


Abstract
Characterizing the pharmacokinetic properties of drug candidates represents an essential task during drug development. In the past, liver microsomes and primary suspended hepatocytes have been extensively used for this purpose, but their relatively short stability limits the applicability of such in vitro systems for drug compounds with low metabolic turnover. In the present study, we used three-dimensional primary human hepatocyte spheroids to predict the hepatic clearance of seven drugs with low to intermediate clearance in humans. Our results indicate that hepatocyte spheroids maintain their in vivo–like phenotype during prolonged incubations, allowing to monitor the depletion of parent drug for 7 days. In contrast, attempts to increase the relative metabolic capacity by pooling hepatocyte spheroids resulted in an immediate fusion of the spheroids followed by hepatocellular de-differentiation processes, demonstrating limited applicability of the pooling approach for quantitative pharmacokinetic studies. The hepatic clearance values obtained from incubations with individual spheroids were in close correlation with the clinical reference data, with six out of seven drug compounds being predicted within a 3-fold deviation and average fold and absolute average fold errors of 0.57 and 1.74, respectively. In conclusion, the hepatocyte spheroid model enables accurate hepatic clearance predictions for slowly metabolized drug compounds and represents a valuable tool for determining the pharmacokinetic properties of new drug candidates as well as for mechanistic pharmacokinetic studies.
SIGNIFICANCE STATEMENT Traditional in vitro systems often fail to predict the hepatic clearance of slowly metabolized drug compounds. The current study demonstrates the ability of primary human hepatocyte spheroids to provide accurate projections on the hepatic clearance of drug compounds with low and intermediate clearance.
Introduction
Appropriate absorption, distribution, metabolism, and excretion (ADME) properties are crucial for selecting drug candidates and avoiding late-stage failures of drug development programs. Therefore, new chemical entities routinely undergo in vitro high-throughput ADME screenings at early drug discovery stages to identify and eliminate compounds with undesirable pharmacokinetic properties, such as short half-life and poor bioavailability. During lead optimization, medicinal chemists further improve the metabolic stability and other pharmacokinetic properties of lead compounds (Obach et al., 1997; Kratochwil et al., 2017). Consequently, a large number of compounds entering preclinical development display low or no turnover in metabolic stability assays typically conducted using liver microsomes in the presence of nicotinamide adenine dinucleotide phosphate (Di and Obach, 2015).
Although prolonged metabolic stability is a highly desirable pharmacokinetic attribute allowing for a once daily dosing regimen, the characterization of such low clearance compounds is extremely challenging for ADME scientists during the later preclinical development, since common in vitro systems as used in screening assays are no longer applicable. The preclinical testing is further challenged by the fact that not only slowly metabolized compounds but also compounds eliminated by nonoxidative metabolism (e.g., phase II conjugation reactions by uridine 5′-diphospho‐glucuronosyltransferases) and pathways beyond hepatic metabolism (e.g., transporter-mediated permeability or extrahepatic metabolism) are preferably selected by the microsomal screening approach (Argikar et al., 2016). For instance, uridine 5′-diphospho‐glucuronosyltransferase–mediated metabolism can be covered in microsomal incubations by adding corresponding cofactors, whereas microsomal fractions lack cytosolic enzymes and are likewise not applicable for compounds with low (rate-limiting) active and/or passive permeability (Kusuhara and Sugiyama, 2009; Camenisch, 2016). The use of primary hepatocytes can partly meet these challenges, as they express the entirety of hepatic drug-metabolizing enzymes and transporters, yet primary hepatocytes rapidly de-differentiate in suspension or 2D monolayer culture, and the activity of enzymes and transporters begins to decrease within a few hours (Di and Obach, 2015). For drug compounds with low metabolic clearance in vitro, this period is too short to achieve sufficient depletion of parent drug compound that would allow for reliable intrinsic clearance (CLint) estimations (Chan et al., 2013; Kratochwil et al., 2017).
Recently, three-dimensional spheroid cultures of primary human hepatocytes have been established allowing the culture of phenotypically stable hepatocytes for several weeks (Lin and Chang, 2008; Messner et al., 2013; Bell et al., 2016). Extensive characterization studies by our laboratory revealed that primary human hepatocyte spheroids maintain their morphology, viability, hepatocellular function, and the activity of drug-metabolizing enzymes over time, closely resembling hepatocytes in vivo (Bell et al., 2016, 2017; Vorrink et al., 2017). Moreover, we have successfully applied hepatocyte spheroids for hepatotoxicity screenings and mechanistic investigations as well as for the modeling of liver diseases (Hendriks et al., 2016; Kozyra et al., 2018; Vorrink et al., 2018; Hendriks et al., 2019; Hurrell et al., 2020).
In the present study, we established primary human hepatocyte spheroids as a novel in vitro–in vivo extrapolation (IVIVE) tool for predicting the hepatic clearance of low and intermediate turnover drugs. After the functional validation of hepatocyte spheroids, the CLint of seven drugs with low and intermediate clearance, which are metabolized by different cytochrome P450 (P450) enzymes, was determined in hepatocyte spheroids from three different donors and compared with clinical reference data. Furthermore, the effect of pooling individual spheroids to enhance the metabolic capacity was investigated with regard to cell morphology, hepatocellular function, and expression of hepatic drug-metabolizing enzymes and transporters.
Materials and Methods
Cell Culture
Cryopreserved primary human hepatocytes were obtained from BioreclamationIVT (USA) or KalyCell (France). Donor information of hepatocyte lots are summarized in Supplemental Table 1. Hepatocytes were thawed according to the manufacturer’s instruction and hepatocyte spheroids were generated as described previously (Bell et al., 2016). In brief, hepatocytes from individual donors were seeded in culture media [Williams' E medium (Life Technologies, Thermo Fisher Scientific) supplemented with 2 mM l-glutamine, 100 units/mL penicillin, 100 μg/mL streptomycin, 100 nM dexamethasone, 5.5 μg/mL transferrin, 6.7 ng/mL sodium selenite, and 10% fetal bovine serum] in 96-well ultra-low attachment plates (Corning, USA) at a density of 1500 viable cells per well. After cell aggregation on day 5, the media was replaced by serum-free culture media, and drug exposures were initiated on day 7.
Media-Loss Assay
The in vitro CLint of seven drugs with low [apixaban (CYP3A4), carbamazepine (CYP3A4), clozapine (CYP1A2), olanzapine (CYP1A2)] and intermediate [nortriptyline (CYP2D6), risperidone (CYP2D6), venlafaxine (CYP2D6/CYP2C19)] clearance was estimated during up to 7 days. Hepatocyte spheroids were incubated in 100 μL serum-free culture media containing either 1 μM apixaban, 1 μM carbamazepine, 1 μM clozapine, 0.1 μM nortriptyline, 1 μM olanzapine, 0.01 μM risperidone, or 1 μM venlafaxine at 37°C and 5% CO2 in a humidified incubator. Nominal drug concentrations were at least 10-fold below the reported Michaelis-Menten constant (Km) (Ring et al., 1996; Linnet and Olesen, 1997; Fogelman et al., 1999; Venkatakrishnan et al., 1999; Yasui-Furukori et al., 2001; Cazali et al., 2003; Wang et al., 2010) to avoid saturation of drug-metabolizing enzymes (Obach, 2001). The DMSO concentration was adjusted to 0.2% in all incubations. Nortriptyline, risperidone, and venlafaxine were exclusively investigated in donor C, which was identified as a CYP2D6 extensive metabolizer (Supplemental Table 1). At designated time points (10 minutes, 4 hours, 24 hours, 48 hours, 72 hours, 96 hours, 120 hours, and 168 hours), 40 μL media from three individual wells was pooled and quenched with 240 μL acetonitrile containing internal standard [either 0.5 μM apixaban-d3 (Toronto Research Chemicals, Canada), 0.5 μM carbamazepine-d8 (Toronto Research Chemicals, Canada), 0.5 μM [13C,2H3]-clozapine (Alsachim, France), 0.05 μM nortriptyline-d3 hydrochloride (Toronto Research Chemicals, Canada), 0.5 μM olanzapine-d3 (CDN isotopes, Canada), 0.005 μM risperidone-d4, (Toronto Research Chemicals, Canada), or 0.5 μM D,L-venlafaxine-d6 (Toronto Research Chemicals, Canada)]. After one freeze-thaw cycle, samples were centrifuged at a speed of 14,000 × g for 15 minutes at 4°C, and drug concentrations were quantified in the supernatant by ultra-performance liquid chromatography–tandem mass spectrometry (UPLC-MS/MS).
UPLC-MS/MS Analysis
Test drugs were analyzed using sample preparation procedures and UPLC-MS/MS instrumentation validated and certified for therapeutic drug monitoring purposes. Samples were mixed with equal volumes of cold acetonitrile/methanol (90:10) containing internal standard and were mixed for 5 seconds, frozen for 10 minutes at −20°C, and centrifuged at a speed of 3100 × g for 10 minutes at 2°C. The samples were injected onto a BEH Shield RP18 column (1.7 µm, 1.0 × 100 mm; Waters) and eluted with a mobile phase gradient of acetonitrile-ammonium acetate buffer (10 mM, pH 4.8) at a flow rate of 0.2 mL/min on an Acquity UPLC system (Waters). Determination of the analyte concentrations was performed using a Quattro Premier mass spectrometer (Waters) operated in positive electrospray ionization multiple reaction monitoring mode using selected transitions for each of the compounds. Calibration standards and quality control samples were prepared in phosphate buffered saline from working solutions containing the respective compounds, and the UPLC gradient was adapted depending on the test drug. The performance of the analytical assays were within the general requirements defined by the US Food and Drug Administration with intraday and interday inaccuracies and imprecisions of <15% for all analytes.
Processing of In Vitro Clearance Data
The in vitro CLint (μL/min/106 cells) was determined from the linear phase of parent drug depletion according to eq. 1 (Obach et al., 1997):
 where ke (h) is the elimination rate constant representing the slope of the linear regression from the ln-transformed percentage of remaining parent drug versus incubation time. Scaling factors for hepatocellularity and liver weight were taken from Smith et al. (2008) and Davies and Morris (1993), respectively. Incubational binding was taken into account by determining parent drug concentrations in media at early time points (i.e., 10 minutes after application of the dosing solution) representing unbound initial drug concentrations in serum-free media.
where ke (h) is the elimination rate constant representing the slope of the linear regression from the ln-transformed percentage of remaining parent drug versus incubation time. Scaling factors for hepatocellularity and liver weight were taken from Smith et al. (2008) and Davies and Morris (1993), respectively. Incubational binding was taken into account by determining parent drug concentrations in media at early time points (i.e., 10 minutes after application of the dosing solution) representing unbound initial drug concentrations in serum-free media.
The hepatic organ clearance (CLh) was predicted using the well-stirred liver model according to eq. 2 (Pang and Rowland, 1977):
 where Qh is the hepatic blood flow (20.7 mL/min/kg) and fub is the unbound fraction in blood (Supplemental Table 2). Clinical pharmacokinetic parameters used to derive the clinical reference CLh data as well as corresponding literature references are provided in Supplemental Table 2. Clinical CLh values were derived from the total oral plasma clearance, taking into account the oral bioavailability and the fractional contribution of renal drug clearance assuming only hepatic and renal drug elimination and a human body weight of 70 kg as described in the Supplemental Material. The classification of low and intermediate clearance was derived based on the corresponding hepatic extraction ratio (Eh) (Eh = CLh/Qh) in humans, where low (CLh < 6.2 mL/min/kg) and intermediate hepatic clearance (6.2 < CLh < 14.5 mL/min/kg) refers to Eh < 0.3 and 0.3 < Eh < 0.7, respectively (Benet and Zia-Amirhosseini, 1995).
where Qh is the hepatic blood flow (20.7 mL/min/kg) and fub is the unbound fraction in blood (Supplemental Table 2). Clinical pharmacokinetic parameters used to derive the clinical reference CLh data as well as corresponding literature references are provided in Supplemental Table 2. Clinical CLh values were derived from the total oral plasma clearance, taking into account the oral bioavailability and the fractional contribution of renal drug clearance assuming only hepatic and renal drug elimination and a human body weight of 70 kg as described in the Supplemental Material. The classification of low and intermediate clearance was derived based on the corresponding hepatic extraction ratio (Eh) (Eh = CLh/Qh) in humans, where low (CLh < 6.2 mL/min/kg) and intermediate hepatic clearance (6.2 < CLh < 14.5 mL/min/kg) refers to Eh < 0.3 and 0.3 < Eh < 0.7, respectively (Benet and Zia-Amirhosseini, 1995).
The statistical significance of parent drug depletion over time was determined using an F-test (GraphPad Prism 9.0.1, Suite, US). Slopes of the concentration-time profiles were considered to be significantly nonzero if a P value of <0.05 was obtained. The accuracy of CLh predictions was assessed by calculating the average fold error (AFE) and absolute average fold error (AAFE) according to eq. 3 and eq. 4, respectively:

 where pred and obs are the predicted and observed CLh values, respectively, and N is the number of data points.
where pred and obs are the predicted and observed CLh values, respectively, and N is the number of data points.
Cell Viability
The viability of hepatocyte spheroids was assessed by determining the cellular ATP content using the CellTiter-Glo Luminescent Cell Viability Assay (Promega) according to the manufacturer’s instructions. Luminescence per spheroid was measured on a MicroBeta LumiJET 2460 Microplate Counter (Perkin Elmer).
Albumin Secretion
The secretion of albumin by hepatocyte spheroids was determined by quantifying the albumin concentration in the cell culture medium using the Human Albumin ELISA Kit (Bethyl Laboratories) according to the manufacturer’s instructions.
Gene Expression Profiling
Total RNA was isolated using the Qiazol Lysis Reagent (Qiagen). RNA was reverse-transcribed into cDNA using SuperScript III reverse transcriptase and analyzed by reverse transcription polymerase chain reaction using a TaqMan Universal mix and Taqman probes (Supplemental Table 3) on a 7500 Fast Real-Time PCR system. Data analysis was performed using the 2-ΔΔCt method and TATA-binding protein as housekeeping gene.
CYP3A4 Protein Expression
Total protein was extracted from hepatocyte spheroids using RIPA Lysis and Extraction buffer (Thermo Fisher Scientific) supplemented with cOmplete protease inhibitor (Roche, Switzerland). The protein content was determined using a Bradford assay, and 25 µg of protein was separated using a 10% Mini-PROTEAN TGX Precast Protein Gel (Bio-Rad Laboratories) and transferred onto an AmershamProtran membrane (GE Healthcare Life Sciences, UK). The membrane was incubated with anti-CYP3A4 (1:1000, Cypex, UK) and antivinculin (1:10,000, Abcam, UK) followed by goat anti-rabbit IgG/horseradish peroxidase (1:10,000, DAKO, Denmark) and was visualized using Super Signal West Femto Chemiluminescent Substrate (Thermo Fisher Scientific, USA).
Results
Viability and Functionality of Hepatocyte Spheroids
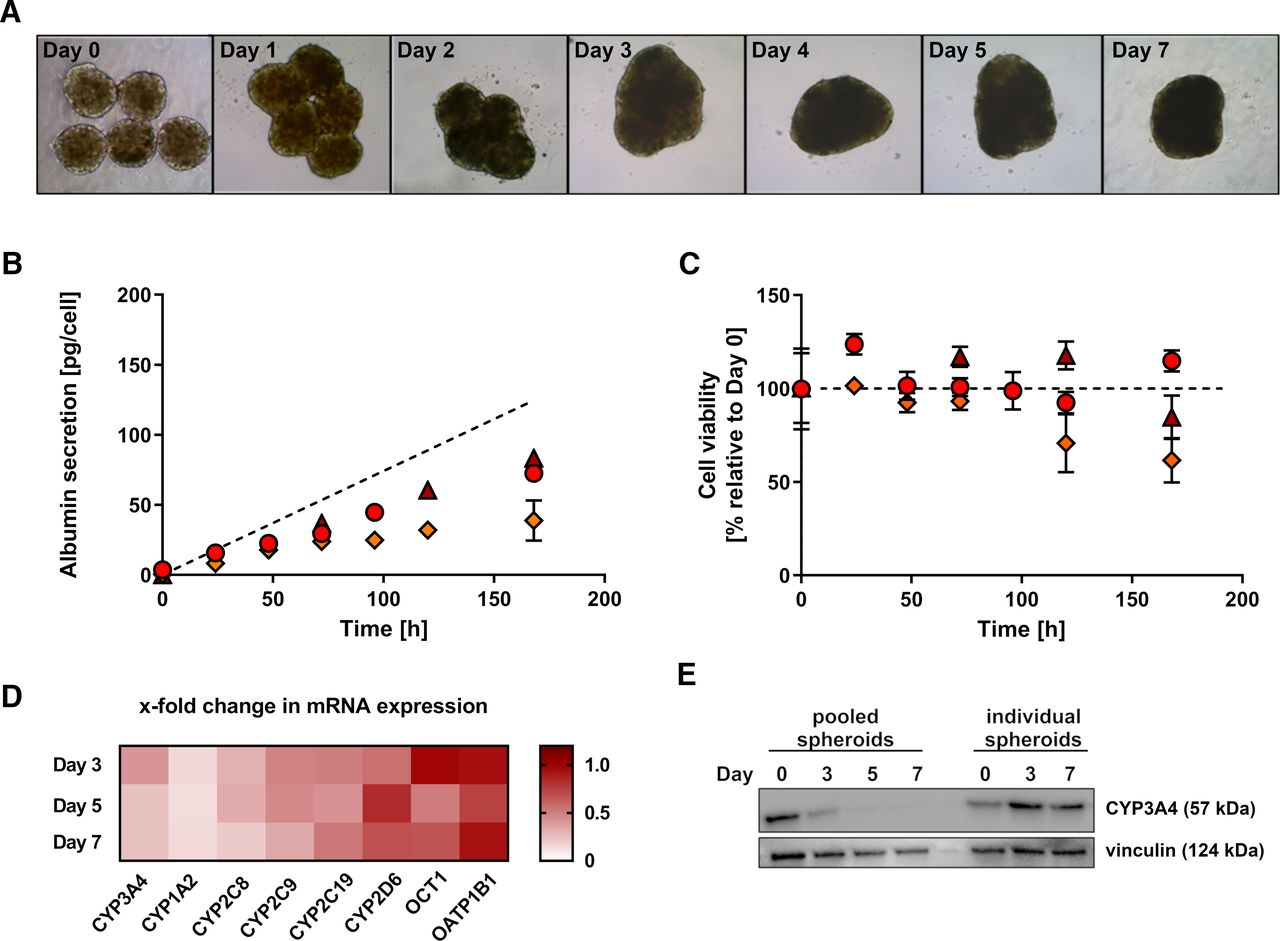
Hepatocyte spheroids have previously been shown to maintain their phenotype and the activity of drug-metabolizing enzymes over several weeks (Bell et al., 2016), enabling chronic drug exposures for hepatotoxicity screening as well as mechanistic investigations (Vorrink et al., 2018; Hendriks et al., 2019). Chronic hepatotoxicity testing in a metabolically active in vitro system requires regular supply with new parent drug compound to substitute for the loss of metabolized drug (i.e., drug exposures every 2–3 days). Yet, clearance experiments do not allow regular media changes to quantitatively monitor the loss of parent drug or the formation of metabolite(s) over time. In particular, investigations of drugs with low clearance require extended incubations for several days exceeding the common period between media changes. To validate persistent cell viability and functionality in the absence of regular nutrient supply, the morphology of hepatocyte spheroids as well as the secretion of albumin as a marker of hepatocellular function were monitored over 7 days. Bright-field images of hepatocyte spheroids indicated no changes in cellular morphology and spheroid size over the investigated period (Fig. 1A). Moreover, the albumin concentrations in the culture media steadily increased over time, suggesting stable albumin production and hepatocellular function of spheroids from all donors (Fig. 1B). The average rates of albumin secretion were 19.9 ± 3.4, 23.3 ± 7.5, and 12.0 ± 5.0 pg/hepatocyte/day for donors A, B, and C, respectively, whereby the albumin secretion rate in vivo is reported to be 17.8 pg/hepatocyte/day (Jang et al., 2015).

Hepatocyte spheroids remain viable and functional during extended incubations. (A) Bright-field images of hepatocyte spheroids from donor A over the time course of 7 days. (B) Media concentration of albumin secreted by hepatocyte spheroids over 7 days. Data points represent mean values of quadruplicates ± S.D. The dashed line represents the albumin secretion rate in vivo [17.8 pg/hepatocyte/day (Jang et al., 2015)]. Δ, donor A; Ο, donor B; ⋄, donor C.
Loss of Hepatocellular Phenotype in Pooled Hepatocyte Spheroids
To enhance the metabolic capacity of hepatocyte spheroids to reduce the assay duration, we hypothesized that pooling of spheroids should increase the depletion of parent drug as well as metabolite formation. Although the amount of enzyme is adjustable in traditional in vitro systems like liver microsomes or hepatocytes (Obach, 2001; Nordell et al., 2013), the presented spheroid system was developed using only 1500–2000 hepatocytes per spheroids to prevent the risk for intraspheroid hypoxia, which is expected in larger spheroids (Lin and Chang, 2008). Thus, we attempted to increase the cell number per incubation by a factor of 10 by pooling hepatocyte spheroids (5 spheroids/50 μL media as opposed to 1 spheroid/100 μL media) after completed spheroid formation on day 7 postseeding and investigated potential changes in the morphology and hepatocellular function over time (Fig. 2).

Loss of hepatocellular phenotype in pooled hepatocyte spheroids. (A) Bright-field images of pooled hepatocyte spheroids from donor B over the time course of 7 days. (B) Albumin secretion of pooled hepatocyte spheroids over 7 days. Data points represent mean values of quadruplicates ± S.D. The dashed line represents the albumin secretion rate in vivo [17.8 pg/hepatocyte/day (Jang et al., 2015)]. (C) Cellular viability (total cellular ATP) of pooled hepatocyte spheroids over 7 days. Data points represent mean values of quadruplicates ± S.D. Δ, donor A; Ο, donor B; ⋄, donor C. (D) Fold change in mRNA expression (relative to day 0) of major hepatic P450 enzymes and uptake transporters in pooled hepatocyte spheroids during 7 days. Data points represent mean values of the measured fold change in mRNA expression in hepatocyte spheroids from donors A, B, C, and D. (E) Western blot of CYP3A4 and vinculin (loading control) in individual and pooled hepatocyte spheroids from donor D during 7 days.
As indicated by bright-field images, the hepatocyte spheroids formed loose aggregates within 24 hours after pooling. After 48 hours, the spheroids entirely fused, forming solid and compact aggregates (Fig. 2A). In line with the changes in spheroid morphology, the albumin secretion per cell decreased about 2-fold in pooled spheroids as compared with individual spheroids (Fig. 2B) with estimated average albumin secretion rates in pooled spheroids from donors A, B, and C being 12.1 ± 0.6, 11.7 ± 2.2, and 7.3 ± 1.6 pg/hepatocyte/day, respectively. In contrast, the cellular ATP content indicated stable cell viability of pooled spheroids from donors A and B during 7 days, whereas the cellular ATP content in spheroids from donor C decreased by about 40% within 7 days after pooling (Fig. 2C). Furthermore, the mRNA expression of major hepatic P450 enzymes and uptake transporters was analyzed during 7 days after pooling (Fig. 2D), indicating a rapid decrease in CYP3A4, CYP2C8, CYP2C9, and CYP1A2 mRNA expression. In contrast, the CYP2C19, CYP2D6, and organic cation transporter 1 mRNA expression decreased less than 2-fold, and no change in the mRNA expression of organic anion transporting polypeptide 1B1 was detected during 7 days. Similarly, the protein expression of CYP3A4, which was analyzed as a representative P450 enzyme, immediately decreased after pooling the spheroids with hardly any CYP3A4 protein detectable by Western blot after 5 and 7 days (Fig. 2E). In contrast, the CYP3A4 protein expression did not decrease in spheroids from the same donor, which were cultured in individual wells.
In Vitro CLint Incubations and Prediction of CLh
To examine the feasibility of CLh predictions, seven test drugs with reported low to intermediate hepatic clearance were incubated with individual hepatocytes spheroids, and the disappearance of parent drug was monitored over time (Fig. 3). The low clearance drugs apixaban, carbamazepine, clozapine, and olanzapine (reported CLh in humans ranging from 0.110 to 3.55 mL/min/kg, Table 1) were incubated with spheroids from donors A, B, and C during 7 days without medium change to allow for sufficient removal of parent drug from the incubations. Significant depletion of three out of four low clearance drugs was observed in hepatocyte spheroids from all three donors, and concentration-time profiles are shown in Fig. 3, A–D. Exception was carbamazepine, for which no significant depletion of parent drug was measured in hepatocyte spheroids from any of the tested donors. Although not significant, a small decline in parent drug concentrations over time was detected in incubations with hepatocyte spheroids from donors A and C, and the obtained in vitro data are shown in Table 1, yet carbamazepine was excluded for the subsequent comparison with clinical reference data and further statistical tests. In contrast, no depletion of carbamazepine was observed in donor B (data not shown), whose concentration-time profile was not considered for the predicted CLh value presented in Table 1.
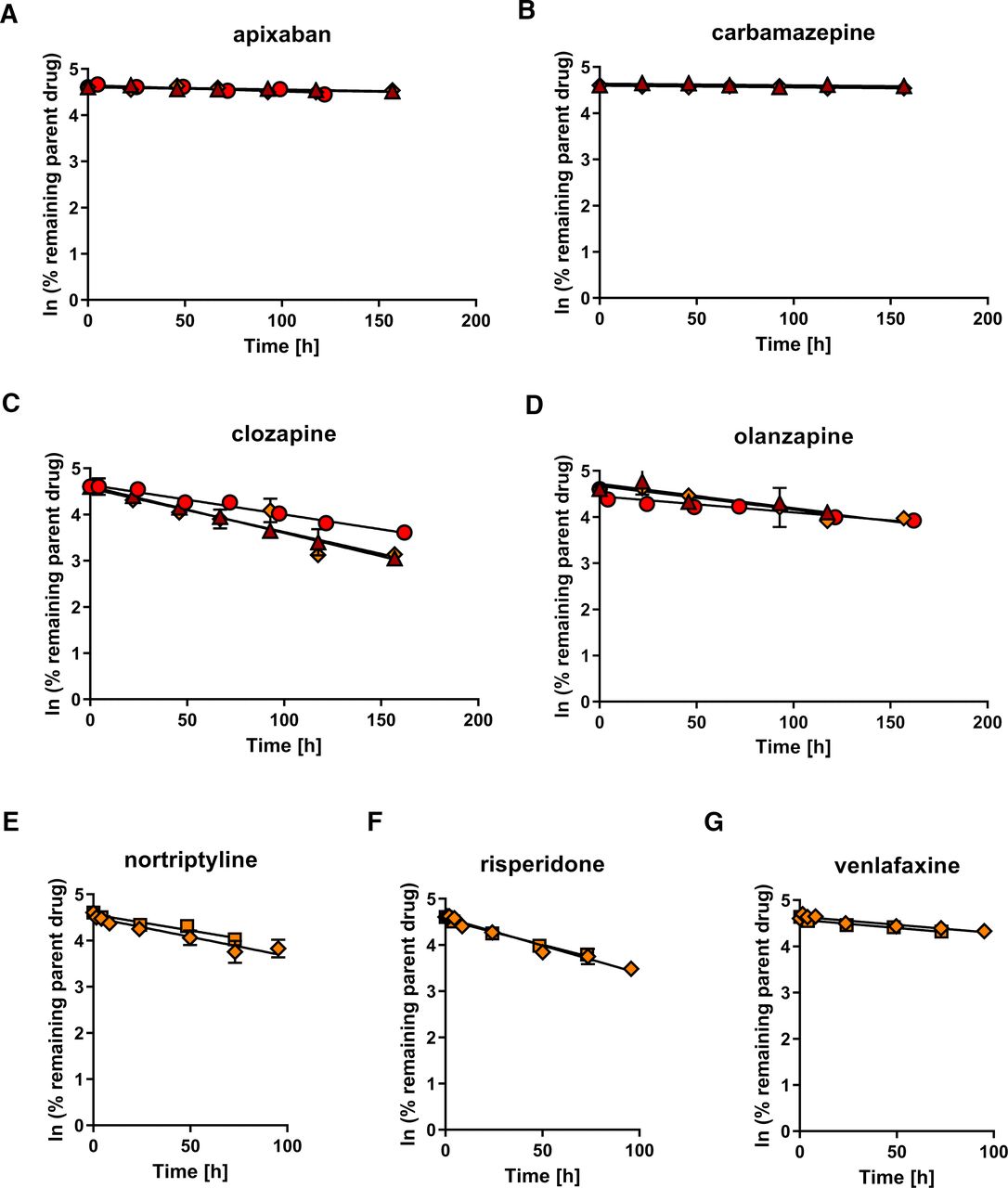
Substrate depletion-time profiles in hepatocyte spheroids. (A-D) Depletion of compounds with low hepatic clearance in individual hepatocyte spheroids from donors A, B, and C. (E–G) Depletion of compounds with intermediate hepatic clearance in individual hepatocyte spheroids from donor C (data obtained in two independent experiments). Parent drug concentrations were quantified in cell culture media, and CLint was estimated from the linear phase of parent drug depletion as described in Materials & Methods. Data points represent mean values of duplicates ± S.D. Δ, donor A; O, donor B; ⋄ and □, donor C.
Predicted CLint and CLh values and clinical reference data
In vitro half-life (t1/2) is expressed in [h]. Clearance values are expressed in [mL/min/kg] and refer to blood elimination. Data represent mean values ± S.D. obtained using hepatocyte spheroids from donors A, B, and C (apixaban, carbamazepine, clozapine, olanzapine) or mean values ± S.D. obtained using hepatocyte spheroids from donor C in two independent experiments (nortriptyline, risperidone, venlafaxine). Literature references for clinical CLh values are provided in Supplemental Table 2.
In addition, the intermediate clearance drugs nortriptyline, risperidone, and venlafaxine (reported CLh in humans ranging from 6.68 to 8.79 mL/min/kg, Table 1) were investigated in hepatocyte spheroids from donor C. All three drug compounds are predominately eliminated by the highly polymorphic CYP2D6 enzyme (Fogelman et al., 1999; Venkatakrishnan et al., 1999; Yasui-Furukori et al., 2001). To facilitate the comparison with clinical CLh reference data, incubations were conducted in donor C only, which was identified to inhere the CYP2D6 extensive metabolizer (wild-type) genotype (Supplemental Table 1). Similar to previous incubations, parent drug depletion was observed for all three test compounds over time (Fig. 3, E–G), yet, the in vitro half-life was generally markedly lower than for the investigated low clearance drugs (Table 1). Accordingly, incubations with the intermediate clearance drugs could be terminated already after 3–4 days.
Next, we performed CLh predictions based on the in vitro CLint data and compared the obtained values with clinical reference data (Table 1). The predicted CLh values ranged from 0.434 to 1.64 mL/min/kg and from 3.00 to 5.59 mL/min/kg for drugs with low and intermediate clearance, respectively. The direct comparison with clinical reference data indicated a close correlation between predicted and observed values with an AFE of 0.57, an AAFE of 1.74, and six out of seven drug compounds predicted within a 3-fold deviation when excluding carbamazepine (Fig. 4).

Comparison of predicted and observed CLh data. Data points represent mean CLh values ± S.D. obtained using hepatocyte spheroids from donors A, B, and C (apixaban, clozapine, olanzapine) or mean CLh values of duplicates ± S.D. obtained using hepatocyte spheroids from donor C in two independent experiments (nortriptyline, risperidone, venlafaxine). The solid line is the line of unity and dotted lines represent 3-fold deviations. api, apixaban; clo, clozapine; nor, nortriptyline; ola, olanzapine; ris, risperidone; ven, venlafaxine.
To investigate potential changes in the metabolic activity after the pooling of hepatocyte spheroids, we monitored the depletion of the low clearance drugs apixaban, carbamazepine, clozapine, and olanzapine in pooled spheroids from donors A, B, and C during 7 days. The drug compounds were cleared from the media to a higher extent in all incubations with pooled spheroids as compared with individual spheroid incubations (substrate depletion-time profiles are shown in Supplemental Fig. 1). Yet, after taking the hepatocyte number into account, the CLint data derived from pooled spheroids were lower, notably underestimating the hepatic clearance of the majority of the investigated compounds. The fold deviation between predicted and observed CLh data ranged from 0.1 to 0.6, and the AAFE increased to 4.95 for the four test drugs (Table 1).
Discussion
Anticipating the clearance of slowly metabolized drugs is particularly challenging, as common in vitro systems feature short-term stability and do not allow to reliably determine the clearance of such compounds (Di and Obach, 2015). Here, we established primary human hepatocyte spheroids as a new IVIVE tool for predicting the hepatic clearance of compounds with low and intermediate metabolic turnover. Hepatocyte spheroids demonstrated phenotypical stability during 7 days allowing for extended incubations to quantify the disappearance of parent drug from the culture media. Using this experimental setup, we were able to determine the in vitro CLint of six low and intermediate clearance drugs from individual spheroids and successfully predicted the hepatic clearance of these compounds within a 3-fold deviation from the clinical reference data (AAFE of 1.74).
The demands for in vitro ADME test systems undergo fundamental changes as current drug discovery programs generate compounds with uncommon physicochemical properties from unprecedented chemical space (Tu et al., 2013). Nonoxidative metabolism and transporter-mediated pathways like sinusoidal uptake or renal and biliary secretion are often involved in the elimination of such compounds. These processes can largely be covered in profiling assays by using more holistic in vitro systems such as primary hepatocytes or cytosolic fractions (Sahi et al., 2010). Furthermore, static hepatic clearance models such as the Extended Clearance Model have been developed, allowing the integration of individual hepatic in vitro process clearances to predict the hepatic drug clearance in humans (Kusuhara and Sugiyama, 2009; Camenisch et al., 2015). Nevertheless, the clearance of compounds with high metabolic stability cannot be estimated in short-term in vitro assays requiring time and cost-intensive allometric scaling approaches (Hutzler et al., 2015). In the present study, we found that hepatocyte spheroids can help to overcome these challenges due to their long-term stability (Fig. 1), allowing for sufficient depletion of parent drug even for compounds with low hepatic clearance such as apixaban (clinical CLh = 0.6 mL/min/kg). Exception was the test drug carbamazepine (clinical CLh = 0.11 mL/min/kg), for which no significant loss could be detected during 7 days of incubation. Although not statistically significant, minor depletion of carbamazepine over time was observed, which likely resulted from metabolic elimination rather than cellular accumulation of carbamazepine, considering the small number of cells in relation to the media volume. For instance, even in the situation of a high cell-to-media drug concentration ratio (Kp) of 100, cellular accumulation would account for the disappearance of only 0.33% of carbamazepine from the media [based on a cell volume of 2.2 μL/106 hepatocytes (Riede et al., 2017)]. Yet, parent drug depletion due to chemical instability can generally not be excluded. To account for nonspecific compound loss, additional incubations in the absence of hepatocyte spheroids could be performed allowing to correct for potential chemical instability of the test compounds.
We previously demonstrated that hepatocytes in spheroid culture maintain the expression of important hepatic genes involved in ADME including phase I and II drug-metabolizing enzymes as well as uptake and efflux transporters on the transcriptomic and proteomic level, mimicking in vivo liver tissue (Bell et al., 2016, 2017). Although the present study did not include drugs being primarily eliminated by non-P450 enzymes or (rate-limiting) transporter-mediated permeability, hepatocyte spheroids are expected to provide equally accurate hepatic clearance projections for such compounds, making them an attractive tool for pharmaceutical companies. Yet, the applicability of hepatocyte spheroids for assessing the metabolic stability of a vast amount of compounds in a drug discovery setting is limited until robotic platforms suitable for automated formation and handling of spheroids are routinely available to decrease the assay costs (Lucendo-Villarin et al., 2020). Furthermore, hepatocyte spheroids preserve the hepatocyte donor’s phenotype and genotype (Bell et al., 2016), which is of great advantage for studying specific mutations (i.e., polymorphic drug-metabolizing enzymes) or liver diseases (Prill et al., 2019; Hurrell et al., 2020). Accordingly, hepatocyte spheroids from single donors do not reflect the diversity in the population, which would require the simultaneous screening of drug compounds with several spheroid batches, whereas microsomal or hepatocyte assays are typically conducted with pooled material from up to 50 and 200 donors, respectively (Izumi et al., 2017). Although hepatocyte spheroids can be produced from multiple donor hepatocyte lots (Kermanizadeh et al., 2019), not every platable hepatocyte lot is suitable for producing spheroids, and the use of multiple donor hepatocyte lots would require careful monitoring to ensure equal incorporation and survival of hepatocytes from all donors in spheroids. Due to these limitations, the application of hepatocyte spheroids in high-throughput screening assays in drug discovery remains challenging, nevertheless, they can be considered as a second tier approach for drug compounds with low turnover when standard in vitro systems have failed, as well as for characterizing compounds with complex enzyme-transporter interplay and other tailor-made mechanistic studies.
The use of commercial long-term in vitro liver systems for pharmacokinetic studies continuously increases, and most often micropatterned cocultures (MPCCs) of primary human hepatocytes with stromal cells are applied. MPCC systems likewise feature long-term stability and have been successfully used for IVIVE of low turnover compounds (Chan et al., 2013; Hultman et al., 2016; Umehara et al., 2020). These studies yielded overall a comparable accuracy with 70%–92% of compounds being predicted within a 3-fold deviation, whereby our spheroid model predicted 86% of compounds within a 3-fold deviation. Nevertheless, it has to be noted that the selection of compounds differed between studies. In particular, Umehara et al. (2020) included a diverse set of uptake transporter and non-P450 substrates, whereas our compound set consisted of P450 substrates. A face-to-face comparison of a harmonized set of compounds and the same hepatocyte lots would be required to directly compare the performance of hepatocyte spheroid and MPCC models. Similarly, another recent study by Kanebratt et al. (2021) applied hepatocyte spheroids to predict the clearance of compounds with low turnover. Despite introducing an additional empirical scaling factor to account for systemic underprediction, their study achieved fewer accurate predictions as indicated by AFE and AAFE values of 0.53 and 2.7, respectively, whereas our study obtained AFE and AAFE values of 0.57 and 1.74, respectively, without applying empirical scaling factors. Furthermore, the study by Kanebratt et al. (2021) indicated a trend toward more pronounced deviation between predicted and observed clearance data for drug compounds with higher clearance, which was not evident in our study. Interestingly, the authors pooled three spheroids per incubation to increase the metabolic capacity and observed comparable CLint data using pooled and individual hepatocyte spheroids. Our attempts to improve the cell-to-media volume ratio provided markedly lower CLint values in pooled spheroids (Table 1). Follow up investigations on the effect of pooling spheroids indicated merging of the individual spheroids within 24–48 hours, which was accompanied by reduced albumin secretion as well as reduced mRNA expression of major hepatic drug-metabolizing enzymes and uptake transporters as well as reduced CYP3A4 protein expression (Fig. 2). The fusion of spheroids likely introduced major rearrangements in the cellular structures resulting in de-differentiation and entire loss of the hepatocellular phenotype. We did not investigate whether the hepatocellular phenotype reverses over time (like during normal spheroid formation), however, hepatocytes in the center of the fused spheroid structure are likely not sufficiently supplied with oxygen and nutrients due to the larger spheroid diameter (Lin and Chang, 2008) and will be susceptible to early cell death. Consequently, we recommend to avoid pooling of spheroids, but to perform extended incubations with individual spheroids to allow for sufficient parent drug depletion. As an alternative, plates with integrated microwells could be considered(Wassmer et al., 2020).
The good performance of our model might result from the ability of hepatocyte spheroids to continuously secrete albumin at in vivo rate (Fig. 1). The presence of albumin (or other protein) in the medium is assumed to support the cellular uptake, a process known as protein-mediated uptake. The underlying mechanism is not fully understood and current hypotheses were reviewed in detail elsewhere (Bowman and Benet, 2018; Bteich et al., 2019). In disagreement with the free-drug hypothesis (Pang and Rowland, 1977), different studies observed that cellular uptake rates decreased less than expected in the presence of plasma protein when considering the unbound drug concentration, and this effect was increased for compounds with higher plasma protein binding (Miyauchi et al., 2018; Poulin and Haddad, 2018; Bowman et al., 2019). Furthermore, underprediction of drug clearance by in vitro approaches was reported to be proportional to the extent of plasma protein binding (Liang et al., 2020). In line with these observations, a recent study applying MPCC reported superior clearance predictions using hepatocyte donors with high albumin secretion rate (Da-Silva et al., 2018), and likely both MPCC and spheroids models profit from the integrated albumin secretion. On the other hand, the increasing concentrations of albumin over time complicate the estimation of intrinsic clearance, as the unbound fraction of drug in the media changes during the incubation. In the present study, initial unbound drug concentrations in the media were determined from supernatant sampled from the assay plate shortly upon applying the dosing solution, assuming complete absence of protein in the media. With regard to the very low albumin concentrations in the media observed after 7 days and the rather small degree of plasma protein binding of the investigated compounds (fraction unbound in plasma ≥0.05, Supplemental Table 2), no effects of albumin on the fraction unbound in the media and obtained CLint estimates are expected for our dataset. To account for the time-dependent increase of albumin for drugs with extensive plasma protein binding, measured drug concentrations in the media could be corrected with the fraction unbound for the respective albumin concentration (estimated using plasma protein binding data).
In conclusion, we demonstrated that primary human hepatocyte spheroids remain phenotypically stable for up to 7 days without medium change allowing for extended incubations to accurately predict the hepatic clearance of drug compounds with low to intermediate hepatic clearance. Hepatocyte spheroids represent a valuable IVIVE tool and are expected to significantly facilitate the characterization of slowly metabolized compounds to improve IVIVE during pharmaceutical development.
Acknowledgments
The authors would like to thank Vlasia Kastrinou-Lampou for experimental support and Dr. Birk Poller for his critical review of this manuscript.
Authorship Contributions
Participated in research design: Riede, Wollmann, Molden, Ingelman-Sundberg.
Conducted experiments: Riede, Wollmann.
Performed data analysis: Riede, Wollmann.
Wrote or contributed to the writing of the manuscript: Riede, Wollmann, Molden, Ingelman-Sundberg.
Footnotes
- Received December 21, 2020.
- Accepted April 15, 2021.
↵1 Current affiliation: PK Sciences, Novartis Institutes for BioMedical Research, Basel, Switzerland.
This work was funded by the Swedish Cancer Society [Grant Agreement 17 0599], the European Research Council (ERC)–Advanced Grant (AdG) project HEPASPHER [Grant Agreement 742020], and the European Union’s Horizon 2020 research and innovation program [Grant Agreement 668353/U-PGx].
None of the authors has any conflict of interest to this study to declare.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AAFE
- absolute average fold error
- ADME
- absorption, distribution, metabolism, and excretion
- AFE
- average fold error
- CLh
- hepatic organ clearance
- CLint
- intrinsic clearance
- Eh
- hepatic extraction ratio
- IVIVE
- in vitro–in vivo extrapolation
- MPCC
- micropatterned coculture
- P450
- cytochrome P450
- UPLC
- ultra-performance liquid chromatography
- UPLC-MS/MS
- UPLC–tandem mass spectrometry
- Copyright © 2021 The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.





{kind=link}
{kind=link}
{kind=link}
{kind=link}