to-plasma concentration ratio (0.56), and Qh is the hepatic blood flow (86.9 l/h) (Davies and Morris, 1993). The combined contribution of fraction absorbed and intestinal first pass (Fa × Fg) was estimated from the equation Fa × Fg = Fh/F. fe (fraction excreted), represents the fractional contribution of each pathway to the extraction of fevipiprant from plasma. Details on active vs. filtration clearance in the kidney are provided in the discussion and Table 3. Red lines represent inhibition mechanisms by cyclosporine (OATP1B3) and probenecid (OAT, UGTs). CLr, m represents the renal clearance of the AG metabolite.
In summary, results from interaction studies with probenecid and cyclosporine revealed the elimination of fevipiprant is dependent on OATP1B1-mediated hepatic uptake, OAT3-mediated renal excretion, and glucuronidation (via UGT1A3, UGT2B7, UGT2B17) (Fig. 4). These parallel elimination pathways result in a low risk of major victim DDI or pharmacogenetic/ethnic variability for this compound (Pearson et al., 2017). This is exemplified in the reported study, in which a perpetrator drug interferes with more than one fevipiprant elimination pathway, but the DDI effect remains weak to moderate.
Acknowledgments
The authors thank Cathy McDonnell (NBS CONEXTS, Dublin, Ireland) for providing medical writing support, which was funded by Novartis Institutes for Biomedical Research, Novartis Pharma AG, Basel, Switzerland, in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
Authorship Contributions
Participated in research design: Weiss, Langenickel, Cain.
Conducted experiments: Cain, Shah, Rahmanzadeh.
Performed data analysis: Weiss, Kulkarni, Shah, Vemula, Poller.
Wrote or contributed to the writing of the manuscript: Weiss, Langenickel, Cain, Kulkarni, Shah, Vemula, Rahmanzadeh, Poller.
Note Added in Proof: The author affiliations were accidentally not included in the Fast Forward version that appeared online February 25, 2021. The author line has now been added.
Footnotes
- Received October 5, 2020.
- Accepted January 29, 2021.
This study was sponsored by Novartis Pharma AG.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.Novartis will not provide access to patient-level data if there is a reasonable likelihood that individual patients could be reidentified. Phase 1 studies by their nature present a high risk of patient reidentification; therefore, patient individual results for phase 1 studies cannot be shared. In addition, clinical data in some cases have been collected subject to contractual or consent provisions that prohibit transfer to third parties. Such restrictions may preclude granting access under these provisions. If codevelopment agreements or other legal restrictions prevent companies from sharing particular data, companies will work with qualified requestors to provide summary information when possible.
All authors apart from T.L. are employees of Novartis, and all hold shares in the company apart from J.V. T.L. was employed by Novartis at the time the study was conducted and eligible to receive stock.

Abbreviations
- AE
- adverse event
- Ae0–24
- cumulative amount of an analyte excreted in urine from zero to 24 hours
- AG
- acyl glucuronide
- AUC
- area under the curve
- AUC0–24
- area under the plasma (or serum or blood) concentration-time curve from time 0 to 24 hours (mass × time/volume)
- AUCinf
- area under the concentration-time curve in plasma from time 0 extrapolated to infinite time
- AUClast
- area under the concentration-time curve in plasma from time 0 to time of last quantifiable concentration
- CI
- confidence interval
- CL
- systemic clearance
- CL/F
- apparent systemic clearance
- CLr
- renal clearance from plasma (volume/time)
- DDI
- drug-drug interaction
- DP2
- prostaglandin D2 receptor 2
- F
- bioavailability
- fu
- unbound fraction
- GFR
- glomerular filtration rate (approximately 7.5 l/h in a healthy subject)
- MR
- metabolite-to-parent drug ratio
- OAT
- organic anion transporter
- OATP
- organic anion–transporting polypeptide
- P-gp
- P-glycoprotein
- PK
- pharmacokinetics
- UGT
- UDP-glucuronosyltransferase
- Vz/F
- apparent volume of distribution
- Copyright © 2021 by The Author(s)
This is an open access article distributed under the CC BY Attribution 4.0 International license.





{kind=link}
{kind=link}
{kind=link}
{kind=link}