



Abstract
KZR-616 is an irreversible tripeptide epoxyketone-based selective inhibitor of the human immunoproteasome. Inhibition of the immunoproteasome results in anti-inflammatory activity in vitro and based on promising therapeutic activity in animal models of rheumatoid arthritis and systemic lupus erythematosus KZR-616 is being developed for potential treatment of multiple autoimmune and inflammatory diseases. The presence of a ketoepoxide pharmacophore presents unique challenges in the study of drug metabolism during lead optimization and clinical candidate profiling. This study presents a thorough and systematic in vitro and cell-based enzymatic metabolism and kinetic investigation to identify the major enzymes involved in the metabolism and elimination of KZR-616. Upon exposure to liver microsomes in the absence of NADPH, KZR-616 and its analogs were converted to their inactive diol derivatives with varying degrees of stability. Diol formation was also shown to be the major metabolite in pharmacokinetic studies in monkeys and correlated with in vitro stability results for individual compounds. Further study in intact hepatocytes revealed that KZR-616 metabolism was sensitive to an inhibitor of microsomal epoxide hydrolase (mEH) but not inhibitors of cytochrome P450 (P450) or soluble epoxide hydrolase (sEH). Primary human hepatocytes were determined to be the most robust source of mEH activity for study in vitro. These findings also suggest that the exposure of KZR-616 in vivo is unlikely to be affected by coadministration of inhibitors or inducers of P450 and sEH.
SIGNIFICANCE STATEMENT This work presents a thorough and systematic investigation of metabolism and kinetics of KZR-616 and related analogs in in vitro and cell-based enzymatic systems. Information gained could be useful in assessing novel covalent proteasome inhibitors during lead compound optimization. These studies also demonstrate a robust source in vitro test system that correlated with in vivo pharmacokinetics for KZR-616 and two additional tripeptide epoxyketones.
Introduction
The proteasome has been validated as a therapeutic drug target through regulatory approval of three compounds for use in the treatment of B-cell neoplasms, such as multiple myeloma and mantle cell lymphoma (Bross et al., 2004; Kisselev et al., 2001; Kisselev et al., 2012; Herndon et al., 2013; Offidani et al., 2014; Gentile et al., 2015). These compounds comprise two chemical classes: the reversible boronic acid derivatives bortezomib (VELCADE) and ixazomib (NINLARO) and the tetrapeptide epoxyketone-based compound carfilzomib (Kyprolis). Unlike bortezomib and ixazomib, carfilzomib is the first of a new generation of irreversible inhibitors that target proteasome subunits with prolonged inhibition and high selectivity (Demo et al., 2007; Kuhn et al., 2007; Zhou et al., 2009; Arastu-Kapur et al., 2011; Harshbarger et al., 2015). Validated with the success of three approved agents, next-generation proteasome inhibitors are now an important class of drugs being investigated for other diseases (Teicher and Tomaszewski, 2015).
The immunoproteasome is a unique form of proteasome mainly expressed in immune effector cells (Miller et al., 2013). The immunoproteasome distinguishes itself from the ubiquitously expressed constitutive proteasome by the presence of three distinct catalytic subunits: low-molecular-mass polypeptide (LMP) 7, LMP2, and multicatalytic endopeptidase complex-like 1. These active site subunits can also be induced in nonimmune cells upon exposure to inflammatory cytokines, such as tumor necrosis factor α (TNF-α) or interferon γ (Ferrington and Gregerson, 2012). Selective inhibition of the immunoproteasome results in a reduction of inflammatory cytokines from immune effector cells and can block disease progression in animal models of several autoimmune diseases, including rheumatoid arthritis, multiple sclerosis, and systemic lupus erythematosus (SLE) (Puttaparthi and Elliott, 2005; Egerer et al., 2006; Groettrup et al., 2010; Basler and Groettrup, 2012; Miller et al., 2013; Basler et al., 2015). KZR-616 (Fig. 1) is a tripeptide epoxyketone that selectively and irreversibly inhibits the LMP7 and LMP2 subunits of the human immunoproteasome; it was selected for clinical development after a thorough medicinal chemistry campaign (Johnson et al., 2018). In human peripheral blood mononuclear cells, KZR-616 blocks inflammatory cytokine production, including TNF-α, granulocyte-macrophage colony-stimulating factor, interleukin-6, and interleukin-23. In lymphocytes, T-cell production of interferon γ, TNF-α, and granulocyte-macrophage colony-stimulating factor and differentiation of activated B-cells to plasma blasts are inhibited by KZR-616 (Muchamuel et al., 2017). Finally, in mouse models of rheumatoid arthritis and SLE, KZR-616 blocks disease progression at well tolerated doses without affecting normal T-cell–dependent immune responses (Muchamuel et al., 2017; Johnson et al., 2018). KZR-616 is currently being evaluated in phase 2 clinical trials in patients with SLE and lupus nephritis.

Chemical structures of KZR-616, KZR-59177, KZR-59240, their diol derivatives, carfilzomib, and oprozomib.
Like carfilzomib, KZR-616 is an exquisitely chemo-selective, irreversible covalent inhibitor of N-terminal threonine proteases, a class of proteases to which the proteasome active site subunits are nearly exclusive (Kisselev et al., 2012). Although the boronic acid–based proteasome inhibitors were shown to have a primary metabolic pathway involving oxidation via classic phase 1 cytochrome P450 (P450) enzymes, carfilzomib is believed to be cleared extrahepatically via peptidase cleavage and epoxide hydrolysis to an inactive diol (Yang et al., 2011; Wang et al., 2013). Oprozomib, a tripeptide epoxyketone analog of carfilzomib, which is currently under clinical investigation in multiple myeloma, was shown to be a substrate of microsomal epoxide hydrolase (mEH) (Wang et al., 2017). Given the structural similarities between KZR-616 and both carfilzomib and oprozomib, we aimed to determine the major and specific metabolic pathways and enzymes involved in metabolism of KZR-616 prior to initiation of clinical trials in chronic disorders. In addition, by conducting a thorough and methodical in vitro experimental approach and assessing pharmacokinetics in large animals, we were able to carry out in vitro to in vivo extrapolation correlations to determine which surrogate absorption, distribution, metabolism, and excretion (ADME) in vitro model system is most informative for development of novel epoxyketone-based clinical candidates.
Materials and Methods
Materials and Cell Growth Conditions.
All chemicals and reagents were obtained from Sigma (St. Louis, MO) and liquid chromatography [mass spectrometry (MS) grade] solvents were obtained from Fisher Scientific (Pittsburgh, PA). KZR-616, deuterated KZR-616 (d8-KZR-616), KZR-59587, KZR-59177, KZR-59240, and KZR-616 analogs were synthesized at Kezar Life Sciences. 2-2-Nonylsulfanyl-propionamide (NSPA), human recombinant mEH, and soluble epoxide hydrolase (sEH) were gifts from Bruce Hammock’s laboratory at the University of California, Davis (Newman et al., 2003; Morisseau et al., 2008; Montellano, 2018). 1-Trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea (TPPU) and 1-aminobenzotriazole (1-ABT) were obtained from Sigma. Liver subcellular fractions including male or female monkey liver microsomes (pool of six; 20 mg/ml), cytosols (pool of six; 10 mg/ml), human liver subcellular fractions including male or female liver microsomes (pool of 10, 20 mg/ml), cytosols (mixed sex; pool of 50, 10 mg/ml), pooled human cryopreserved hepatocytes (100 donors, mixed sex) and culture media (K8400), and monkey cryopreserved hepatocytes (five donors of male cynomolgus monkey, PPCH 2000) and culture media (20-1-0526) were purchased from Sekisui Xenotech (Kansas City, KS). Human hepatocytes were incubated at 37°C in an environment of 95% air/5% CO2.
Metabolic Stability in Liver Microsomes in the Presence and Absence of NADPH.
Reactions were assessed in pooled human (H) and monkey (M) liver microsomes (LMs) with or without NADPH. Test compound and probe substrates were mixed with HLM (0.5 mg/ml) and MLM (0.25 mg/ml) with or without 1.2 mM NADPH in 0.1 M potassium phosphate buffer (pH 7.4) containing 3.3 mM MgCl2. Mixtures were kept at 37°C for up to 60 minutes, and reactions were terminated at indicated timepoints by addition of two volumes of acetonitrile containing 0.1% formic acid (FA) and 25 nM of internal standard (IS) d8-KZR-616. The disappearance of test compound and their diol metabolites and the metabolites of probe substrates were monitored using liquid chromatography with tandem mass spectrometry (LC-MS/MS) or liquid chromatography with UV detection (LC-UV) methods as described below. Each experimental reaction was conducted in triplicate. The intrinsic clearance (Clint) was displayed by the rate of parent compound disappearance. Testosterone (50 µM) and cis-stilbene oxide (SO; 50 µM) were used as probe substrates for P450 and mEH, respectively.
Metabolic Stability in Monkey Hepatocytes.
Pooled cryopreserved monkey hepatocytes were thawed and resuspended in culture media. KZR-59177, KZR-616, and KZR-59240 (1 µM) and reference substrates (50 µM testosterone, cis-SO and trans-SO) were incubated in duplicate with monkey hepatocytes (0.25 × 106 cells/ml) in a 37°C cell culture incubator with 95% air and 5% CO2 for 40 minutes. Reactions were terminated at various time points by addition of two volumes of acetonitrile containing 0.1% FA and 25 nM of IS d8-KZR-616. Samples were centrifuged, and KZR-616, KZR-59177, and KZR-59240; their diol metabolites KZR-59587, KZR-59165, and KZR-59433; and the metabolites of probe substrates were quantified by LC-MS/MS and LC-UV as described below. Testosterone (50 µM), cis-SO (50 µM), and trans-SO (50 µM) were used as probe substrates for P450 and epoxide hydrolase (EH), respectively.
P450 Phenotyping in LM.
KZR-616 was incubated in HLMs in the absence and presence of known chemical inhibitors for the six major P450 isoforms: furafylline (CYP1A2, 10 µM), montelukast (CYP2C8, 5 µM), sulfaphenazole (CYP2C9, 5 µM), benzylnirvanol (CYP2C19, 1 µM), quinidine (CYP2D6, 1 µM), and ketoconazole (CYP3A4/5, 1 µM). Reaction mixtures contained a final concentration of 1 µM KZR-616, 0.2 mg/ml HLM protein, and 1 mM NADPH in 100 mM potassium phosphate (pH 7.4) buffer with 3.3 mM MgCl2. The extent of metabolism was calculated as the disappearance of KZR-616 after incubation of 15 and 30 minutes compared with the 0-minute control reaction incubations. The formation of KZR-59587 (KZR-616 diol) was monitored as well.
Liver Microsome Binding.
MLMs (0.25 mg/ml) and HLMs (0.5 mg/ml) were preincubated with 50 µM of NSPA in 0.1 M potassium phosphate buffer (pH 7.4) for 10 minutes followed by addition of KZR-616, KZR-59177, KZR-59240, and chlorpromazine (control) at a final concentration at 1 µM and incubation at 37°C for 30 minutes. Incubated solutions were centrifuged at 85,000 rpm for 4 hours at 37°C, aliquots (30 µl) of the supernatants were added with equal volume of compound-free blank LM residues, and aliquots (30 µl) of the LM residues were added with equal volume of compound-free blank supernatants. Samples were quenched with 150 µl of internal standard solution in acetonitrile/methanol (1:1) containing 0.1% FA. After centrifugation, the supernatants of all samples were subjected to LC-MS/MS analysis. The percentage of test compound bound to protein is calculated by the following equation: % Free = (concentration of the test compound in supernatant/concentration of the test compound in LM residues) × 100%; % Bound test compound = 100% − % Free
Plasma Protein Binding.
KZR-616 binding to cynomolgus monkey plasma binding was conducted by equilibrium dialysis. Dialysis plates were prepared by adding 350 µl phosphate buffer (pH7.4) to the buffer chamber and 200 µl of 5 µM KZR-616 and controls (phenacetine; quinidine and warfarin) to the Red side. The plates were sealed and incubated at 37°C for 5 hour at 100 rpm on an orbital shaker. Duplicate analysis samples were prepared by mixing 50 µl plasma sample (from Red side) with 50 µl of blank buffer and 50 µl buffer sample (from buffer chamber) with 50 µl of blank plasma. Samples were quenched with 150 µl of internal standard solution in acetonitrile/methanol (1:1) containing 0.1% FA. After centrifugation, the supernatants of all samples were subjected to LC-MS/MS analysis.
Pharmacokinetics in Monkeys.
All of the animal studies were carried out in accordance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the US National Institutes of Health and were approved by the Institution’s Animal Care and Use Committee or local equivalent (Institute of Laboratory Animal resources, 1996). KZR-616, KZR-59177, and KZR-59240 were administrated to cynomolgus monkeys as single subcutaneous administrations. A total of 16 male, non-naïve, and nonfasted animals were randomly divided into three groups (four animals/group) receiving a dose of 3 mg/kg of the test compounds in aqueous 10% polysorbate (PS-80). Blood samples were collected prior to dosing and at 2, 5, 10, 20, and 30 minutes and 1, 2, 4, 8, and 24 hours postdose. Plasma samples were obtained by centrifuging at 3000 rpm at 4°C for 5 minutes. Plasma samples were quenched by addition of three volumes of acetonitrile containing 0.1% FA and 25 nM of IS d8-KZR-616. Parent and diol metabolite levels were quantified by LC-MS/MS as described below.
Metabolism via Recombinant Human EH.
Reactions were conducted in 0.1 M Tris-HCl buffer for mEH (pH 9.0) and sEH (pH 7.5) containing 0.1 mg/ml fatty acid–free bovine serum albumin (BSA). A series of enzyme incubation solutions was prewarmed for 5 minutes at 37°C before the addition of 1 µM carfilzomib or KZR-616. Enzyme concentrations were 2, 4, 8, and 10 µg/ml for mEH and 1, 10, 25, 50, and 100 µg/ml for sEH. Reactions were terminated after 60 minutes by addition of two volumes of acetonitrile containing 0.1% FA and 25 nM of IS d8-KZR-616. Diol formation was quantified by LC-MS/MS. Trans-SO (50 µM) and cis-SO (50 µM) were used as probe substrates for sEH and mEH, respectively, and diol formation was quantified by LC-UV as described below.
Metabolic Profiling of KZR-616 in Human Hepatocytes.
Pooled cryopreserved human hepatocytes were thawed and resuspended in cell culture media. Reactions containing hepatocytes (2 × 106 cells/ml) and KZR-616 (10 µM) were conducted in cell media in a 24-well cell culture plate at a 37°C cell culture incubator with 95% air and 5% CO2 for 2 hours. Reactions were terminated by addition of four volumes of acetonitrile containing 0.1% FA. A separate aliquot of hepatocytes (2 × 106 cells/ml) was quenched with four volumes of acetonitrile and then spiked with KZR-616 (10 µM) as a control sample. The resulting mixture was centrifuged at 4000 rpm for 15 minutes. Supernatants were dried under an N2 stream, and the resultant residue was reconstituted with 30% acetonitrile (v/v) before LC-MS/MS analysis. Metabolite identification was conducted using an Applied Bio-systems mass spectrometer (API 4000 QTrap) with Shimadzu LC-20. Chromatographic separation was performed using a Kinetex 2.6 µ C18 100A column (3.0 mm × 50 mm) with mobile phases (A: water containing 0.1% FA; B: acetonitrile containing 0.1% FA) at a flow rate of 0.6 ml/min. The mobile phase step increased from 5% (at 0.3 minutes) to 95% B (at 12 minutes). The total run time was 15 minutes.
Metabolism in Human Hepatocytes.
Pooled cryopreserved human hepatocytes (0.5 × 106 cells/ml) were preincubated in culture media containing 0.1% DMSO with varying concentrations of inhibitors of EHs or P450 (10 µM NSPA for 5 minutes; 200 µM TPPU and 500 µM 1-ABT for 30 minutes) in a 37°C cell culture incubator with 95% air and 5% CO2. After preincubation, KZR-616 (1 µM) and reference substrates (50 µM testosterone, cis-SO, and trans-SO) were incubated in triplicate in the presence or absence (1-ABT, NSPA, and TPPU, respectively, in a 37°C cell culture incubator for 1 hour. After 1, 10, 20, 30, 40, and 60 minutes as indicated, reactions were terminated by addition of two volumes of acetonitrile containing 0.1% FA and 25 nM of IS d8-KZR-616. Samples were centrifuged, and KZR-616, the diol, and metabolites of reference substrates were quantified by LC-MS/MS and LC-UV as described below.
Metabolite Profiling of KZR-616 in Human Plasma.
Plasma samples were collected from patients with SLE receiving weekly subcutaneous administration of KZR-616 for 13 weeks (NCT03393013). Samples from patients receiving a KZR-616 dose of 75 mg, the maximum administered dose, were analyzed. Each sample was AUC pooled from the time points of blood withdraw (predose, 0.0833, 0.25, 0.5, 1, 2, 4, 8 hours postdose) on day 29 from one of the four subjects (110-004, 112-021, 113-002, and 116-012). Each plasma sample (400 µl) was extracted with three volumes of ACN/MeOH (1:1) containing 0.1% FA and then centrifuged at 3000 rpm for 15 minutes. The supernatant from each sample was concentrated under a nitrogen stream and reconstituted in 200 µl of 30% ACN/MeOH (1:1) containing 0.1% FA. Each sample was injected into the liquid chromatography (LC)-MS system for separation and identification. Chromatographic separation was performed using a Waters XBridge C18, 3.0 × 150 mm, 3.5 µm with mobile phases (A: water containing 0.1% FA; B: acetonitrile containing 0.1% FA) at a flow rate of 0.8 ml/min. The mobile phase step increased from 3% at 0.3 minutes to 35% B at 25 minutes and to 90% B at 26 minutes. The total run time was 35 minutes. Metabolite identification was conducted using an AB Sciex Triple TOF API6600 mass spectrometer equipped with a Shimadzu Nexera UPLC system. Metabolite structures were assigned based on accurate mass (±5 mDa) determination of a parent ion from a full-scan TOF MS followed by triggered MS/MS fingerprints.
Reaction Kinetics.
Reaction kinetics of KZR-616 were conducted in female and male HLMs (0.5 mg/ml) and MLMs (0.2 mg/ml) in 0.1 M potassium phosphate buffer (pH 7.4) with 0.1 mg/ml BSA at 37°C for 30 minutes. The kinetics of KZR-616 were also studied in human hepatocytes in hepatocyte maintenance medium at 37°C for 30 minutes. Reaction kinetics of KZR-616 were determined in 4 µg/ml recombinant human mEH at 37°C for 30 minutes in 0.1 M Tris-HCl buffer (pH 9.0) containing 0.1 mg/ml BSA. After prewarming enzymes for 5 minutes, KZR-616 (0–1000 µM) was added to initiate the reaction. For kinetic study of carfilzomib, incubations were optimized and conducted using 2 µg/ml prewarmed recombinant human mEH at 37°C for 20 minutes in 0.1 M Tris-HCl buffer (pH 9.0) containing 0.1 mg/ml BSA.
NSPA was assessed in female and male HLMs (0.5 mg/ml) and MLMs (0.25 mg/ml), human hepatocytes (0.5 × 106 cells/ml), and recombinant human mEH (4 µg/ml) to determine IC50 values on KZR-616 (10 µM) utilizing varying concentrations of NSPA (0–100 µM) in the reaction buffers of the enzymes at 37°C for 15–30 minutes. NSPA was also assessed in recombinant human mEH to determine the IC50 on carfilzomib epoxide hydrolysis. Briefly, prewarmed recombinant human mEH (2 µg/ml) was incubated with 1 µM CFZ in a range of NSPA concentrations (0–10 µM) at 37°C for 20 minutes. Reactions were terminated by addition of two volumes of acetonitrile containing 0.1% FA and 25 nM of IS d8-KZR-616, and the concentrations of diol derivatives of KZR-616 and carfilzomib were quantified using LC-MS/MS methods. Each set of data was fit to a simple Michaelis-Menten kinetics model using nonlinear regression data analysis (GraphPad Prism 7.04.). Each experimental reaction was conducted in duplicate. Cis-SO (50 µM) incubation was conducted in parallel as a positive control.
Epoxide Hydrolase Inhibition Activity.
In vitro inhibition potential of KZR-616 on mEH and sEH was investigated in LM and recombinant EH. Cis-SO and trans-SO were used as probe substrates for mEH and sEH, respectively. For testing of KZR-616 as an inhibitor of mEH, prewarmed pooled HLMs (0.5 mg/ml), MLMs (0.25 mg/ml), and recombinant mEH (4 µg/ml) were incubated with cis-SO (50 µM) in the absence or presence of 100 µM KZR-616 in the reaction buffers at 37°C for 30 minutes. For study of KZR-616 as an inhibitor of sEH, prewarmed pooled human hepatic cytosol (0.5 mg/ml) and recombinant sEH (100 µg/ml) were incubated with trans-SO (50 µM) in the absence or presence of 100 µM KZR-616 in 0.1 M potassium phosphate buffer (pH 7.4) containing 0.1 mg/ml BSA and in 0.1 M Tris-HCL buffer (pH 7.4) containing 0.1 mg/ml BSA, respectively. Reactions were quenched with two volumes of cold acetonitrile, and the concentrations of diol derivatives of cis-SO and trans-SO were quantified by using LC-UV methods as described below.
LC-MS/MS and LC-UV Quantification.
LC-MS/MS methods were developed for quantifying carfilzomib, KZR-616, KZR-59177, KZR-59240, the P450 probe substrate (testosterone) and their diol derivatives using an AB Sciex 5500 Q-Trap mass spectrometer equipped with an electrospray ionization source. Multiple reaction monitoring was used with following mass transitions for each compound m/z (parent > product ion): 587.4 > 371.2, 605.1 > 389.2, 571.3 > 389.6, 589.7 > 150.1, 607.3 > 391.2, 719.8 > 402.2, 737.9 > 402.2, 289.3 > 109.1, 305.1 > 269.1, and 594.9 > 371.2 for KZR-616, KZR-59587 (diol), KZR-59177, KZR-59165 (diol), KZR-59240, KZR-59433 (diol) carfilzomib, testosterone, 6β-testosterone, and IS d8-KZR-616, respectively. Chromatographic separation was achieved using YMC-pack pro C18 (3 µ, 3.0 mm × 50 mm) (A) water and (B) acetonitrile containing 0.1% FA at flow rate of 0.5 and 1.0 ml/min. For the flow rate at 0.5 ml/min, the mobile phase started at 5% B for 0.5 minutes, progressed linearly to 95% acetonitrile in 2.3 minutes, held at 95% B for 0.7 minutes, and returned to 5% B with 0.1 minutes. For the flow rate at 1.0 ml/min, the mobile phase started at 10% B for 0.5 minutes, which was followed by a linear change up 95% acetonitrile in 1.7 minutes, held at 95% B for 0.5 minutes, and returned to 5% B with 0.1 minutes.
Quantification of cis-SO, trans-SO, and corresponding metabolites was fulfilled using a LC-30AD system coupled with a SPD-20AV detector. Chromatographic separation was achieved using Kinetex reverse phase C18 (1.7 µ, 2.1 mm × 50 mm) column with mobile phase (A) water and (B) acetonitrile containing 0.1% FA at flow rate of 0.5 ml/min. The mobile phase started at 10% B for 1.0 minute and gradually increased to 95% acetonitrile in 5 minutes, held at 95% B for 1.0 minute, and returned to 10% B with 0.1 minutes.
A calibration curve was established for each test compound by plotting its peak area ratio for each compound against IS (d8-KZR-616) versus the corresponding concentrations of each compound across a calibration range (1–5000 ng/ml) and fitting with a linear regression equation. The calibration curve was then used to calculate the concentration of each test compound.
Kinetic Analysis and IC50 Data Processing.
The kinetics of epoxide hydrolysis of KZR-616 were obtained from duplicate experiments with KZR-616 in a concentration range from 0 to 1000 µM. Km and Vmax were calculated by fitting the diol (KZR-59587) formation data into the Michaelis-Menten equation using GraphPad Prism software. IC50 was calculated by using nonlinear fit of log (inhibitor) versus response-variable slope. The IC50 data were validated only when the data lay within the inhibitor concentration range.
GraphPad Prism was used for the statistical analysis. All data are expressed in mean ± S.D. format. For the kinetic and IC50 analyses, 95% CI (confidence interval) threshold with goodness of fit (R2 > 0.99) were used.
Results
Characterization of the Metabolism of KZR-616 and Its Analogs in Liver Microsomes and Monkey Hepatocytes.
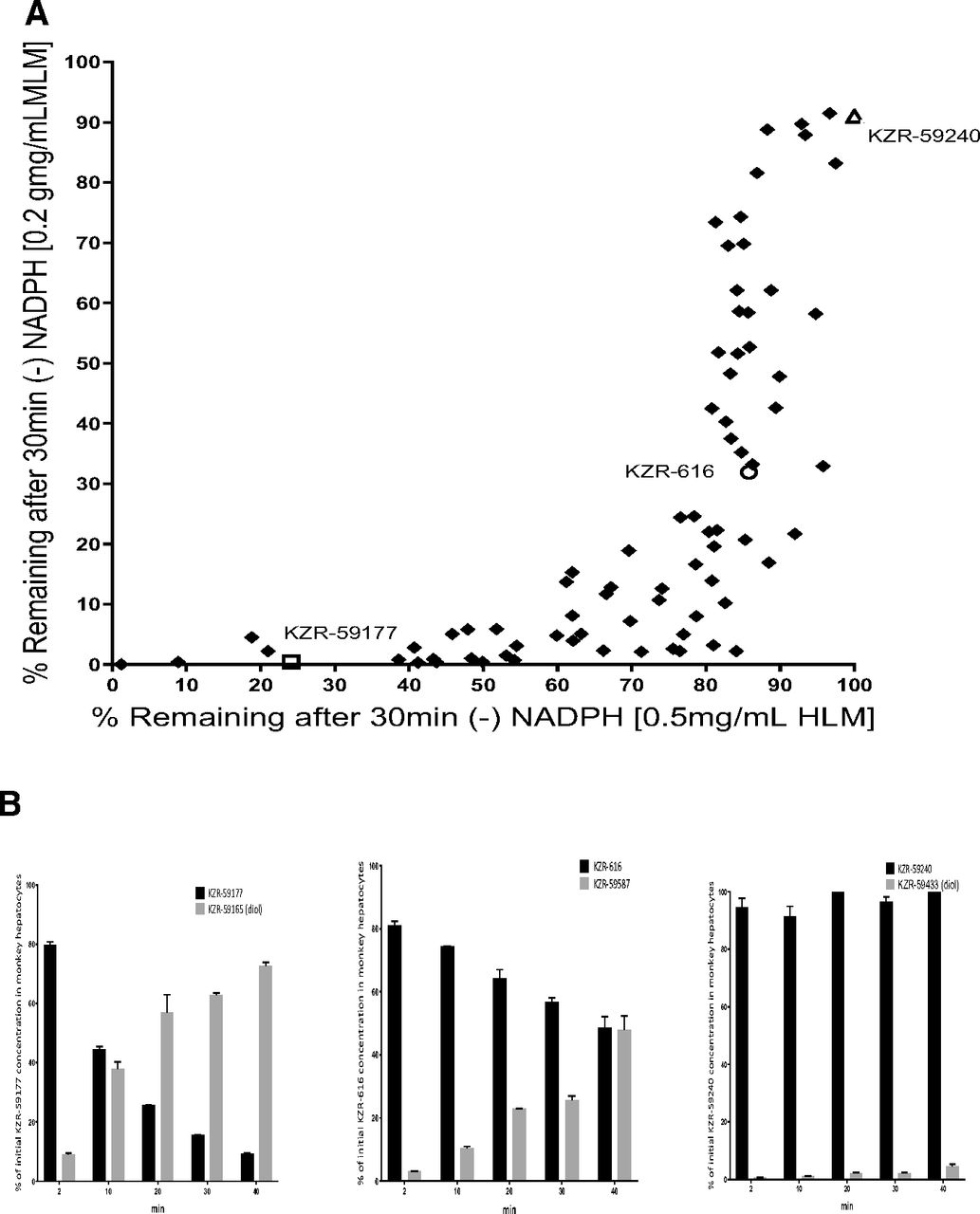
KZR-616 was selected from a medicinal chemistry effort that involved the synthesis of over 400 hundred tripeptide epoxyketones (patents: McMinn D, et al. Patent Application US9657057B2; Patent Application US10647744B2). To further understand the structure-activity relationship of this chemotype, the metabolic stability of a subset of 79 potent and selective analogs was evaluated in an in vitro model system utilizing HLM and MLM, which exhibit high levels of both mEH and P450 activity. Testosterone, cis-SO, and trans-SO were used as probe substrates for P450 (Supplemental Fig. 1), mEH, and sEH (Supplemental Fig. 2), respectively. Epoxide hydrolase stability was assessed in mixtures of compound and LM in the absence of NADPH to prevent P450 activity. As shown in Fig. 2A, a wide range of epoxide hydrolysis stability across the 79 tested compounds was noted in both HLM and MLM incubations. KZR-616, KZR-59177 (an unstable analog), and KZR-59240 (a stable analog) were chosen as tool compounds to explore P450-mediated metabolism pathways in LM. The intrinsic clearance of KZR-616, KZR-59177, and KZR-59240 increased significantly with the addition of NADPH in the LM incubations, suggesting that P450 enzymes play the dominant role in metabolism in this experimental system (Table 1).

(A) Metabolism of KZR-616 and analogs in human and monkey liver microsomes in the absence of NADPH. Percentages of detected residual KZR-616, KZR-59177, and KZR-59240 and a total of 79 analogs in HLMs (0.5 mg/ml) or MLMs (0.25 mg/ml) in the absence of NADPH. Initial concentrations of KZR-616 and analogs were 1 µM. Residual concentrations of KZR-616 and analogs were quantified using LC-MS/MS assays. Data are presented as mean ± S.D. from triplicate incubations. (B) Metabolism of KZR-616 and analogs in monkey hepatocytes. Percentage of KZR-616, KZR-59177, and KZR-59240 remaining and their diol formation from initial parents in monkey hepatocytes (0.25 × 106 cells/ml) after 40-minute incubation. The initial concentration of parents was 1 µM. Concentrations of parents and their diols were quantified using LC-MS/MS analysis. Data are presented as mean ± S.D. from duplicate incubations.
In vitro metabolic stability of KZR-616 and analogs in HLMs and MLMs in the presence or absence of NADPH
Predicted t1/2 and Clint were obtained from in vitro assays. Data are presented as mean ±
S.D. (n = 3). The conc. of test compound in the assays was 1 µM. LM binding (%) of test compound was conducted using ultracentrifugation.
We next determined the effect of microsomal binding on the intrinsic clearance rates of three test compounds. The % bound average was found to be 7.4% and 12.3% for KZR-616, and 21.3% and 20.4% for KZR-59240 in monkey and human LM mixtures, respectively. The % bound average for KZR-59177 was not applicable because of stability issues in the LM mixtures that led to negative % bound values. Overall, the difference of the % bound values between KZR-616 and KZR-59240 was small in respective human and monkey LM and, as such, the intrinsic clearance of KZR-616 and KZR-59240 (Table 1) can be considered reliable.
To evaluate the metabolic stability of these three compounds, we also selected cell-base in vitro model using monkey hepatocytes, which exhibit high levels of both mEH and P450 activity. As shown in Fig. 2B, quantification of disappearance of KZR-59177, KZR-616, and KZR-59240 and their diol formations were performed using synthesized standards. The recovery of parents and diols were 82.0, 96.4, and 100% for KZR-59177, KZR-616, and KZR-59240, respectively, after 40-minute incubations. These quantitative results suggest that epoxide hydrolysis was the major metabolic pathway for the three tool compounds in monkey hepatocytes.
P450 phenotyping demonstrated that CYP3A4/5 was the major enzyme involved in the microsomal metabolism of KZR-616, with CYP2C8 playing a minor role in its metabolism. Formation of the KZR-616 diol, KZR-59587, was not affected by the presence of P450 inhibitors, demonstrating that other enzymes, such as epoxide hydrolases, play important roles in diol formation (Supplemental Table 1).
Pharmacokinetics of KZR-616, KZR-59177, and KZR-59240 in Monkeys.
Given the apparent difference in metabolic stability seen in vitro, we next studied the pharmacokinetics (PK) of KZR-616, KZR-59177, and KZR-59240 in cynomolgus monkeys after single subcutaneous administration of 3 mg/kg. As shown in Table 2, total exposures (area under the curve from zero to infinity, AUCinf) were highest with KZR-59240, followed by KZR-616 and KZR-59177. The diol derivatives were determined to be the major metabolites for each compound, and the AUC ratios (diol: parent) were 3.40 ± 0.81, 3.35 ± 1.20, and 0.78 ± 0.46 for KZR-616, KZR-59177, and KZR-59240, respectively. The PK of both parent and diol for each compound was consistent with the stability results in MLM in the absence of NADPH, in which the P450 enzymes were not activated. Interestingly, administration of KZR-59240, which was resistant to diol formation in vitro, resulted in a significant level of diol formation (AUC ratio of diol/parent: 0.78 ± 0.46) in monkeys.
PK parameters of KZR-616 and analogs after a single subcutaneous dose at 3 mg/kg in monkeys
Involvement of Human mEH and sEH on KZR-616 Epoxide Hydrolysis.
KZR-616 is structurally related to carfilzomib, which is also metabolized to the inactive diol in vivo (Table 2). Since epoxide hydrolysis was identified to be the major metabolic pathway of KZR-616 in vivo, we assessed the contribution of mEH and sEH on diol formation from KZR-616 and carfilzomib using recombinant enzymes. Formation of the diol derivatives of KZR-616 (Fig. 3A) and carfilzomib (Fig. 3B) increased with an increase of mEH from 2 to 10 µg/ml, but no significant diol derivatives were detected for either compound in the presence of recombinant sEH up to 200 µg/ml. Epoxide hydrolysis of KZR-616 was also investigated in human and monkey liver microsomes (containing mEH) and hepatic cytosols (containing sEH). Probe substrates cis-SO (50 µM) and trans-SO (50 µM) were used as controls for mEH and sEH activity, respectively (Supplemental Fig. 2). Similar to findings using recombinant enzymes, diol formation increased with increasing liver microsomal concentration, but minimal diol formation was seen in incubations with hepatic cytosols (Fig. 3, C and D). Taken together, these data indicate that the epoxide hydrolysis of KZR-616 and carfilzomib is mediated predominantly by mEH and that sEH plays little to no role in the metabolism of these compounds.

Epoxide hydrolysis of KZR-616 and carfilzomib in recombinant mEH and sEH; epoxide hydrolysis of KZR-616 in HLMs, MLMs, and human and monkey cytosol. (A) Epoxide hydrolysis of KZR-616 in recombinant human mEH (2–10 µg/ml) and sEH (20–100 µg/ml). (B) Epoxide hydrolysis of carfilzomib (CFZ) in recombinant human mEH (2–10 µg/ml) and sEH (20–200 µg/ml). (C) Epoxide hydrolysis of KZR-616 in HLM (0.25–1 mg/ml) and human cytosol (0.4–1 mg/ml). (D) Epoxide hydrolysis of KZR-616 in MLM (0.2–1 mg/ml) and monkey cytosol (0.1–1 mg/ml). The concentrations of KZR-616, carfilzomib, and their diols were quantified using LC-MS/MS assays. Data are presented as mean ± S.D. from triplicate incubations.
KZR-616 Metabolism in Human Hepatocytes and Plasmas.
Next, we quantitatively evaluated the disappearance of KZR-616 and the formation of KZR-59587 and additional metabolites in human hepatocytes. The metabolites identified following exposure of KZR-616 for 2 hours is shown in Fig. 4-3A. Direct epoxide hydrolysis was the major pathway of KZR-616 in this system. Only trace levels of additional metabolites, including from demethylation and oxidation, and the combination of oxidation and epoxide hydrolysis were detected. The MS/MS spectrum of each metabolite was displayed in Fig. 4-1.



The MS/MS spectrum of identified metabolites. The metabolites identified from the 2-hour incubations in human hepatocytes (2.0 × 106 cells/ml). (4-2) KZR-616 metabolism in human hepatocytes and effect of 1-ABT, NSPA and TPPU. (A) Percentage of KZR-616 remaining in human hepatocytes in the presence or absence of 1-ABT. (B) Formation of diol KZR-59587 in human hepatocytes in the presence or absence of 1-ABT. (C) Percentage of KZR-616 remaining in human hepatocytes in the presence or absence of NSPA and TPPU. (D) Formation of diol KZR-59587 in human hepatocytes in the presence or absence of NSPA and TPPU. The concentration of cryopreserved human hepatocytes in incubations was 0.5 × 106 cells/ml. The initial concentration of KZR-616 was 1 µM. The concentrations of inhibitors were 0.5 mM, 10 µM, and 200 µM for 1-ABT, NSPA, and TPPU, respectively. Concentrations of KZR-616 and KZR-59587 were quantified using LC-MS/MS analysis. Data are presented as mean ± S.D. from duplicate incubations. (4-3) The metabolic pathways of KZR-616 in human hepatocytes and plasmas. (4-3A) The metabolite identification after exposure of KZR-616 (10 µM) in pooled cryopreserved human hepatocytes (2 × 106 cells/ml) for 2 hours. (4-3B) The metabolite identification in human plasma from four patients (0–8 hours postdose) receiving a 75-mg dose of KZR-616.
We used 1-ABT (a pan-P450 inhibitor), NSPA (an inhibitor of mEH), and TPPU (an inhibitor of sEH) to evaluate these metabolic pathways. There was no difference in the rate of clearance of KZR-616 upon P450 inhibition (Fig. 4-2) with intrinsic clearance values of 16.5 ± 0.306 and 18.6 ± 0.400 µl/min/106 cells in the presence and absence of 1-ABT, respectively. Recovery of KZR-616 and diol was around 100% over the 60-minute time course. Similarly, elimination of KZR-616 and formation of diol was not affected by concentrations of TPPU up to 200 µM. In contrast, NSPA affected both the loss of parent and formation of diol, indicating that mEH plays the primary role in the metabolism of KZR-616 in human hepatocytes.
The metabolites identified in human plasma from four patients (0-8 hours postdose) receiving a 75-mg dose of KZR-616 are shown in Fig. 4-3B. KZR-59587 (diol) was identified to be the major metabolite. KZR-59587 and its parent KZR-616 were quantified as peak area percentage of total peak area of KZR-616, and all observed metabolites were 33.8 and 61.2; 45.8 and 49.0; 47.9 and 46.5; and 43.8 and 51.8, respectively for the four patients (Table 4). Other metabolites, including the isomer of KZR-59587, oxidation, hydrolysis, and dehydrogenation, and double oxidation of KZR-616 were all less than 1% of total peak area of KZR-616 and metabolites.
Summary of kinetic parameters from Michaelis-Menten analysis of epoxide hydrolysis of KZR-616 (data represent mean ± S.D.; duplicate incubations; efficiency defined as Vmax/Km).
Summary of KZR-616 and proposed metabolites observed in human plasma
Kinetics of KZR-616 Epoxide Hydrolysis.
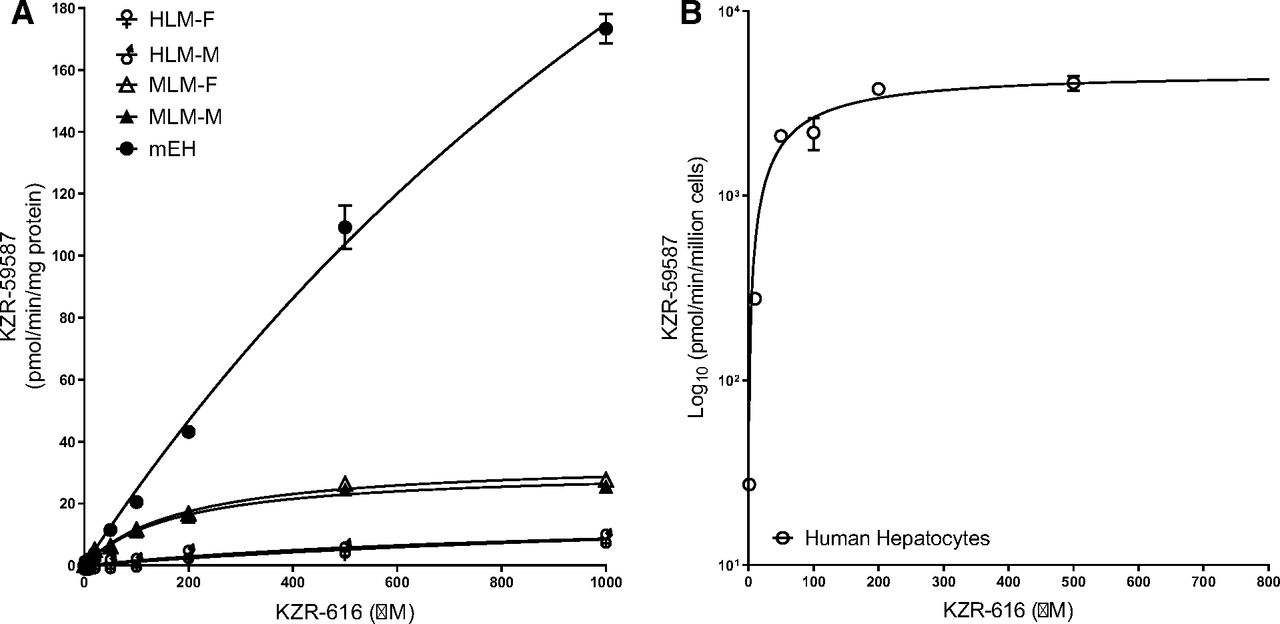
Next, we determined the reaction kinetics of diol formation using human recombinant mEH, HLMs, and human hepatocytes across a wide range of KZR-616 concentrations (0–1000 µM) (Fig. 5A, and 5B). Optimized enzyme concentrations and incubation times were applied to ensure that initial mEH-mediated hydrolysis was in the linear product formation range for accurate estimation (Fig. 6). Inhibition of epoxide hydrolysis of KZR-616 and cis-SO by NSPA was evaluated in parallel. NSPA inhibited mEH-mediated hydrolysis for KZR-616 with mean IC50 values ranging from 0.409 to 0.826 µM across the different test systems (Fig. 7). From these experiments, we were able to obtain kinetic constants (Km), maximum velocities (Vmax) and efficiency (Vmax/Km) of KZR-616 epoxide hydrolysis (Table 3). As expected, the greatest metabolic efficiency was observed with recombinant mEH. Metabolic efficiencies of female/male human and monkey liver microsome were 0.33%–0.45% and 4.5%–4.6% of recombinant mEH, respectively, suggesting that mEH levels are lower in HLMs versus MLMs.

Kinetics of epoxide hydrolysis of KZR-616 in HLMs, MLMs, recombinant human mEHs, and human hepatocytes. (A) Kinetics of diol KZR-59587 formation in male and female HLMs, MLMs, and recombinant human mEHs were performed at 37°C for 30 minutes. (B) Kinetics of the formation of KZR-59587 in human hepatocytes cells at 37°C for 30 minutes. The Km and Vmax values were estimated by fitting the curve to the Michaelis-Menten equation. Data represent mean ± S.D. from duplicate incubations.

Inhibition of epoxide hydrolysis of KZR-616 in HLMs, MLMs, human hepatocytes, and recombinant human mEHs by NSPA. (A) Inhibition of mEH activity by NSPA in male and female HLMs. (B) Inhibition of mEH activity by NSPA in male and female MLMs. (C) Inhibition of mEH activity by NSPA in human hepatocytes. (D) Inhibition of mEH activity by NSPA in recombinant human mEH. The IC50 values were estimated using nonlinear regression data analysis. Data are presented as mean ± S.D. from duplicate incubations.

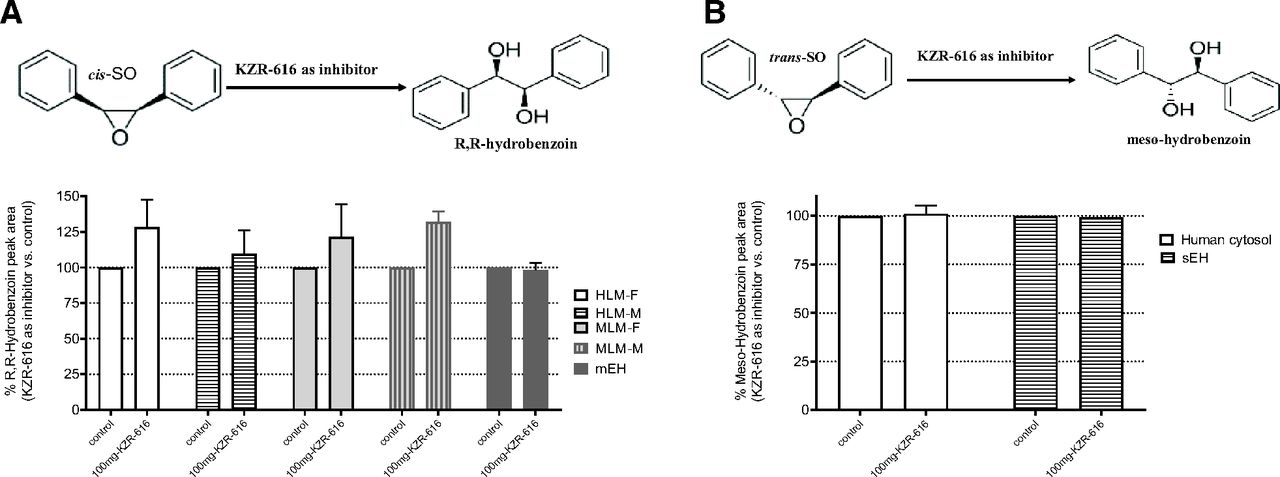
Inhibition of epoxide hydrolysis of probe substrates cis-SO and trans-SO by KZR-616. (A) Scheme of cis-SO metabolism and inhibition of cis-SO hydrolysis by KZR-616. (B) Scheme of trans-SO metabolism and inhibition of trans-SO hydrolysis by KZR-616. The diol derivatives of cis-SO and trans-SO (R,R-hydrobenzoin and mesoydrobenzoin) were quantified by using LC-UV methods.
Finally, we sought to determine whether KZR-616 could inhibit EH activity in recombinant enzymes, LMs, or intact cells. Epoxide hydrolysis of cis-SO by mEH and epoxide hydrolysis of trans-SO by sEH were unaffected by the presence of KZR-616 at concentrations up to 100 µM (Fig.).
Discussion
Targeted covalent inhibitors provide several advantages over conventional reversible inhibitors, including increased efficacy and selectivity (Leung et al., 2017). Peptide epoxyketone-based proteasome inhibitors, such as carfilzomib and oprozomib, can induce prolonged pharmacodynamic effects despite rapid clearance and short systemic exposure in vivo due in part to covalent modification of proteasome active sites (Yang et al., 2011; Wang et al., 2013). These compounds are primarily metabolized to the inactive diol via epoxide hydrolysis, which renders the molecule inactive. Given this nontraditional metabolic pathway, there are currently no well characterized in vitro test systems to study the metabolic properties of this class of compounds. KZR-616 is a novel peptide epoxyketone that was designed to selectively bind the immunoproteasome active sites LMP7 and LMP2 and is currently being evaluated in phase 2 clinical trials in autoimmune disorders (Johnson et al., 2018). We studied KZR-616 and its analogs in multiple in vitro test systems ranging from recombinant enzymes to live cell incubation and compared those data with clearance values after administration to nonhuman primates in an effort to determine optimal in vitro test systems for future medicinal chemistry campaigns.
In this work, we attempted to develop ADME assays that can be used for screening for irreversible covalent protease inhibitors. During the drug discovery stage, a challenge we encountered was to find an optimum in vitro test system that can correlate with in vivo PK results. Three tool compounds (KZR-616, KZR-59177, and KZR-59240) were used to study their metabolic stabilities in human and monkey liver microsomes (LMs) in the presence and absence of NADPH. We found that the Clint of these compounds was driven by P450 plus EH in the presence of NADPH but only by EH in the absence of NADPH and that CYP3A4/5 was the major isoform of P450 driving metabolism in the presence of NADPH in LM. However, PK data revealed that their diol derivatives were the predominant metabolites, indicating that mEH play a major role in the in vivo and that P450-mediated metabolism is not involved. This implies that the in vitro LM test system does not generate a correlated or indicative metabolic result.
By using monkey hepatocytes, we found that diol derivatives were the major metabolites for these compounds, indicating that the mEH play a major role in the hepatocyte system and correlating with PK results. The clinical candidate KZR-616 was further investigated in human hepatocytes. The metabolism profiling of KZR-616 in human hepatocytes indicated that the diol KZR-59587 generated from mEH metabolic pathway was the major metabolite of KZR-616 and only trace levels of other metabolites from oxidation and dehydrogenation pathways. Using the inhibitors of P450 and EHs to study KZR-616 metabolism in human hepatocytes also concluded that mEH-mediated metabolic is the major pathway. Similar to the monkey system, metabolism profiling of human plasma samples from patients receiving KZR-616 revealed the diol KZR-59587 as the major metabolite. All other metabolites were all less than 1% of total of the parent and the observe metabolites. Therefore, the monkey systems both in vitro and in vivo were predictive of the ADME properties of KZR-616 in humans.
Liver microsomes are a widely used in vitro test system for high-throughput ADME screening in lead compound discovery programs. However, concerns related to the correlation with in vivo study results and overestimation of P450 activity have recently been reported (Tingle and Helsby, 2006; Brown et al., 2007). In the presence of NADPH, a necessary cofactor for P450 activity in LM preparations, KZR-616 and its analogs were rapidly cleared, but the recovery of parent and diol was poor (∼50% in 1 hour), indicating that there were other metabolic pathways involved. However, in the absence of NADPH, the stability with varying degrees was observed across 79 related compounds. Diol products were also the major metabolite found after administration of KZR-616 and two other structurally related compounds to monkeys. The PK exposures measured in vivo for these compounds correlated well with the observed stability in LM incubations in the absence of NADPH. Interestingly, for KZR-59240, which was stable in LM incubations and showed minimal diol formation, significant formation of the diol metabolite (KZR-59433) was measured in vivo. The discrepancy between the metabolite detection from standard LM cultures in the absence of NADPH and in vivo pharmacokinetic studies indicates that EH enzymatic activities might be lost during microsome preparation or suppressed by other enzymes in LM test systems.
To overcome the major deficiency of using LM to study peptide epoxyketone metabolism, we used cryopreserved human hepatocytes as an in vitro test system. Intact hepatocytes contain both phase I and II metabolism enzymes and likely provide a more comprehensive system to correlate in vitro test results with in vivo PK (McGinnity et al., 2004; Newman JW et al., 2005; Brown et al., 2007). Similar to what was observed in PK studies, the predominant metabolite of KZR-616 found after incubations with intact hepatocytes was the diol. As demonstrated using enzyme-specific probe substrates, hepatocytes contain both sEH and mEH, and we used isoform-specific inhibitors to phenotype KZR-616 metabolism. Diol formation was blocked by the mEH inhibitor NSPA, but neither TPPU (an sEH inhibitor) nor ABT-1 (a pan-P450 inhibitor) had an impact on diol formation or loss of parent compound. Thus, using human hepatocytes we were able to match the metabolite profile seen in vivo as well as determine the specific enzyme responsible for diol formation from KZR-616.
Epoxide hydrolases are a ubiquitously expressed family of enzymes found in mammals (de Waziers et al., 1990; Enayetallah, et al., 2006; Morisseau, 2013; Gautheron and Jéru, 2020) and are the primary pathway for the detoxification of compounds containing an epoxide residue (Decker et al., 2009; Kitteringham et al., 1996). There are two major isoforms: mEH, encoded by the EPHX1 gene (Hartsfield et al., 1998; Kerr et al., 1989; Vaclavikova et al., 2015) and localized predominantly in the endoplasmic reticulum, and sEH, encoded by the EPHX2 gene and confined mainly to cytoplasm (Larsson et al., 1995). Both are highly expressed in mammalian hepatocytes (Gill and Hammock, 1980; Coller et al., 2001). Using human hepatocyte cells, we determined that mEH is the primary enzyme-mediating metabolism of KZR-616 and its analogs. Enzymatic kinetic constants for KZR-616 epoxide hydrolysis were determined using recombinant enzymes, LMs, and in intact cells. These studies revealed that the EH-mediated metabolic efficiency of LM was <5% of the recombinant enzyme and that hepatocytes are a more efficient cell-based system for epoxide hydrolysis. Given that mEH also mediates metabolism of carfilzomib anted oprozomib, this represents a common pathway for metabolism of this class of compound. It is noteworthy that unlike KZR-616, both carfilzomib and oprozomib show peptidase hydrolysis as an additional metabolic pathway in vivo (Wang et al., 2013; Wang et al., 2017). It is also interesting that in vitro plasma stability studies of CFZ and KZR-616 in monkeys (unpublished data) at 37°C for 6 hours did not produce any peptide cleavage metabolites, and the epoxide hydrolysis metabolites (diol) were below Limit of quantification for both compounds, indicating lacking both peptidase and mEH in commercial monkey plasmas.
In conclusion, through a thorough analysis of in vitro test systems and analysis of pharmacokinetic data, the predominant metabolic pathway of KZR-616 was determined to be epoxide hydrolysis via the action of mEH. These data suggest that the PK of KZR-616 is unlikely to be affected by coadministration of P450 and sEH inhibitors/inducers and that KZR-616 is unlikely to alter epoxide hydrolysis of other mEH and sEH substrate drugs. Given the widespread tissue distribution and the efficiency of KZR-616 metabolism measured with recombinant enzyme, it would be difficult to saturate this process in vivo. This suggests a low risk for altered exposure to KZR-616 upon coadministration of known or suspected mEH inhibitors.
The work presented here describes a series of in vitro and cell-based enzymatic metabolism and kinetic investigations for peptide epoxyketone analogs. Despite some described limitations, hepatocytes served as a good in vitro test system to assess the metabolic profiles of KZR-616 and other peptide epoxyketones and could be useful in assessing novel covalent proteasome inhibitors during lead compound optimization.
Acknowledgments
We thank Celia Economides, Kiruthi Palaniswamy, Michelle Greenman, and Mark Shiller from Kezar Life Sciences for reviewing the manuscript and helpful suggestions.
Authorship Contributions
Participated in research design: Fang, Kirk, Wang.
Conducted experiments: Fang, Johnson, Anderl, Muchamuel, McMinn, Wang.
Contributed new reagent or analytic tools: Morisseau, Hammock.
Performed data analysis: Fang, Wang.
Wrote or contributed to the writing of the manuscript: Fang, Morisseau, Hammock, Kirk, Wang.
Footnotes
- Received November 9, 2020.
- Accepted June 24, 2021.
↵1 Current affiliation: University of California, Davis, California.
The work was supported by Kezar Life Sciences.
The authors declare that they have no competing interests.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- 1-ABT
- 1-aminobenzotriazole
- ADME
- absorption, distribution, metabolism, and excretion
- AUC
- area under the curve
- BSA
- bovine serum albumin
- Clint
- intrinsic clearance
- d8-KZR-616
- deuterated KZR-616
- EH
- epoxide hydrolase
- FA
- formic acid
- H
- human
- HLM
- human liver microsome
- IS
- internal standard
- LC
- liquid chromatography
- LC-MS/MS
- LC with tandem mass spectrometry
- LC-UV
- LC with UV detection
- LM
- liver microsome
- LMP
- low-molecular-mass polypeptide
- M
- monkey
- mEH
- microsomal epoxide hydrolase
- MLM
- monkey liver microsome
- MS
- mass spectrometry
- NSPA
- 2-nonylsulfanyl-propionamide
- P450
- cytochrome P450
- PK
- pharmacokinetics
- sEH
- soluble epoxide hydrolase
- SLE
- systemic lupus erythematosus
- SO
- stilbene oxide
- TNF-α
- tumor necrosis factor α
- TPPU
- 1-trifluoromethoxyphenyl-3-(1-propionypiperidin-4-yl) urea
- Copyright © 2021 by The Author(s)
This is an open access article distributed under the CC BY-NC Attribution 4.0 International license.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}