


Abstract
Bile acids have been known for decades to aid in the digestion and absorption of dietary fats and fat-soluble vitamins in the intestine. The development of gene knockout mice models and transgenic humanized mouse models have helped us understand other functions of bile acids, such as their role in modulating fat, glucose, and energy metabolism, and in the molecular regulation of the synthesis, transport, and homeostasis of bile acids. The G-protein coupled receptor TGR5 regulates the bile acid induced alterations of intermediary metabolism, whereas the nuclear receptor FXR regulates bile acid synthesis and homeostasis. However, this review indicates that unidentified factors in addition to FXR must exist to aid in the regulation of bile acid synthesis and homeostasis.
SIGNIFICANCE STATEMENT This review captures the present understanding of bile acid synthesis, the role of bile acid transporters in the enterohepatic circulation of bile acids, the role of the nuclear receptor FXR on the regulation of bile acid synthesis and bile acid transporters, and the importance of bile acids in activating GPCR signaling via TGR5 to modify intermediary metabolism. This information is useful for developing drugs for the treatment of various hepatic and intestinal diseases, as well as the metabolic syndrome.
Introduction
The chemical structures of bile acids were elucidated in the 1930s. Bile acids are produced in the liver, stored in the gallbladder, and secreted into the upper part of the small intestine after a meal. In the small intestine, bile acids help in the breakdown and absorption of fats and fat-soluble vitamins. The role of bile acids in the breakdown of fats was the dominant theme in bile acid research for a long time. Bile acids are absent in invertebrates but are present in all known vertebrate species (Haslewood, 1965, 1967). Although our knowledge of the biology of bile acids gradually increased over a span of fifty years or so after the elucidation of their chemical structures, the current state of understanding of the role of bile acids as metabolic sensors, as well as bile acid homeostasis and its molecular regulation has rapidly developed since the discovery of receptors for bile acids in 1999. It is now known that bile acids signal through two different types of receptors, one belonging to the nuclear receptor family and the other belonging to the G-protein coupled receptor family. Major advances in our understanding of the regulation of bile acid synthesis, transport, metabolism, and role of the microbiome in determining bile acid homeostasis and composition have been facilitated by the development of various gene knockout mice models and transgenic humanized mouse models.
Chemistry and Functions of Bile Acids
Bile acids are amphipathic molecules; that is, they have both polar and nonpolar regions that confer both hydrophilic (water-loving) and lipophilic (fat-loving) properties. Figure 1 illustrates that bile acids are saturated (no double or triple bonds), hydroxylated C24 steroid carboxylic acids derived from cholesterol (C27). Steroids contain a cyclopentanophenanthrene carbon skeleton; i.e., a phenanthrene (three fused benzene rings) with a carboxylic cyclopentane ring. Most steroids are alcohols and are called sterols (e.g., cholesterol, ergosterol, estradiol). In contrast, bile acids have an acid (‒COOH) group to their side chains; hence bile acids are steroid carboxylic acids. The steroid nucleus of bile acids has four fused carbon rings consisting of three 6-carbon rings, one 5-carbon ring with carbon #24 (C-24) of the side chain being part of the carboxylic acid (–COOH) group. All bile acids have a hydroxyl group (OH) group in the 3-position. The OH group on C-12, when present, is also in the α position. Other carbon atoms (C-6 and C-7) can have the OH group in either the α or β position.

The structure of C27 cholesterol with the carbon atoms numbered (top) and C24 bile acid (bottom) showing the carbon atoms where hydroxylations can occur. C-3 and C-12 always have the OH group in the α position. Other carbon atoms (C-6 and C-7) can have the OH group in either α or β position.
In mammals, most C24 bile acids are 3α-OH, 5β-H; that is, the C-3 OH is in α position, and the C-5 H is in β position. The 3α-OH, 5β-H C24 bile acids are called isobile acids. The corresponding epimers 3β-OH (C-3 epimer) and 5α-H (C-5 epimer) C24 bile acids are called allobile acids. Allobile acids have a flat structure, with the A and B rings of the steroid nucleus on the same plane in contrast to regular (iso) bile acids in which the A and B rings are almost perpendicular (Wahlström et al., 2016). Allobile acids are present in certain reptilian and marine species; allobile acids are normally absent in healthy human adults but are present in the fetus, newborn, and pregnant woman, and in hepatic injury. So, they might serve as biomarkers for liver disease, but further characterization in humans is required (Wahlström et al., 2016; Shiffka et al., 2017, 2020).
The amphipathic nature of bile acids is due to the presence of a hydrophilic side (α-face, concave lower side) and a hydrophobic (less hydrophilic) side (β-face, convex upper side). The hydroxyl groups oriented toward the α-side together with the carboxylic acid on the side chain confer hydrophilic character to bile acids. Ursodeoxycholic acid (UDCA), a C-7β epimer of chenodeoxycholic acid (CDCA) and β-muricholic acid (β-MCA), a C-7β epimer of α-muricholic acid (α-MCA) are exceptions because they both have 7β-OH but are more hydrophilic than their corresponding 7α-OH containing epimers; i.e., CDCA and α-MCA, respectively. The hydrophobic methyl groups at C-18 and C-19 are oriented toward the β-side. Consequently, bile acids exhibit a great deal of surface activity, forming small micelles in aqueous solutions (Monte et al., 2009).
It should be remembered that in terms of true solubility and “water-loving” character, bile acids are more hydrophilic than hydrophobic. It is the degree of hydrophilicity that may be different, that is, more hydrophilic versus less hydrophilic. In other words, the so-called “hydrophobic” bile acids are still more hydrophilic than lipophilic. In the following text, the expressions hydrophobic or less hydrophilic bile acids does not mean bile acids that are not soluble in water or are lipophilic, but those that are less hydrophilic compared with the most hydrophilic bile acid species.
Primary bile acids are synthesized in the liver from cholesterol. In humans, primary bile acids are cholic acid (CA), a 3α,7α,12α-trihydroxy bile acid, and CDCA, a 3α,7α-dihydroxy bile acid (Fig. 2). In mice, the primary bile acids are CA, α-MCA (3α,6β,7α-trihydroxy) and β-MCA (3α,6β,7β-trihydroxy). Both α-MCA and β-MCA are synthesized from CDCA and are both 6β-hydroxylated; hence, they are referred to as 6-OH bile acids (Fig. 2). Secondary bile acids are formed in the intestine by the action of bacteria on primary bile acids (discussed later). UDCA (3α,7β) is a primary bile acid in members of the Ursidae family (bears and pandas); hence the prefix “Urso” in the name. UDCA is also formed in mice from CDCA by the epimerization of the 7α-OH of CDCA to 7β-OH, but in humans UDCA is formed from CDCA by intestinal bacteria. Therefore, UDCA is a primary bile acid in mice, but a secondary bile acid in humans.

Primary bile acids in human and mouse. In humans, primary bile acids are cholic acid (CA; 3α,7α,12α-trihydroxy) and chenodeoxycholic acid (CDCA; 3α,7α-dihydroxy). In mice, the primary bile acids are CA, α-muricholic acid (α-MCA) and β-muricholic acid (β-MCA). Both α-MCA and β-MCA are synthesized from CDCA, and they differ in the epimerized C-7 hydroxyl group; α-MCA is 7α-hydroxy and β-MCA is 7β-hydroxy. In addition, they are also 6β-hydroxylated; hence, they are also referred to as 6-OH bile acids.
Well-established functions of bile acids are (I) aiding in lipid digestion and absorption, (II) decreasing cholesterol levels by converting them into bile acids, (III) increasing bile flow, (IV) enhancing the excretion of cholesterol, (V) stimulating biliary phospholipid secretion, (VI) regulating its own synthesis, and (VII) regulating energy metabolism (https://www.ncbi.nlm.nih.gov/books/NBK549765/).
Bile Acids and Fat Breakdown
The long-known physiologic role of bile acids (as bile salts) is to help in the digestion and absorption of fats and fat-soluble vitamins in the small intestine. In healthy subjects, depending on age, postprandial serum bile acid concentrations increase 2- to 5-fold compared with fasting levels (Linnet, 1983; Salemans et al., 1993). Bile acids act as detergents, emulsifying fats to form micelles facilitating the digestion of fats by pancreatic lipase and subsequent absorption. Lipid digestion is an interfacial process in which the surface area of lipid droplets influences the binding of lipase and thus digestion; smaller droplets provide larger surface area and more efficient digestion by lipase (Armand et al., 1999). In large fat globules, hydrophilic pancreatic lipase cannot reach the core of the globules and can only cleave ester bonds of the triglycerides on the surface of the globules. Therefore, large fat globules need to be broken down to a smaller size to facilitate lipid digestion. When stomach chyme is pushed through the pyloric canal into the duodenum, the emulsion particles are subjected to strong shearing forces that tear the liquid interfaces apart. The emulsion particles that are mixed with bile in the upper small intestine are generally less than 0.5 μm in diameter (Carey et al., 1983). Bile acids are amphipathic and have high surface activity; hence they can mix with larger fat globules, coat fat droplets, and increase the ionization of the interface that facilitates emulsification (Sarkar et al., 2016). Emulsification reduces the particle size and increases the interfacial area of fat droplets. The process eventually leads to a breakup of the bulk oil phase in the presence of low interfacial tensions. Because of the increase in the surface area of the emulsified fat, the lipase digests the ester bonds efficiently. In humans, the total bile acid concentration during lipid digestion remains constant at 2.5–10 mM despite multiple enterohepatic cycles of the bile acid pool (Carey et al., 1983).
Various Bile Acids Have Different Abilities to Enhance Lipid Absorption
Hydrophobic (less hydrophilic) bile acids are more effective in increasing cholesterol absorption compared with more hydrophilic bile acids. Wang et al. (2003) fed mice with chow or chow supplemented with 0.5% (by weight) each of CA, CDCA, deoxycholic acid (DCA), dehydrocholic acid, hyodeoxycholic acid (HDCA), UDCA, ursocholic acid (UCA), ω-muricholic acid (ω-MCA), hyocholic acid (HCA), α-MCA, and β-MCA, and determined cholesterol absorption using a dual-isotope ratio method (Fig. 3). The authors found that compared with chow (mean cholesterol absorption 37%), feeding hydrophilic bile acids, such as β-MCA, resulted in low cholesterol absorption (mean: 11%), whereas feeding a “hydrophobic” (less hydrophilic) bile acid, such as CA, resulted in high cholesterol absorption (mean: 63%). The authors concluded that the balance of hydrophobic and hydrophilic bile salts in bile plays an important role in regulating intestinal cholesterol absorption. The authors hypothesized that bile acids that are more hydrophilic might reduce the solubility of cholesterol by inducing phase separation of the sterol from mixed micelles to a coexisting liquid crystalline vesicle phase, resulting in the reduction of intestinal cholesterol absorption (Fig. 3).

The effect of hydrophobicity of bile acids on the percentage of cholesterol absorption when fed to mice. The figure is based on estimated mean values from Wang et al. (2003). Moving counterclockwise from β-MCA, the bile acids have increasing hydrophobicity, except for CDCA, which in mice is readily converted to hydrophilic α-MCA and β-MCA. Compared with chow (mean: 37%), the cholesterol absorption was significantly lower by hydrophilic bile acids, such as β-MCA and α-MCA (mean: about 11–12%). Cholesterol absorption increased significantly by hydrophobic (less hydrophilic) bile acids, being highest by CA (mean: 63%). DHCA, a hydrophilic bile acid, increased cholesterol absorption to 45% ± 6% because it was biotransformed mainly to the more hydrophobic taurocholate and taurodeoxycholate.
Bile Acid Concentrations in Liver, Serum, and Bile
Using a high-performance liquid chromatography-tandem mass spectrometry (LC-MS/MS) method, Alnouti et al. (2008) quantified six major bile acids, as well as their taurine conjugates in the serum, liver, and bile of mice. The major findings are shown in Table 1. Similar findings were reported by Huang et al. (2011). There is a marked increase in the concentration of bile acids from the serum to liver (about 300-fold) and from liver to bile (about 175-fold) in mice. Thus, the bile acid concentration in bile is enriched over 50,000-fold compared with serum. Tables 1 and 2 show the concentrations of total bile acids in serum, liver, and bile in mouse and humans, respectively (Alnouti et al., 2008). When bile was collected over 2 hours, the initial bile acid concentration was 32 mg/ml, and at the end of 2 hours, it was 19 mg/ml.
Concentrations of total BA in mouse serum, liver, and bile
Concentrations of total BA in human serum, liver, and bile
Figure 4 shows the relative abundance of various bile acids and their conjugates in plasma (top panel A), liver (middle panel B), and bile (bottom panel C). In the plasma of mice (Fig. 4A), about 61% of bile acids are conjugated and about 66% are primary bile acids. Of the conjugated bile acids, 47% is taurine-conjugated cholic acid (TCA), 16% tauro α+β-MCA (Tα+β-MCA), and 27% tauro ω-MCA (Tω-MCA). In liver (Fig. 4B), almost all the bile acids (94%) are conjugated, and most (84%) are primary bile acids. Of the conjugated bile acids in the liver, over half (56%) is TCA, 25% is Tα+β-MCA, and 8% is Tω-MCA. The bottom panel (Fig. 4C) indicates that almost all (>99%) of the bile acids excreted into bile are in the conjugated form, are primary bile acids (95%), and over half is TCA (56%), followed by taurine conjugated β-muricholic acid (32%) and taurine conjugated α-muricholic acid (6%). In mice, the enzyme that drives the amino acid conjugation of bile acids is specific for taurine, which explains the overwhelming proportion of taurine-conjugated bile acids in mice (Falany et al., 1994). In contrast, bile acids in humans are 70%–75% glycine conjugates and 25%–30% taurine conjugates (Linnet, 1983).

Relative abundance of various bile acids and their conjugates in the plasma (A; top panel), liver (B; middle panel) and bile (C; lower panel) of control mice. It also shows the relative abundance of total taurine-conjugated bile acids and total unconjugated bile acids; primary bile acids (1°BAs) and secondary bile acids (2°BAs); as well as various individual taurine–conjugated bile acids in plasma, liver, and bile of mice (see text for details). The figure has been created based on the control mice data from the laboratory of C.D. Klaassen. The prefix T before bile acid name indicates the taurine conjugate of specific bile acids (e.g., TCDCA: Taurochenodeoxycholic acid; TUDCA: Tauroursodeoxycholic acid; TLCA: Taurolithocholic acid).
Bile acids excreted into the small intestine are almost entirely conjugated. At the end of the small intestine, transporters exist that remove most of the conjugated bile acids from the intestinal lumen and transport them into the portal blood. The fraction of bile acids that are not removed by these transporters enter the large intestine (colon), where the primary bile acids are converted into secondary bile acids by the intestinal bacteria, which remove the amino acid conjugate (deamidation) and the 7-OH group of bile acids. These secondary bile acids are thought to be reabsorbed in the colon by passive diffusion. The unconjugated/secondary bile acids are about one-third of the bile acids in the serum. Such a high proportion of deconjugated bile acids in the serum may reflect the fact that a fraction of the deconjugated bile acids absorbed from the large intestine into the portal circulation may escape the liver at first pass. However, this proportion is lower in the hepatocytes as the hepatocytes convert the secondary bile acids back into primary bile acids. After the secondary bile acids are transported into the hepatocytes, they are conjugated with taurine, and the 7-position is rehydroxylated in some species, such as mice, but not in humans (The 7-rehydroxylation does not occur in humans). This conversion of secondary to primary bile acids is sometimes referred to as bile acid repair. Compared with liver, the proportion of unconjugated/secondary bile acids is further reduced in bile. The bile acid efflux transporter, known as bile salt export pump (BSEP), has a higher affinity for conjugated than unconjugated bile acids (Mita et al., 2006).
Figure 5 depicts the amount of total bile acids as well as taurine-conjugated bile acids and unconjugated bile acids in serum, liver, gallbladder, small intestine, and large intestine (Fu and Klaassen, 2013). The percent of total bile acids is highest in the small intestine, followed by large intestine, gallbladder, liver, and serum. Almost all the total bile acids in the liver, gallbladder, and small intestine are taurine conjugated, and conjugated bile acids are absorbed from the distal ileum. Conversion of primary to secondary bile acids occurs in the large intestine containing the microbiota. Hence, unconjugated bile acids are higher in the large intestine.
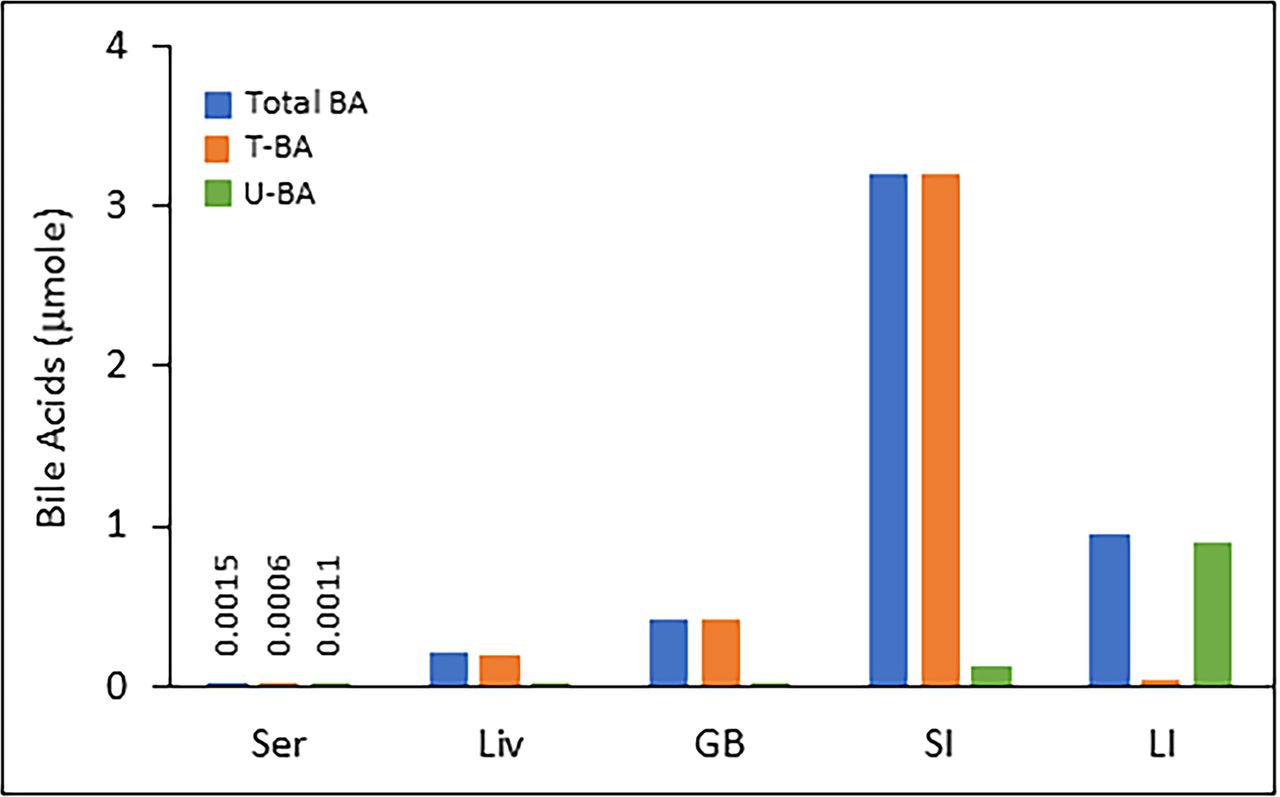
Total bile acids as well as taurine–conjugated bile acids and unconjugated bile acids in serum, liver, gallbladder, small intestine, and large intestine. The proportion of total bile acids (as a percentage) is highest in the small intestine, followed by the large intestine, gallbladder, liver, and serum. Almost all of the total bile acids in the liver, gallbladder, and small intestine are taurine–conjugated bile acids. The figure has been created based on the control mice data from the laboratory of C.D. Klaassen.
The average bile acid content in humans is about 3–5 g (Dowling, 1972), but the range might be wider, such as 1–6 g (Mok et al., 1977). About 0.5 g of bile acid is lost per day during enterohepatic circulation and is replenished by de novo synthesis in the liver to maintain a constant bile acid pool. The bile acid concentration gradient between systemic serum, portal serum, liver tissue, and hepatic bile was reported to be 1:3:80:2600 in individuals with normal liver function (Lindblad et al., 1977). The authors reported human bile acid concentrations in various tissue compartments (Table 2). A similar bile acid concentration in systemic serum of healthy individuals was reported by Luo et al. (2018).
Bile Acid Synthesis
The production of bile acids in the liver is highest in the perivenous (centrilobular) hepatocytes, that is, the hepatocytes surrounding the central vein (Twisk et al., 1995). Cholesterol, which is hydrophobic and uncharged, is converted to bile acids that are organic anions. Synthesis of bile acids represents the major pathway for cholesterol catabolism; thus, the rate of bile acid formation is an important determinant for cholesterol homeostasis (Twisk et al., 1995). Synthesis of bile acids accounts for nearly 50% of the daily turnover of cholesterol (Insull, 2006).
The overall pathway of bile acid synthesis is complex. Figure 6 shows the structural changes that cholesterol undergoes to produce bile acids. It involves various enzymes in multiple cellular compartments including the cytosol, endoplasmic reticulum, mitochondria, and peroxisomes. There are two major pathways of bile acid synthesis: the classic (neutral) pathway and the alternative (acidic) pathway. Figure 7 depicts the major steps of bile acid synthesis by both the classic and alternative pathways. The classic pathway produces most of the bile acids in mice and humans (Russell, 2003). In the classic pathway, the sterol nucleus of cholesterol is modified before the oxidative cleavage of its side chain. In contrast, the alternative pathway of bile acid synthesis begins with an initial hydroxylation on the side chain of cholesterol followed by 7α-hydroxylation of the sterol nucleus.

Overall change in conformation that cholesterol undergoes to produce bile acids.
Classic (Neutral) Pathway
The classic pathway is referred to as the “neutral” pathway because the steroid nucleus modifications occur before the side‐chain oxidation; therefore, most of the intermediates in this pathway do not have a carboxylic acid group until later in the pathway when cytochrome P450 (CYP) 27A1 catalyzes oxidation of the side chain (Fig. 7).
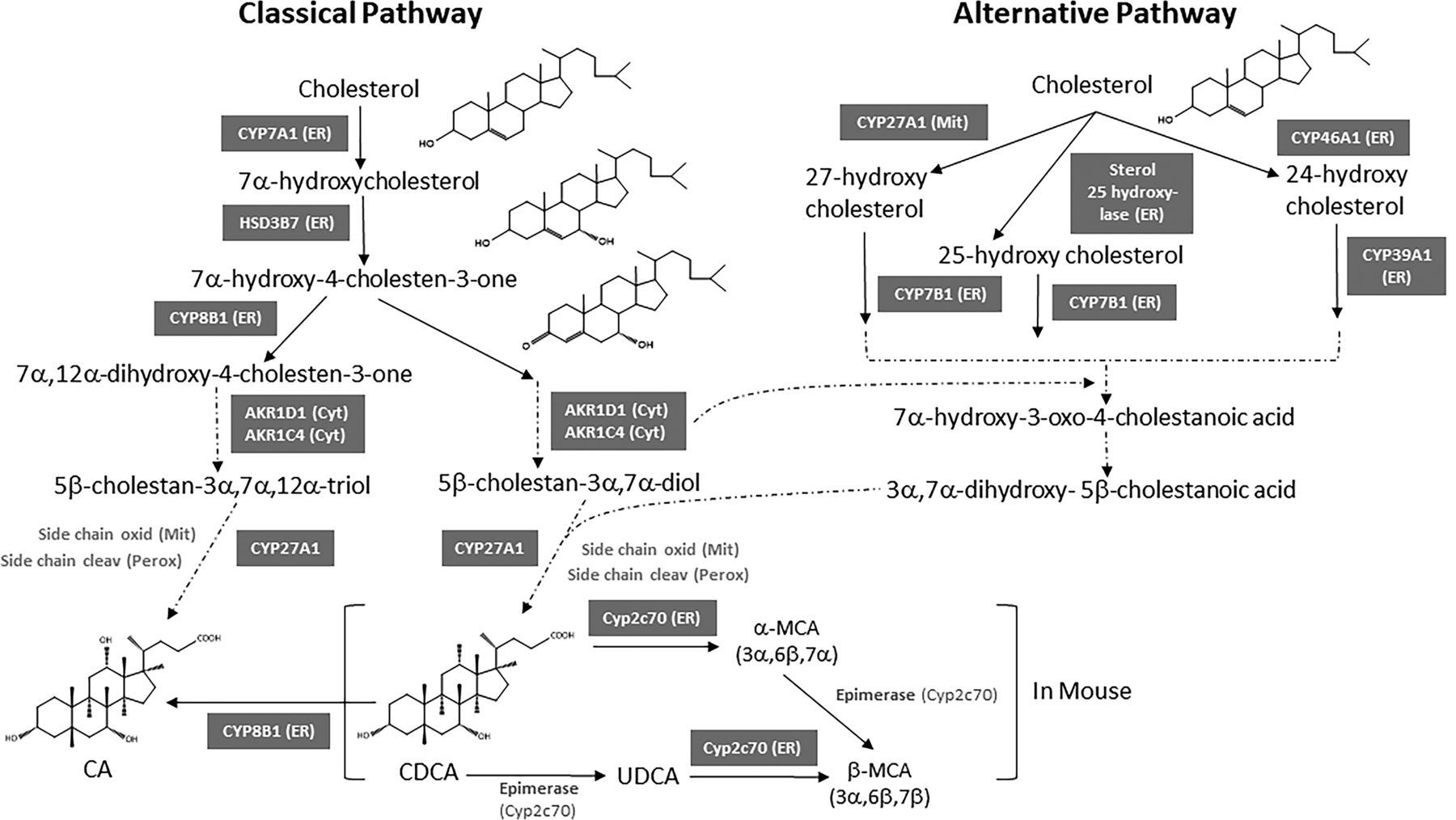
Bile acid biosynthesis pathways. Both the classic and the alternative pathways are shown.
The classic pathway operates in the liver (Agellon, 2008). In both humans and mice, the classic pathway begins with the hydroxylation of cholesterol at the C7 position by microsomal cholesterol 7α-hydroxylase (CYP7A1), which is the rate-limiting enzyme of the pathway. CYP7A1 was first cloned and characterized from rat liver by Noshiro et al. (1989). The resulting 7α-hydroxycholesterol is converted to 7α-hydroxy-4-cholesten-3-one (named C4) by the microsomal hydroxysteroid dehydrogenase named HSD3B7 (3β-hydroxy-Δ5-C27-steroid dehydrogenase). HSD3B7 then catalyzes the epimerization of the 3β-hydroxyl group of cholesterol to the 3α-hydroxyl of bile acids (Shea et al., 2007). C4 is the common precursor for synthesis of CA and CDCA. The serum C4 concentration is now used as a biomarker for the rate of bile acid synthesis. C4 is metabolized by microsomal sterol 12α-hydroxylase (CYP8B1) to become CA, and without the action of CYP8B1 it becomes CDCA. Therefore, the activity of the CYP8B1 determines the ratio of CA to CDCA. Hydroxylation of C4 at the C12 position by CYP8B1 produces 7α,12α-dihydroxy-4-cholesten-3-one, which goes through NADPH-dependent reductions at the C5 and C3 positions by aldo-keto reductase 1D1 (AKR1D1) and 1C4 (AKR1C4), respectively. Reduction of the C3 double bond by AKR1C4 produces 3α,7α,12α-cholesten-5β-triol. This is followed by CYP27A1-catalyzed side chain oxidation (from ‒OH to ‒COOH) in mitochondria to form 3α,7α,12α-trihydroxycholestanoic acid; and 3α,7α-dihydroxycholestanoic acid without CYP8B1. These bile acid intermediates are activated to form bile acid-CoA thioesters by peroxisomal long chain acyl-CoA synthase (or bile acid-CoA synthase, BACS), which enter peroxisomes via a peroxisomal transporter, an ATP-binding cassette (ABC) D3 for β-oxidation reactions to cleave a propionyl-CoA to form cholyl-CoA and chenodeoxycholyl-CoA, respectively. These bile acid-acyl-CoAs are conjugated to an amino acid taurine or glycine to form conjugated bile acids by bile acid-CoA: amino acid N-acyltransferase (BAAT) (Fig. 7). Conjugated bile acids are secreted into bile.
Human and mouse CYP8B1 were cloned and characterized by Gåfvels et al. (1999). Recently, using a fission yeast-based expression system, Fan et al. (2019) have shown that human CYP8B1 can directly convert CDCA to CA by catalyzing 12α-hydroxylation. However, the relevance of this observation in vivo needs to be established (Fig. 7).
Alternative (Acidic) Pathway
The alternative pathway of bile acid synthesis is also referred to as the “acidic” pathway because the cholesterol side‐chain oxidation occurs early in the pathway and before the modifications to the steroid nucleus. C27 bile acids and oxysterols formed in various cells in the body are transported to the liver to ultimately produce C24 bile acids in hepatocytes. For example, almost all the 24-hydroxycholesterol in the liver originates from the brain (Agellon, 2008). Likewise, 27-hydroxycholesterol is the most abundant oxysterol in the plasma of mice (Li-Hawkins et al., 2000) and humans (Dzeletovic et al., 1995). It is synthesized from cholesterol by sterol 27-hydroxylase in multiple tissues including the liver (Fig. 7).
The acidic pathway is initiated by mitochondrial/microsomal C-24, C-25, or C-27 sterol hydroxylases that hydroxylate cholesterol to form 24-, 25-, and 27-hydroxycholesterol, respectively, which are then rapidly 7α-hydroxylated by microsomal oxysterol 7 -hydroxylase (CYP7B1) (Pandak and Kakiyama, 2019). In the mammalian central nervous system, sterol 24-hydroxylase (CYP46A1) hydroxylates cholesterol to form 24-hydroxycholesterol in the membranes of the smooth endoplasmic reticulum of neurons (Russell et al., 2009) (Fig. 7). Based on the observation that CYP46A1 and CYP27A1 knockout mice have no apparent alterations in bile acid synthesis, along with the finding that cholesterol 7α-hydroxylase deficient mice have a bile acid content that is 50% of normal because these mice cannot convert cholesterol directly into bile acids, it was concluded that the alternative pathway contributes to about 50% of the bile acids (BA) pool in mice (Schwarz et al., 1998; Schwarz et al., 2001). It is interesting to note that female mice have little or no CYP7B1 (Fu et al., 2012) but do not have decreased BA levels. Analysis of the bile acids in a human subject with cholesterol 7α-hydroxylase deficiency suggests that the alternative pathway contributes to about 5%–10% of the bile acid pool in humans (Pullinger et al., 2002).
Figure 7 shows that the products of the upstream reactions that are part of the alternative pathway feed into the downstream reactions shared by the classic pathway. The alternative pathway for the synthesis of oxysterols exists in numerous tissues. Side-chain oxidation is followed by 7α-hydroxylation of the sterol nucleus by microsomal oxysterol 7α-hydroxylase (CYP7B1) in most tissues. Table 3 compares the classic and the alternative pathway of bile acid synthesis. CYP27A1 was first cloned and characterized from rabbit (Andersson et al., 1989), and CYP7B1 was first cloned and characterized from rat brain (Stapleton et al., 1995). It is important to note that both CYP7A1 and CYP7B1 add a 7-hydroxyl group to their respective substrates; however, CYP7A1 in the classic pathway is a highly specific cholesterol 7α-hydroxylase, whereas CYP7B1 in the alternative pathway is an oxysterol 7α-hydroxylase. CYP7B1 prefers 25-hydroxycholesterol and 27-hydroxycholesterol as substrates, whereas CYP39A1 sterol 7α-hydroxylase is selective for 24-hydroxycholesterol (Agellon, 2008). Table 3 shows the major differences between the classic and the alternative pathway, and Table 4 shows the main differences between human and mouse bile acids.
Comparison between the classic and the alternative pathways of BA synthesis
Main differences between human and mouse BAs
Setchell et al. (1998) described the clinical presentation of a 10-week-old boy with severe cholestasis, cirrhosis, and liver failure, all due to defects in bile acid synthesis. There was a deficiency in microsomal CYP7B1 in the alternative pathway. Gene sequence analysis revealed a C→ T transition mutation in exon 5 affecting codon 388, thereby creating a premature stop codon and a nonfunctional enzyme. Setchell et al. (1988) had previously reported that early human fetal (16–19 weeks) gallbladder bile is enriched in CDCA and metabolites not found in adults. Based on the predominance of CDCA in the bile, the authors proposed a possible dominance of the alternative pathway in early development. In contrast to humans, CYP7B1 in mice develops slowly during development. Cui et al. (2012) showed that Cyp7b1 mRNA was low in mice before 20 days of age and then increased markedly to adult levels.
Observations from Knockout (Null) Mouse Models in Which Different Genes of the Classic and Alternative Bile Acid Synthesis Pathways Are Inactivated
Five null mouse models are discussed here, CYP7A1-null, CYP7B1-null, CYP8B1-null, CYP27A1-null, and HSD3B7-null.
CYP7A1-Null
CYP7A1-null (Cyp7a1–/–) mice were genetically engineered by removing exons 3–5 of the gene resulting in a total absence of a functional CYP7A1 enzyme (Ishibashi et al., 1996). Most of the pups arising from crosses of the CYP7A1-null mice died before weaning (3–4 weeks after birth). CYP7A1-null pups defecated fatty stools indicating fat malabsorption (steatorrhea). However, if the pregnant mother’s chow and water were supplemented with both CA and vitamins beginning from gestation day 12 and continued through weaning, CYP7A1-null pups survived (Ishibashi et al., 1996). Once past the weaning stage (3–4 weeks after birth), CYP7B1 expression begins and the alternative pathway of bile acid synthesis is activated, compensating for the absence of CYP7A1 activity (Schwarz et al., 1996; Fu and Klaassen, 2013). Interestingly, Erickson et al. (2003) established their own colony of CYP7A1-null mice. By breeding vendor-supplied CYP7A1-null mice and selecting for pups that did not need vitamin and cholate supplementation, the authors were able to increase the survival to >65% in their CYP7A1-null mice colony. Erickson et al. (2003) speculated that the differential survival of the two groups of CYP7A1-null mice, one needing vitamin and cholate supplementation to survive as pups whereas the others did not, suggested a “leaky” phenotype, that is, the presence of a compensatory mechanism in those pups that survived without vitamin and cholate supplementation. Nevertheless, the complete absence of CYP7A1 activity in CYP7A1-null mice, particularly early in life, causes several phenotypic abnormalities compared with the wild type due to altered lipid homeostasis. The role of CYP7A1 in human bile acid homeostasis has been demonstrated in population studies where an association between high plasma cholesterol concentrations and polymorphisms at the CYP7A1 locus, such as the A→C transversion polymorphism at position –204 of the promoter (–204A>C) was observed (Couture et al., 1999). Ferrell et al. (2016) showed that CYP7A1-null mice in C57BL/6J genetic background survive well and are protected from high-fat-diet induced metabolic disorder.
CYP7B1-Null
CYP7B1-null (Cyp7b1–/–) mice have no adverse phenotype and are indistinguishable from wild-type mice (Li-Hawkins et al., 2000). Bile acid content and composition, plasma triglycerides and cholesterol concentrations are all normal in male and female CYP7B1-null mice. The cholesterol content of major tissues was not altered. However, plasma and tissue levels of 25- and 27-hydroxycholesterol, two oxysterol substrates of CYP7B1, were markedly elevated in CYP7B1-null mice. In contrast to mice, defects in the CYP7B1 gene in humans give rise to two different disease phenotypes: liver failure in children due to defects in bile acid synthesis and progressive neuropathy (spastic paraplegias) in adults. Lack of CYP7B1 activity results in an elevation of serum oxysterols and other bile acid intermediates, and a corresponding absence of mature bile acids. The spastic paraplegias are associated with lower limb spasticity, and weakness and a corresponding axonal degeneration of motor neurons in the spinal cord (Stiles et al., 2009). It is interesting to note that female mice have very low constitutive expression of CYP7B1 enzyme compared with males and yet they have normal cholesterol and bile acid levels (Li-Hawkins et al., 2000).
Using control (B6/129 genetic background), CYP7B1-null and CYP27A1-null liver cells, Kakiyama et al. (2019) reported a new pathway of bile acid synthesis initiated in liver mitochondria; the pathway begins with the formation of 24-hydroxycholesterol (24HC). This was shown by an increased expression of the steroidogenic acute transport protein (StarD1) that facilitated an increased transport of cholesterol into mitochondrial CYP27A1. In control mice livers, there was a marked downregulation of oxysterol 7α-hydroxylase (CYP7B1), a marked increase in 26-hydroxycholesterol (26HC), and the formation of a vital regulatory oxysterol, 24-hydroxycholesterol (24HC). In CYP7B1-null mice liver, however, a marked increase (13-fold versus control) of 24HC was observed in the StarD1 over expressed mice. No detectable levels of 24HC and 26HC were present in the CYP27A1-null liver.
CYP8B1-Null
In both humans and mice, the CYP8B1 gene is devoid of introns (Gåfvels et al., 1999). Disruption of the Cyp8b1 gene to create CYP8B1-null (Cyp8b1–/–) mice resulted in a complete absence of CA (Li-Hawkins et al., 2002). Because CDCA in mice is converted to MCA, the bile acid pool in these mice is dominated by MCA. In CYP8B1-null mice, the feedback downregulation of CYP7A1 activity is lost causing expansion of the bile acid content and alterations in cholesterol metabolism. Wild-type female mice typically have a 35%–40% larger bile acid pool compared with males due to the increased production from the CYP7A1 pathway. This sexually dimorphic trait was maintained in the CYP8B1-null mice. Despite a larger bile acid content in female mice, the absorption of dietary cholesterol and lipids is reduced (steatorrhea), suggesting that the composition of the bile acid pool is more important than its size (Li-Hawkins et al., 2002). Subsequent studies further confirmed better lipid profile, glucose homeostasis, and improved liver health in CYP8B1-null mice. CYP8B1-null mice are protected from the following conditions: (1) metabolic impairments, hepatic inflammation, and fibrosis (Patankar et al., 2018); (2) western diet-induced weight gain and hepatic steatosis due to impaired fat absorption (Bertaggia et al., 2017); and (3) weight gain on a high fat diet and liver enlargement/steatosis/serum LDL-cholesterol (Bonde et al., 2016). CYP8B1-null mice also have increased fecal excretion of cholesterol (Bonde et al., 2016) and improved glucose homeostasis because of increased glucagon-like peptide-1 (GLP-1) (Kaur et al., 2015). In mice fed a high cholesterol diet, siRNA-mediated knockdown of CYP8B1 expression was found to significantly decrease steatosis, hepatic lipid content, and hepatic lipid accumulation. Increased clearance of previously accumulated hepatic cholesterol was also observed. The authors concluded that the data demonstrate that inhibition of CYP8B1 could be a viable therapeutic target (Chevre et al., 2018). In human bile, CA and CDCA occur in a molar ratio of approximately 2:1 (Wang et al., 2005).
An absence of functional CYP8B1 results in the absence of CA. For example, in naked mole rats, the CYP8B1 gene has been completely removed by a genomic deletion resulting in a total absence of CA. In both African and Asian elephants, the CYP8B1 gene has accumulated multiple inactivating mutations resulting in a nonfunctional CYP8B1 gene and a total absence of CA (Sharma and Hiller, 2018). Shinde et al. (2019) stated that the loss of CYP8B1 gene in certain species indicates recurrent changes in the selection landscape that is associated with changes in dietary lipid content. However, there are many exceptions, such as phylogenetically closer herbivorous species that show divergence in bile acid production, such as horse and white rhinoceros (both in order Perissodactyla); horse produces both CA and CDCA, but white rhinoceros does not produce either. In contrast, there are phylogenetically more distant herbivorous species that show similarity in bile acid production, such as elephant (order Proboscidea) and manatee (order Sirenia); both do not produce either CA or CDCA. It appears that the loss of CYP8B1 occurred independently in many lineages during mammalian evolution.
CYP27A1-Null
CYP27A1-null (Cyp27a1–/–) mice have normal plasma levels of cholesterol and normal appearance (Rosen et al., 1998), but the bile acid content is reduced. In CYP27A1-null mice, the bile acid concentration in bile was 0.9 mg/ml compared with 8 mg/ml in controls, with CA being the predominant bile acid. Additionally, the excretion of fecal bile acids was reduced by 80% (78 μg/g in CYP27A1-null mice compared with 430 μg/g in wild type), as determined by the analysis of an equal amount of daily fecal excretion. The reduced synthesis of bile acids in CYP27A1-null mice leads to reduced absorption of cholesterol, which may lead to a compensatory increased synthesis. CYP27A1-null mice have a compensatory upregulation of CYP7A1 expression due to the decrease in negative feedback suppression, resulting in a 4- to 10-fold increase in the concentration of circulating 7α-hydroxycholesterol compared with the wild-type mice (Rosen et al., 1998). CYP27A1-null mice also have hypertriglyceridemia, hepatomegaly, adrenomegaly, and increased hepatic fatty acid synthesis, indicating a more global role of CYP27A1 in lipid homeostasis than previously known (Repa et al., 2000). In humans, mutations in the CYP27A1 gene cause cerebrotendinous xanthomatosis (CTX), which is a rare genetic metabolic disorder of cholesterol and bile acid metabolism causing systemic and neurologic abnormalities (https://www.omim.org/entry/606530; last accessed: June 9, 2021). Patients with CTX have accumulated cholesterol and a bile alcohol, cholestanol. However, CYP27A1-null mice do not have CTX phenotypes.
Rizzolo et al. (2019) produced CYP7A1-CYP27A1 double-null mice models by crossing homozygous CYP7A1-null and homozygous CYP27A1-null mice. The double-null mice, despite lacking two crucial enzymes in bile acid synthesis pathway, still produced bile acids in the liver. Although there was a 40% reduction in bile acid production in the liver, the bile acid pool composition was similar compared with the wild-type mice.
HSD3B7-Null
The development of hydroxysteroid dehydrogenase HSD3B7-null mice (Hsd3b7 –/–) (Shea et al., 2007) demonstrated that the stereochemistry of the C-3 OH group of bile acids is important in maintaining the functional and regulatory properties of bile acids in mice and presumably in other species. The HSD3B7 enzyme catalyzes two reactions required for the epimerization of the 3β-hydroxyl group of cholesterol to the 3α-hydroxyl of bile acids. Predictably, the HSD3B7-null mice cannot epimerize the 3β-OH to 3α-OH. In these mice, the bile acid pool is dominated by 3β-OH bile acids, and 90% of these mice die within 3 weeks of birth when maintained on standard feed and water. These mice excrete 2–3 times more bile acids and sterols in the feces compared with wild-type littermates. Consequently, intestinal cholesterol absorption in HSD3B7-null mice is reduced to < 9% of the control mice. Additionally, farnesoid-X-receptor (FXR)-mediated negative feedback regulation is also disrupted. Cholesterol 7α-hydroxylase and sterol 12α-hydroxylase mRNAs are elevated fourfold compared with the wild-type mice. Both these enzymes are normally repressed by bile acids acting through FXR. The expression of FXR target genes, such as small heterodimer partner (SHP) mRNA in the liver, and SHP and fibroblast growth factor 15 (FGF15) mRNAs in the gut are significantly decreased. In the liver, SHP mRNA expression is decreased about 50%, whereas in the gut, the expression of both SHP and FGF15 mRNAs are decreased about 90%. The role of FXR and fibroblast growth factor- (FGF) 15 in bile acid homeostasis is discussed in detail later. Mutations inactivating HSD3B7 gene cause a recessive form of neonatal liver failure in humans (Shea et al., 2007); that is, individuals with homozygous recessive genotype have clinical presentation of the disease phenotype.
Formation of MCA in Mice by CYP2C70 and Observations from CYP2C70-Null Mice
In mice, CDCA (3α,7α) is 6β-hydroxylated by CYP2C70 to produce β-MCA (3α,6β,7α). UDCA (3α,7β), which is the 7β-epimer of CDCA, is a primary bile acid in mice and is produced from CDCA by the epimerase action of CYP2C70 (Fig. 7). β-MCA (3α,6β,7β) is synthesized by two pathways—one by epimerization of the OH group on C-7 (from 7α to 7β) of α-MCA and the other by 6β-hydroxylation of UDCA (Takahashi et al., 2016; de Boer et al., 2017; Honda et al., 2020). de Boer et al. (2017) reported that CYP2C70 catalyzes both the 6β-hydroxylation and C-7 epimerization reactions. Takahashi et al. (2016) used a Cyp2c gene cluster knockout mouse model and found the complete absence of both α-MCA and β-MCA in the liver of these mice. Because the mouse Cyp2c gene cluster contains 16 genes, the authors analyzed several individual recombinant Cyp2c genes, including Cyp2c70, by expressing them in HepG2 cells. The analysis revealed that CYP2C70 produced α-MCA from CDCA and β-MCA from UDCA, respectively. Recently, Honda et al. (2020) generated Cyp2a12 and Cyp2c70 single and double knockout mice and confirmed the finding of Takahashi et al. (2016) that CYP2C70 converts CDCA to α-MCA and UDCA to β-MCA. Mouse Cyp2a12 converts DCA, a secondary BA, to primary BAs as evidenced by the accumulation of DCA in Cyp2a12-null mice (Honda et al., 2020). However, the accumulation of UDCA was found to be much lower than CDCA in Cyp2c70 knockout mice, suggesting that most β-MCA is synthesized from CDCA via α-MCA. Therefore, epimerization of α-MCA to produce β-MCA appears to be the predominant pathway rather than producing β-MCA directly from UDCA (Guo and Chiang, 2020; Honda et al., 2020).
Consistent with the role of CYP2C70 in producing α- and β-MCA from CDCA, inactivating liver-specific CYP2C70 function resulted in mice with a human-like bile acid composition. Surprisingly, with a decrease in the FXR antagonist β-muricholic acid and an increase in the FXR agonist CDCA, there was no increase in FXR and decrease in bile acid concentrations in the mice; rather, there was an increase in bile acid concentrations and even hepatotoxicity. There was a blunted FXR-induced cholesterol disposal due to decreased transintestinal cholesterol excretion (de Boer et al., 2020). This finding is consistent with an earlier report by the same authors showing that the pharmacological activation of FXR in mice resulted in a significant increase in β-MCA and a corresponding enhanced cholesterol removal via stimulation of transintestinal cholesterol excretion.
Conjugation of Bile Acids
The terminal step in bile acid synthesis involves the addition of an amino acid, usually glycine or taurine, through an amide linkage to the C-24. Conjugation of bile acids decreases bile acid toxicity and increases bile acid solubility for secretion into bile. Bile acids recycled to the liver via enterohepatic circulation are activated by reacting with coenzyme A (the –COOH group of bile acid reacts with CoASH) forming a bile acid-coenzyme A thioester (BACO-SCoA). The reaction is catalyzed by BACS (gene symbol SLC27A5). The BACO-SCoA then reacts with the amino acid (taurine or glycine) forming the conjugated bile acids; the reaction is catalyzed by a cytosolic Bile Acid-CoA‒amino acid N-acyltransferase (BAAT). The conjugation reaction is very efficient. The two steps, shown briefly, proceed as follows.
BACOOH + CoASH + ATP → BACO-SCoA + AMP + PPi … (catalyzed by BACS)
BACO-SCoA + Amino acid (Tau/Gly) → Conjugated bile acid + CoASH … (catalyzed by BAAT)
Figure 8 illustrates the structure of CA after conjugation with glycine to form glycine-conjugated cholic acid (glycocholate) and taurine to form TCA. In mammals, the bile acid side chain is primarily conjugated with taurine or glycine but there exists remarkable species difference. The difference arises due to the affinity of the species-specific BAAT enzyme for taurine or glycine, resulting in the conjugation pattern ranging from almost exclusively glycine conjugates in rabbits and guinea pigs to almost exclusively taurine conjugates in sheep, dogs, and mice (Li and Dawson, 2019). In humans and rats, both glycine and taurine conjugates are formed. In mice, the BAAT enzyme that catalyzes the amino acid conjugation of bile acids is specific for taurine, which explains the overwhelming proportion of taurine-conjugated bile acids in mice (Falany et al., 1997). Interestingly, many primates, such as chimpanzee, baboon and rhesus monkey, also show preferential conjugation of bile acids with taurine, whereas humans produce 70%–75% glycine conjugates and 25%–30% taurine conjugates (Schwenk et al., 1978; Linnet, 1983).
Solubility of Unconjugated and Conjugated Bile Acids
Conjugation of bile acids decreases the pKa, increases the solubility in polar solvents, and decreases the solubility in nonpolar solvents (Carey, 1984). For example, the pKa values for unconjugated bile acids in water is about 5 and that for glycine conjugated bile acids is about 3.7 (Fini and Roda, 1987). Therefore, glycine conjugates are moderately strong acids. For comparison, the pKa of acetic acid is 4.7. In the literature, the reported range of pKa of unconjugated bile acids is mostly 5–6 and that of glycine-conjugated bile acids is 3–4 . Taurine-conjugated bile acids are stronger acids with pKa of 1–2 (Kullak-Ublick et al., 2000). Thus, at pH 7.0 unconjugated bile acids have about a 100-fold higher concentration in the water-soluble ionized form (A‒) than the lipid-soluble unionized or protonated form (HA), about a 10,000-fold higher concentration in the water-soluble than lipid-soluble form for the glycine-conjugated bile acids, and at least 100,000-fold higher concentration in the water-soluble than lipid-soluble form for the taurine-conjugated bile acids.
Hence, most bile acids exist in the water-soluble ionized form at the slightly alkaline pH of the intestinal chyme. As they are ionized, they exist as salts with Na+ and other cations. Thus, they should be termed bile salts, but by tradition they are often referred to as bile acids. The pKa values of bile acids imply that extremely small amounts of bile acids are in the lipid-soluble form to be absorbed passively from the large intestine.
Experimental data show that the aqueous solubility of different bile acids (HA) varies widely, ranging from 5 × 10‒8 M for lithocholic acid (LCA) to 1.6 × 10‒3 M for UCA at 37°C. Fully dissociated sodium bile salts are highly soluble in water, attaining molar concentrations as high as 1 to 2 M (Carey, 1984). The critical micellar concentration (CMC) is the concentration of surfactants above which micelles form and all additional surfactants added to the system will form micelles. Therefore, the higher the hydrophobicity of the surfactant, the lower the CMC value. For bile acids, which are surfactants, the CMC values vary from about 0.6 mM to about 10 mM (Carey, 1984). Using microcalorimetric titration, Simonović and Momirović (1997) determined the CMC values of CA, DCA, and CDCA in phosphate buffer (pH: 7.92) to be about 18, 5, and 7 mM, respectively, indicating that DCA is the most hydrophobic (least hydrophilic) of these three bile acids. Micelles formed by the ionized bile salt (A‒) species can solubilize the unionized (protonated) bile acid (HA) species (Carey, 1984).
Sulfation of Bile Acids
Normally bile acid sulfation is a minor pathway. However, cholestatic conditions significantly increase bile acid sulfation that aids in increased clearance of bile acids through urine and bile. Sulfation is a phase II conjugation reaction catalyzed by sulfotransferases (SULTs). SULTs catalyze the transfer of a sulfonate group (SO3‒) from the universal sulfonate donor, 3′-phosphoadenosine 5′-phosphosulfate (PAPS), to a hydroxyl, or amino, or carboxylic acid group of an acceptor molecule (substrate). The resulting conjugates carry a permanent negative charge and are highly water soluble. As a result, the pKa values of bile acid-sulfates are lower than 1 (https://foodb.ca/compounds/FDB023028; last accessed: June 6, 2021).
Although “sulfonation” is the chemically accurate term to describe this conjugation, “sulfation” is used for historical reasons (Alnouti, 2009). Bile acid sulfation is catalyzed by cytosolic SULTs. The formation of bile acid-sulfated metabolites for excretion by humans was first reported by Palmer (1967). More than 50% of LCA is present in human bile in the sulfated form (Palmer and Bolt, 1971). Bile acid-sulfates are excreted into bile but are also excreted in urine. Biliary excretion of bile acid-sulfates results in their ultimate elimination in the feces because bile acid-sulfates are not well absorbed from the intestine. There are some conflicting reports in the older literature on the proportion of bile acid-sulfate species in humans, but it is now recognized that the primary sulfation site in humans is the 3α-OH whereas that in mice is the 7α-OH, creating bile acid-3-sulfate and bile acid-7-sulfate species, respectively (Zhang and Klaassen, 2010; Chaudhari et al., 2021). Table 5 shows the approximate pKa values of unconjugated and various conjugated bile acids. In humans, bile acid-3-sulfation is catalyzed by SULT2A1 (Radominska et al., 1990), whereas in mice bile acid-7-sulfation is carried out by SULT2A1 and 2A8 (Feng et al., 2017). Unlike bile acids sulfated at the C-3 position, C-7-sulfates are resistant to hydrolysis and metabolism by the intestinal microbiota, which prevents the C-7-sulfates from being absorbed from the intestine (Robben et al., 1986). Among the secondary bile acids, DCA and LCA predominate in the feces. More than half of LCA in humans is sulfated. Some sulfated LCA is excreted in bile to be ultimately excreted in the feces, whereas some sulfated LCA is effluxed by the liver into the sinusoidal blood by multidrug resistance-associated protein (MRP) 3 and MRP4 for renal excretion (Alnouti, 2009). For example, in cholestasis the bile flow is decreased; this triggers a cellular adaptive response excreting larger amounts of sulfated bile acids in the sinusoidal blood for renal excretion (Rius et al., 2003; Halilbasic et al., 2013).
Approximate pKa values of unconjugated and various conjugated BAs
Specific Transporters Enable the Enterohepatic Circulation of Bile Acids
Enterohepatic circulation provides the bile acid recycling path that involves the liver and the intestine. Enterohepatic circulation of bile acids involves the following events: (I) transport of bile acids by the BSEP from hepatocytes into bile canaliculi. Bile canaliculi drain into bile ductules, then bile ducts, and ultimately into the duodenum through the gallbladder (in animals with a gallbladder, such as mice, humans) or directly into the duodenum (in animals without a gallbladder, e.g., rats, horses, deer, whales). Nevertheless, in animals with a gallbladder, a significant amount of bile still flows directly into the duodenum without being stored in the gallbladder; (II) conversion of primary bile acids into secondary bile acids by the intestinal microbiota; (III) uptake of bile acids from the distal ileum into the enterocytes by the apical sodium-dependent bile acid transporter (ASBT); (IV) transfer of bile acids from the apical to the basolateral membrane of the enterocytes by the ileal bile acid-binding protein (IBABP); (V) transport of bile acids into the portal blood by the organic solute heterodimeric organic solute transporter (OST) α and β heterodimer; (VI) uptake of bile acids from (liver) sinusoidal blood into the hepatocytes by the sodium (Na+)-taurocholate cotransporting polypeptide (NTCP) and organic anion transporting polypeptides (OATPs); (VII) repetition of the cycle over and over again. Figures 9 and 11 depict the enterohepatic circulation path and the main bile acid transporters involved. This recycling mechanism plays an essential role in maintaining the bile acid pool and normal bile flow, as well as the bile acid and cholesterol homeostasis.
In humans, about 95% of bile acids are recycled from the gut back into the liver during each cycle of the enterohepatic circulation, and the 5% that is lost through feces is replaced by new synthesis from cholesterol. A bile acid pool of about 3 g consisting of approximately 40% CA, 40% CDCA, 20% DCA, and trace amount of LCA, is recycled 4 to 12 times a day. Bile acids lost in the feces (about 0.5 g/d) are replenished by de novo synthesis in the liver to maintain a constant bile acid pool. This amounts to about a total of 0.5 g of bile acids synthesized/d (Russell, 2003; Chiang, 2013). Direct assessment of (recycled) daily hepatic bile acid secretion into the duodenum in humans yielded a value of ∼12 g/d (Lefebvre et al., 2009).
Cholehepatic Shunt
In addition to the enterohepatic circulation of bile acids between the liver and the intestine, there is a separate shorter path for the recycling of bile acids between hepatocytes and cholangiocytes, which is referred to as the cholehepatic shunt. The cholangiocytes are exposed to high concentrations of bile acids at their apical membrane that faces the flowing bile through bile ductular lumens. In humans, the apical membranes of cholangiocytes have the uptake transporters ASBT and OATP1A2, whereas the basolateral membranes of the cholangiocytes have the efflux transporters Ostα–Ostβ heterodimer and MRP3 (Pauli-Magnus and Meier, 2006). Bile acids transported by the cholangiocytes recycle via the peribiliary plexus back to the hepatocytes for resecretion into bile canaliculus. The cholehepatic shunt prevents the accumulation of bile acids in cholangiocytes, thereby reducing the toxic effects of accumulating bile acids on the cholangiocytes.
Cholehepatic shunt plays an important role in increasing bile flow. Unconjugated bile acids in canalicular bile are protonated by H2CO3 generating protonated bile acids and bicarbonate ion (HCO3−). Protonated bile acids are more lipophilic, hence are absorbed by the bile duct epithelial cells and returned to the hepatocytes. These bile acids are subsequently resecreted into bile. Each cycle of absorption and resecretion induces increased bile flow (choleresis) and the generation and secretion of more bicarbonate-rich fluid. An example of hypercholeretic bile acid is norUDCA, which is secreted in part in unconjugated form (Boyer, 2013).
Efflux of Bile Acids from the Hepatocytes into Bile Canaliculi
Bile acids are excreted primarily as glycine and taurine conjugates from the hepatocytes across the canalicular membrane by BSEP (gene symbol ABCB11 in humans; Abcb11 in rodents). BSEP was originally termed “sister of P-glycoprotein (SPGP)” as it is structurally related to P-glycoprotein (Table 6). BSEP transports both conjugated and unconjugated bile acids but has much higher affinity for conjugated bile acids (Mita et al., 2006). Almost all bile acids in bile are conjugated. Strautnieks et al. (1998) reported that the progressive familial intrahepatic cholestasis type 2 (PFIC2) phenotype in humans is caused by mutations in the BSEP gene, suggesting that BSEP is the major canalicular conjugated bile acid export pump in humans. The loss of BSEP function leads to hepatocellular bile acid accumulation and overload in the liver, causing PFIC2 and the benign recurrent intrahepatic cholestasis type 2 (BRIC2) (Stieger et al., 2007). PFIC2 progresses to cirrhosis and requires liver transplantation, whereas BRIC2 is less severe and associated with intermittent episodes of cholestasis. Kagawa et al. (2008) showed that the phenotypic differences in PFIC2 and BRIC2 correlate well with the stability of the BSEP protein. The BSEP mutations associated with PFIC2 produce BSEP protein that is rapidly degraded, resulting in impaired bile acid secretion. Both these diseases are characterized by liver injury, which can progress to cirrhosis, hepatic failure, hepatocellular carcinoma, and death. The severity of the disease depends on the extent of functional impairment of BSEP. Functional impairment of BSEP also predisposes women to intrahepatic cholestasis of pregnancy (Lam et al., 2007).
Rodent and human BSEPs
Observations from BSEP-Null Mice
Wang et al. (2001a) developed BSEP-null (Spgp–/–) mice. Surprisingly, BSEP-null mice did not show any signs of cholestasis except for a slight reduction in bile flow. Cholestasis involves a marked reduction in bile flow, and bile acid secretion is a major driving force of bile flow. Thus, functional inactivation of BSEP in mice, in contrast to humans, does not exhibit any severe phenotype, such as severe cholestasis or progressive liver injury. This is because mice can detoxify hydrophobic (less hydrophilic) bile acids by hydroxylation producing polyhydroxylated bile acids and use alternative mechanisms of bile acid transport (Lam et al., 2005). Interestingly, BSEP-null mice have a sharply reduced secretion of hydrophobic bile acids (5%–6% of the wild type) but maintain nearly normal amounts of the more hydrophilic bile acids, such as α-MCA and β-MCA, and greatly increased amounts of tetrahydroxy bile acids (18% of total biliary bile acids) that are not normally detected in wild-type control mice. Tetrahydroxy bile acids are produced by hydroxylation of bile acids, such as 12α-hydroxylation of MCAs, and they help avoid cholestasis-induced severe liver damage. The presence of tetrahydroxylated bile acids had previously been reported in humans with cholestasis. For example, CDCA, CA, and DCA can be hydroxylated and transformed into tri- or tetrahydroxylated bile acids in patients with cholestasis. CA, which is already a trihydroxy bile acid can be further hydroxylated at the C-1 or C-6 position, producing a tetrahydroxylated bile acid (Bremmelgaard and Sjövall, 1980). Despite the absence of a typical cholestasis phenotype, increased hydroxylation of bile acids in BSEP-null mice is consistent with a cholestatic phenotype, which is attenuated by a greatly increased secretion of polyhydroxylated bile acids into the bile.
Polyhydroxylated bile acids (e.g., tetrahydroxylated bile acids) are less injurious and are exported into the canalicular space by the multidrug resistance [MDR/P-glycoprotein (P-gp)] 1, and MRP2 (Wang et al., 2009; Megaraj et al., 2010). Table 6 summarizes some information on rodent and human BSEPs.
Observations from MDR1-Null and MRP2-Null Mice
Because BSEP-null mice maintain substantial canalicular bile acid secretion and do not develop severe cholestasis, this suggests the presence of an alternative transport mechanism that maintains bile acid secretion into bile canaliculi and allows these mice to remain physiologically relatively normal. These alternative canalicular transport systems involve P-glycoprotein/ (P-gp) MDRP1 and MRP2.
MDR1/P-gp-Null Mice
Because the expression of both P-gp isoforms in mice, MDR1A (gene symbol: Abcb1a) and MDR1B (gene symbol: Abcb1b), was markedly increased in BSEP-null mice, Wang et al. (2009) postulated that P-gp might play a compensatory role in BSEP-null mice in maintaining the alternative bile acid transport process. The authors crossed MDR1A/1B-double knockout (Abcb1a/1b–/–) mice with BSEP-null mice to create a triple knockout mouse model lacking functional BSEP as well as MDR1A and MDR1B. These mice exhibited signs of cholestasis, including histologic changes typical of cholestatic stress, blockage of bile flow, signs of hepatic inflammation, elevated plasma bilirubin, and mortality. Earlier, Lam et al. (2005) demonstrated that plasma membrane vesicles isolated from a cell line expressing high levels of Chinese hamster P-gp1 (an MDR1 ortholog) was capable of transporting bile acids, albeit with one-fifth the affinity compared with BSEP. This work demonstrated for the first time that MDR1/P-gp in mice could transport bile acids, thus playing a compensatory role in BSEP-null mice in maintaining an alternative bile acid transport process.
MRP2-Null Mice
MRP2 is an efflux transporter that transports glucuronides, glutathione, and sulfate conjugates into bile (Jedlitschky et al., 2006). Mice with a functional BSEP but lacking a functional MRP2 (gene symbol: Abcc2) are not likely to have any major impairment of bile acid transport, as demonstrated by serum bile acid concentrations in MRP2-null (Abcc2‒/‒) mice. Although bile acid concentrations were higher in the serum of some MRP2-null mice, interanimal variation was high and not statistically different from control mice (Chu et al., 2006). However, functional disruption of MRP2 in both humans and rats causes conjugated hyperbilirubinemia. The expression of MRP2 (as well as MDR1A, MDR2, and MRP3) is markedly induced in BSEP-null mice (Lam et al., 2005). To determine the role of MRP2 in the canalicular transport of bile acids, Megaraj et al. (2010) characterized the transport of two isomers of a tetrahydroxylated bile acid 6-OH-taurocholic acid (6-OH-TC) (3α,6α,7α,12α and 3α,6β,7β,12α) by membrane vesicles overexpressing mouse MRP2, MDR1A, or BSEP. They found that all these transporters could transport 6-OH-TC, but MRP2 showed the highest affinity and capacity for 6-OH-TC transport. They also studied the biliary excretion of 6-OH-TC in wild-type and MRP2-null mice in the presence or absence of a P-gp inhibitor. They also used a P-gp and breast cancer resistance protein transporter inhibitor (GF120918, aka Elacridar) to rule out any potential role of breast cancer resistance protein in the transport. These experiments by Megaraj et al. (2010) demonstrated that MRP2 plays an important role for the lost function of BSEP in mice. The relative affinity for 6-OH-TC transport was in the order of MRP2 > P-gp > BSEP, suggesting that MRP2 likely plays a beneficial role in cholestasis in mice.
In summary, BSEP transports most bile acids, whereas unusual bile acids, such as sulfated bile acids or tetrahydroxylated bile acids, are transported by other ABC transporters, such as MDR1 and MRP2 (Dawson et al., 2009). MRP3 and MRP4 also play an important role in the basolateral efflux of sulfated bile acids into the systemic circulation for renal excretion (Zelcer et al., 2003; Kullak–Ublick et al., 2004). Additionally, when BSEP function is compromised, MDR1 and MRP2 play a compensatory role in canalicular bile acid transport.
Coupling of BA Secretion with Phospholipid and Cholesterol Secretion
Plasma membranes of animal cells contain four major phospholipids‒ phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylserine (PS), and sphingomyelin, which together account for over half of the lipids in most biologic membranes. PC and sphingomyelin are predominant in the outer leaflet of the plasma membrane, whereas PE and PS are predominant in the inner leaflet. Such asymmetric phospholipid distribution is the result of their active translocation from one leaflet to the other. Transporters that move lipids from the outer leaflet to the inner leaflet (Outer → Inner) of the plasma membrane are called flippases, whereas those that move lipids from the inner leaflet to the outer leaflet (Inner → Outer) are called floppases. The flippase and floppase functions are necessary to maintain the lipid asymmetry of biologic membranes. This asymmetry can be undone by scramblases (Coleman et al., 2013).
Because of the detergent properties of bile acids, continuous exposure of bile acids results in injury to bile canaliculi and biliary epithelium. Within the liver, bile canaliculi lined by hepatocytes form the interconnecting conduits that propel the secreted bile. Bile canaliculi empty their contents (secreted by the hepatocytes) into tiny bile ductules (<15 μm) (Boyer, 2013). These tiny bile ductules progressively merge to form small intrahepatic bile ducts. The small intrahepatic bile ducts further merge to form large intrahepatic bile ducts that, in turn, coalesce to form the extrahepatic bile ducts. The epithelia of the intrahepatic bile ductules and ducts are lined by cholangiocytes; whereas the epithelia of the extrahepatic bile ducts are lined by columnar epithelial cells. The complex and highly branched intrahepatic bile duct system is called the biliary tree. The junction between the intrahepatic bile duct system and the hepatocytes is called the Canal of Hering, which is lined in part by hepatocytes and in part by cholangiocytes. The Canal of Hering represents the anatomical and physiological link between the biliary tree and the hepatocyte bile canalicular system, which extends within the hepatic lobules. Cholangiocytes, also called bile duct cells, form the lining of the biliary epithelium in the biliary tree, and are exposed to biliary solutes secreted into the biliary tree. Cholangiocytes are classic epithelial cells but are highly heterogeneous both in structure and function (Boyer, 2013).
Phospholipids provide protection to bile canaliculi and biliary epithelium (hence cholangiocytes lining them) from bile acids (Davit-Spraul et al., 2009; Morita and Terada, 2014). Phospholipids also aid bile acids to solubilize cholesterol. Several phospholipid and cholesterol transport proteins, such as MDR3/MDR2 (phospholipid transporter); ABC transporters ABCG5/ABCG8 (G5/G8; cholesterol transporter); and ATP8B1 (phospholipid transporter) participate in the release of phospholipid and cholesterol into the bile canaliculi. ATP8B1, a P4-type ATPase (gene symbol ATP8B1) functions as a membrane lipid flippase to mediate the inward translocation of PS and PE from the outer to the inner (cytoplasmic) leaflets of the plasma membrane. P-type ATPases are transmembrane proteins that couple ATP hydrolysis to the efflux of ions and lipids.
The human MDR3 (gene symbol ABCB4) is a floppase that translocates PC from the inner to the outer leaflet of the canalicular membrane of hepatocytes (Oude Elferink and Paulusma, 2007). Translocation of PC to the outer leaflet allows it to be extracted into the canalicular lumen by bile acids moving through bile canaliculi. Inactivating mutations of ABCB4 leads to progressive familial intrahepatic cholestasis type 3 (PFIC3), which is a chronic cholestatic condition. In addition, affected individuals may be prone to gallstone formation (Sundaram and Sokol, 2007).
Observations from Mdr2-Null and G5/G8-Null Mice
In mice, MDR2 (gene symbol Abcb4) is the ortholog of human MDR3. MDR2-null (Abcb4‒/‒) mice develop cholestasis, hepatic lesions, injury to the bile canaliculi, and portal inflammation. In homozygous null (Abcb4‒/‒) mice, the bile is almost completely devoid of PC whereas in heterozygous null (Abcb4+/‒) mice, the PC content of the bile is 60% of that in the wild-type mice (Smit et al., 1993). MDR2 in mice does not transport cholesterol, which is transported by G5/G8. Nevertheless, cholesterol excretion in MDR2-null mice is severely impaired because the absence of PC makes the bile acid micelles poor cholesterol solubilizers (Oude Elferink and Paulusma, 2007). The role of PC in solubilizing cholesterol in bile is further evidenced by G5/G8-null mice (with both Abcg5 and Abcg8 genes disrupted). Wittenburg and Carey (2002) suggested that the G5/G8 heterodimer translocates cholesterol from the inner to the outer leaflet of the canalicular membrane. Thus, G5/G8-null mice have extremely low biliary cholesterol concentrations, but biliary phospholipids and bile acid concentrations are not altered (Yu et al., 2002).
ATP8B1 is a P4-type ATPase and another membrane lipid translocator. It acts as a flippase that translocates PS and PE from the outer to the inner leaflet of the plasma membrane to maintain the asymmetric lipid distribution. This lipid asymmetry plays an important protective role and makes the membrane more resistant to bile acids. Because PS is not translocated from the outer to the inner leaflet in ATP8B1-null mice, it is readily extracted by bile salts. Hence, ATP8B1-null mice show enhanced biliary excretion of PS and cholesterol (Paulusma et al., 2006).
The current model of canalicular lipid extraction depicts that bile salts transported by BSEP in the bile canaliculi reach a concentration in the bile above the critical micellar concentration and form micelles. MDR3 in humans (MDR2 in mice) translocates PC from the inner to the outer leaflet of the canaliculi so that it can be extracted by bile salt micelles to form mixed micelles. Likewise, G5/G8 translocates cholesterol from the inner to the outer leaflet and allows it to be extracted in the same manner as PC by bile salt micelles. Mixed micelles of bile salts and phospholipids extract and solubilize cholesterol more efficiently than simple bile salt micelles. In this model, ATP8B1 translocates PS and PE from the outer to the inner leaflet of the canalicular membrane of the hepatocyte, thereby increasing the relative content of cholesterol in the outer leaflet, which makes the membrane resistant to bile salts (Oude Elferink and Paulusma, 2007).
Gut Microbiota and the Conversion of Primary Bile Acids to Secondary Bile Acids
Bile acids encounter high concentrations of gut microbiota in the distal ileum and large intestine and undergo a variety of bacterial transformations. Most of the bacteria (94%) are Proteobacteria, Firmicutes, Actinobacteria and Bacteroidetes. Firmicutes and Bacteroidetes make up more than 90% of the overall gut microbiota (Ramirez-Perez et al., 2017). The Firmicutes phylum contains more than 200 different genera; some important genera are Lactobacillus, Bacillus, Clostridium, Enterococcus, and Ruminicoccus. Bacteroidetes consists of predominant genera such as Bacteroides and Prevotella. The Actinobacteria phylum is mainly represented by the Bifidobacterium genus (Rinninella et al., 2019). Data from the MetaHit and the Human Microbiome Project have identified 2172 species in humans belonging to 12 different phyla (Li et al., 2014; Hugon et al., 2015). Of the 2172 species, 386 anaerobic species have been found in mucosal regions, such as the oral cavity and the GI tract (Hugon et al., 2015). By around 2.5 years of age, the composition, diversity, and functional capabilities of the microbiome resemble those of the adult microbiome (Koenig et al., 2011; Rodriguez et al., 2015). Although the gut microbiome is relatively stable in healthy individuals, life events can cause perturbations (Dethlefsen and Relman, 2011).
Bile acids become substrates for microbial biotransformation in the distal part of small intestine and mainly in the large intestine (Ridlon et al., 2006; Li and Dawson, 2019). Microbial biotransformation produces secondary bile acids via several reaction pathways, such as deconjugation, dehydrogenation (oxidation), dihydroxylation (oxidation), and epimerization. Deconjugation reactions by microbial bile salt hydrolase removes the amino acid (taurine or glycine) and produces unconjugated bile acids. Hydroxysteroid dehydrogenases are bacterial enzymes that act on the 3-, 7-, and 12-OH groups of bile acids to catalyze epimerization and oxidation/reduction. Among bile acid metabolizers, bile acid 7-dehydroxylating bacteria are particularly important because they carry out the 7-dehydroxylation of the primary bile acids to produce 7-dehydroxylated secondary bile acids (e.g., 7α-dehydroxylation of CA and CDCA to produce DCA and LCA, respectively), and 7-dehydroxylated bile acids represent most of the secondary bile acids (Marion et al., 2019). In mice, ω-MCA is a major metabolite of β-MCA produced by gut microbiota (Eyssen et al., 1983). Dehydrogenation reactions can produce over 20 metabolites through oxidation and epimerization around C-3, C-7, and C-12 (Ridlon et al., 2006).
Among the secondary bile acids, DCA and LCA predominate in the feces. More than half of LCA in humans is sulfated. Some sulfated LCA is excreted in bile to be ultimately excreted in the feces, whereas some sulfated LCA is effluxed by the liver into the sinusoidal blood by MRP3 and MRP4 for renal excretion. In cholestasis, larger amounts of sulfated bile acids are excreted into the sinusoidal blood for renal excretion. In humans, LCA can be further metabolized in the intestine by CYP3A4 into more hydrophilic (hence less toxic) HCA and UDCA, although UDCA is also thought to be formed by bacterial action (Araya and Wikvall, 1999). The ability to produce bile acid-sulfate conjugates is species-dependent. For example, humans and chimpanzees efficiently sulfate LCA, but baboons and rhesus monkeys poorly sulfate LCA.
Some Gut Microbiota Are Harmed by Bile Acids
Some bile acids can inhibit bacterial growth. The inhibitory effect of bile acids on bacterial growth was observed with Bacteroides, Clostridia, Lactobacillus, Bifidobacteria, Escherichia coli, Enterococcus fecalis, and 7α-dehydroxylating bacteria, such as Clostridium scindens, Clostridium hylemonae, and Clostridium hiranonis (Tian et al., 2020). The cytotoxic effect of bile acids is associated with their amphipathic nature. Bile acids can disrupt bacterial cell membranes, damage DNA, alter the conformation of proteins, and chelate iron and calcium. Therefore, bacteria colonizing the intestinal tract, such as Lactobacillus, Bifidobacterium, and Bacillus can repair bile acid-induced damage. Bacteria chronically exposed to bile acids overexpress proteins that help counteract the negative effects of bile acids, such as transporters that efflux bile salts, and enzymes that modify the global metabolism (Tian et al., 2020). Using a combination of flow cytometry, growth rate measurements (OD600), nuclear magnetic resonance and mass spectrometry based metabolomics to investigate the effects of bile acids on individual bacteria and cecal bacterial community, Tian et al. (2020) showed that (1) unconjugated bile acids possess more potent antibacterial activity than conjugated bile acids and (2) gram-positive bacteria are more sensitive to bile acids than gram-negative bacteria.
Uptake of Bile Acids from the Intestine into the Ileal Enterocytes
The uptake of bile acids into the enterocytes is mediated by ASBT, which belongs to the SLC10 family of solute carrier proteins and requires sodium cotransport for its activity. ASBT (gene symbol SLC10A2; Slc10a2 in rodents) is expressed on the apical membrane of enterocytes and mediates the absorption of bile acids from the ileum. ASBT was first cloned from hamster ileum and was called ileal Na+/bile acid cotransporter (Wong et al., 1994). However, ileal Na+/bile acid cotransporter is also expressed in kidney; therefore, ASBT is a more appropriate name and is more frequently used. ASBT is electrogenic with a Na+:BA transport stoichiometry of 2:1. In addition to the distal ileum, ASBT is also expressed at high levels in renal proximal tubules and biliary epithelium (Balakrishnan and Polli, 2006).
Primary bile acid malabsorption is an intestinal disorder in humans associated with diarrhea, steatorrhea (excretion of large amounts of fat in the stool), and reduced plasma cholesterol levels. Oelkers et al. (1997) identified the following ASBT polymorphisms in a family with congenital primary bile acid malabsorption: an allele with A171S missense mutation and a mutated splice donor site for exon 3; an allele with L243P and T262M missense mutations. The L243P and T262M mutations, singly or together, abolished bile acid transport as demonstrated by studies in transfected COS cells. A similar bile acid diarrhea and steatorrhea are also found after ileal resection surgery. In patients, terminal ileal resection causes a reduction in ASBT, resulting in a reduction in the absorption of bile acids. Shorter length ileal resection causes diarrhea, whereas resections greater than 100 cm also produce steatorrhea. In patients with active Crohn’s disease with an increased prevalence of bile acid malabsorption, ASBT expression is reduced by half (Mottacki et al., 2016). Pharmacological inhibition of ASBT is being explored as a therapy for constipation, dyslipidemia, atherosclerosis, type 2 diabetes mellitus, non-alcoholic fatty liver disease, and cholestatic liver diseases. Some of the ASBT inhibitors in clinical trials are Elobixibat, Odevixibat, Maralixibat, Linerixibat, and Volixibat (van de Peppel, et al., 2020). Table 7 summarizes some information on rodent and human ASBTs.
Rodent and human ASBTs
Observations from ASBT-Null Mice
ASBT-null mice (Slc10a2‒/‒) have impaired intestinal bile acid absorption and, consequently, increased fecal bile acid loss (Dawson et al., 2003). No ileal histologic or ultrastructural changes were noted in ASBT-null mice, and fecal bile acid excretion was increased 10- to 20-fold. Because of the poor bile acid absorption and the increased bile acid loss in feces, bile acid synthesis is increased, but the bile acid content is still decreased by 80% and selectively enriched in CA, reflecting an increased contribution of the CYP7A1/CYP8B1 pathway (Dawson et al., 2003). Because the bile acid pool in mice is rich in hydrophilic bile acids and taurine conjugates, mice appear to be more dependent on ASBT function than humans (Li and Dawson, 2019).
ASBT is also found in the apical membrane of cholangiocytes that form the epithelial lining of bile ducts. Here ASBT plays an important role in cholehepatic shunting in which bile acids, after being secreted from hepatocytes, are reabsorbed by cholangiocytes and cycled back to hepatocytes. Cholehepatic shunting is triggered by bile duct obstruction and cholestasis. During cholehepatic shunting, taurine-conjugated bile acids are transported by ASBT, whereas unconjugated bile acids are thought to directly permeate through the membrane (Glaser and Alpini, 2009).
Transport of Bile Acids from the Apical to the Basolateral Side of Enterocytes
The small intestine contains three intracellular lipid binding proteins: the liver fatty acid binding protein (LFABP; gene symbol Fabp1), the intestinal fatty acid binding protein (IFABP; gene symbol Fabp2), and the (IBABP; gene symbol Fabp6) (Agellon et al., 2002). IBABP is expressed in the enterocytes along with ASBT and OSTα‒OSTβ. IBABP can bind both fatty acids and bile acids, but it binds bile acids with higher affinity. IBABP is involved in the apical to basolateral transport of bile acids.
Observations from IBABP-Null Mice
Praslickova et al. (2012) developed an IBABP-null (Fabp6‒/) mouse model. The IBABP-null mice had a decreased bile acid content, but there were sex dimorphic effects on the retention and excretion of bile acids. Using exogenously administered radiolabeled bile acid (3[H]TCA) to trace the transit and fate of bile acids in the small intestine, it was found that female IBABP-null mice had an increased excretion of bile acids in the feces, whereas IBABP-null males had an increased retention of bile acids in the small intestine. The authors concluded that the loss of IBABP impairs the intestinal transit of bile acids in male mice, but female mice have greater dependence on IBABP for the proper transfer of bile acids across the enterocytes. The authors also concluded that IBABP may not be an absolute prerequisite for the transfer of bile acids within enterocytes; however, it is required for efficient transport of bile acids from the intestinal lumen (apical part of the enterocyte) to the portal circulation (basolateral part of the enterocyte) (Praslickova et al., 2012).
Efflux of Bile Acids from the Enterocytes into the Portal Circulation
Bile acid efflux from enterocytes into the portal circulation is mediated by the heterodimeric complex of OSTα and OSTβ. OSTα and OSTβ (gene symbols: SLC51A and SLC51B in humans; Slc51a and Slc51b in rodents) were first cloned in the marine vertebrate, the little skate Raja erinacea (Wang et al., 2001b). By injecting Xenopus oocytes with the cRNA for both OSTα and OSTβ, individually and together, the authors noted that the OSTα‒OSTβ heterodimer functions as a transporter but not the homodimer. The heterodimer transports taurocholate, estrone sulfate, digoxin, and prostaglandin E2. The transport is sodium independent. Based on the information from human and mouse genome sequencing and hypothetical human and mouse proteins from the databases, Seward et al. (2003) identified putative human and mouse OST homologs. The human OSTα and OSTβ, and mouse OSTβ cDNAs were cloned using RT-PCR-based strategy. The cDNA for the putative mouse OSTα was obtained from a commercial source. Mammalian OST proteins were found to functionally complement the evolutionarily divergent OST proteins of Raja erinacea (Seward et al., 2003). In the small intestine, the OSTα‒OSTβ heterodimer is expressed on the basolateral membrane of enterocytes, and it is the key transporter that effluxes bile acids from enterocytes into the portal circulation to maintain the enterohepatic circulation (Ballatori et al., 2005; Dawson et al., 2005). Table 8 summarizes some information on rodent and human OSTα and OSTβ.
Skate, mouse, and human OSTα and OSTβ
In addition to OSTα–OSTβ, MRP3 (gene symbol Abcc3) has also been proposed as a candidate ileal basolateral bile acid carrier based on indirect evidence, such as basolateral membrane localization and the ability to transport bile acids in vitro. However, studies using MRP3-null mice found no apparent defects in intestinal bile acid absorption (Rao et al., 2008).
Observations from OSTα‒OSTβ-Null Mice
OSTα-null (Slc51a ‒/‒) mice were developed to study the in vivo functions of this unusual transporter (Rao et al., 2008; Ballatori et al., 2008). In OSTα-null mice, OSTβ protein was also markedly reduced in addition to the loss of OSTα protein, demonstrating that the expression of α and β subunits are coordinately regulated so that the loss of OSTα resulted in a dramatically reduced expression of OSTβ. Because of the loss of OSTα–OSTβ heterodimer expression, bile acids accumulate inside the enterocytes, causing damage to the enterocytes. As a result, OSTα-null mice have morphologic changes in the ileum secondary to enterocyte injury (Rao et al., 2008; Ballatori et al., 2008). Total bile acid pool from liver, gallbladder, and small intestine is 90% lower, and serum bile acid pool is 60% lower in OSTα-null mice compared with controls (Ballatori et al., 2008). Unlike ASBT-null mice that have a selective enrichment of CA in the bile acid pool, reflecting an increased contribution of the CYP8B1 pathway, the composition of the bile acid pool is not altered in OSTα-null mice (Dawson et al., 2003; Rao et al., 2008). Fecal cholesterol excretion is increased about fourfold in OSTα-null mice, and the same trend is observed in ASBT-null mice (Dawson et al., 2003; Rao et al., 2008). When OSTα-null mice were fed CA, the fecal excretion of CA was increased about fivefold by day 2. This indicated not only a reduction of OSTα–OSTβ mediated CA transport to the portal circulation, but also a reduction in the uptake of CA into the enterocytes by ASBT, strongly suggesting a corresponding reduced ASBT expression (Rao et al., 2008). A reduction in ASBT expression secondary to the loss of OSTα–OSTβ expression suggests the existence of a sensing mechanism that prevents the accumulation of bile acids in enterocytes, thereby protecting them from structural damage.
By examining intestinal fibroblast growth factor 15 (FGF15) expression in wild-type, OSTα-null, FXR-null, and OSTα‒FXR-double null mice, Lan et al. (2012) found that the changes in bile acid homeostasis in OSTα-null mice were due to the altered gut-liver bile acid signaling through the FXR‒FGF15‒FGFR4 pathway (discussed in more detail later under Observations in FXR-null mice).
The importance of OSTα‒OSTβ in human intestinal bile acid transport has been demonstrated by two brothers bearing an inherited OSTβ-deficiency. The clinical presentation of these pediatric patients included congenital chronic diarrhea, fat-soluble vitamin deficiency, and features of cholestatic liver disease (Sultan et al., 2018). The authors performed whole exome sequencing and identified a homozygous single nucleotide deletion, which is in codon 27 in the first coding exon of the OSTβ gene. This deletion resulted in a frameshift and corresponding formation of a premature stop in codon 50. Cells containing the truncated OSTβ protein have a reduced expression of the OSTα partner protein and markedly reduced taurocholate uptake.
Fate of Bile Acids in the Colon
The absorption of bile acids from the large intestine is generally thought to occur by diffusion. However, some studies suggest an active transport of bile acids. ASBT is most highly expressed in the distal ileum, with negligible expression in the colon. Anderle et al. (2005) studied the expression profiles of several drug and nutrient carrier mRNAs along the anterior-posterior axes of the mouse intestine. The mouse gene expression data were compared with publicly available gene expression data for the human small intestine and the colon. The authors found that the expression of transporters changed most significantly between the ileum and colon. Higher levels of expression were found in the small intestine than in the colon. Interestingly, the authors found that most transporters were similarly expressed in mice and humans. Among the bile acid transporters, Asbt mRNA expression was found to be higher in ileum than jejunum, but Asbt mRNA expression was also detected in the colon. Colonic expression of ASBT was confirmed by Hruz et al. (2006) in normal human subjects. The authors reported ASBT mRNA expression throughout the colon, although the expression was low. Using transepithelial taurocholate uptake, Weihrauch et al. (2006) observed colonic uptake of bile acids in weanling rabbits, although the magnitude of colonic uptake was less than ileal uptake. More recently, Fu et al. (2016) studied the expression of xenobiotic metabolizing genes in the liver and intestine using RNA sequencing. The authors reported that 54% of the genes were expressed most highly in the liver, 21% in the duodenum, 4% in the jejunum, 6% in the ileum, and 15% in the large intestine. Genes in the same family usually exhibited highly different tissue-specific expression patterns, and many were almost exclusively expressed in one tissue and minimally expressed in others.
Another interesting observation was made by Dekaney et al. (2008), who conducted ileocecal resection in normal, FXR-null and germ-free mice. Using a combination of immunochemical detection of proteins and quantitative RT-PCR of mRNA, the authors observed the expression of ASBT, IBABP, and OSTα/β in the colon after ileocecal resection. A similar expression was not observed in germ-free mice, indicating a role of intestinal bacteria in the expression of these transporters. Additionally, the increased expression of these transporters was significantly attenuated in FXR-null mice, suggesting a role for FXR. The authors suggested that the absence of ASBT-mediated absorption of bile acids in the distal ileum exposes the colonic mucosa to a high colonic bile acid load, and the abnormal expression of bile acid transporters may have a protective role against such high bile acid load. These adaptations may partially compensate for the lack of bile acid absorption from the distant ileum.
These findings underscore that unlike the traditional wisdom that active bile acid absorption does not take place in the colon, small amounts of bile acid absorption likely take place in the colon. Deconjugated hydrophobic (less hydrophilic) bile acids are in a more lipid-soluble form and are probably absorbed through diffusion back into the portal circulation in the colon.
Uptake of Bile Acids from the Sinusoidal Blood into the Hepatocytes
Bile acids are taken up in hepatocytes across the sinusoidal (basolateral) membrane by NTCP and organic anion transporting polypeptides 1B2 (OATP1B2). The transport by NTCP is Na+-dependent, while the transport by OATPs is Na+-independent. Hepatocyte uptake of bile acids through the enterohepatic circulation is not 100% efficient. Bile acids not immediately taken up by the hepatocytes spill over into the systemic circulation. Thus, the systemic circulation becomes enriched in those bile acids, having less hepatic uptake. These bile acids are again presented to the liver through hepatic arterial blood and/or hepatic portal blood and are ultimately taken up. Therefore, at any given point of time the systemic blood contains a collection of bile acids that were not initially taken up by the liver.
Uptake by NTCP
NTCP (gene symbol SLC10A1 in humans; Slc10a1 in rodents), is the primary transporter for the uptake of conjugated bile acids across the hepatic sinusoidal membrane from serum into hepatocytes (Fig. 11). NTCP-mediated bile acid transport is Na+-dependent, in which bile acids and Na+ are cotransported in the same direction. Therefore, NTCP is a symporter. The preferred substrates of NTCP are both glycine- and taurine-conjugated bile acids. NTCP also transports unconjugated bile acids, which are moderate to weak substrates (Meier, 1995). Table 9 summarizes some information on rodent and human NTCPs.
Rodent and human NTCPs
Observations from NTCP-Null Mice
In NTCP-null mice (Slc10a1–/–), serum bile acid concentrations remain normal in the majority (60%–75%) of the animals. Only a subset of NTCP-null mice has markedly elevated serum bile acid levels, particularly conjugated bile acid concentrations (millimolar concentration compared with controls, which were <20 µM) (Slijepcevic et al., 2015). The results in NTCP-null mice indicate the existence of a parallel bile acid uptake system in mice. Feeding UDCA to NTCP-null mice resulted in serum bile acid accumulation in the null mice that previously had normal serum bile acid concentrations. This demonstrates the existence of a Na+-independent bile acid uptake system that operates close to its maximum capacity in NTCP-null mice and can be easily saturated (Slijepcevic et al., 2015).
In contrast to NTCP-null mice, humans that inherited NTCP deficiency have nearly a 100-fold increase in serum bile acid concentrations, primarily conjugated bile acids (Vaz et al., 2015). Likewise, NTCP inhibition by myrcludex B, a drug used to block the uptake of viruses into the liver, resulted in a 19-fold increase in total serum bile acids, whereas conjugated bile acids increased more than 100-fold (Blank et al., 2018). These reports underscore a crucial role of NTCP in the hepatic uptake of bile acids, especially conjugated bile acids, in humans.
Uptake by OATPs
The first OATP (gene symbol SLCO in humans; Slco in rodents, e.g., SLCO1B1, Slco1b2) cloned was rat OATP1A1 (previous name OATP1), which is a Na+-independent organic anion transporter that can transport bile acids in vitro (Jacquemin, et al., 1994). By injecting Oatp1a1 mRNA into Xenopus oocytes and expressing OATP1A1 protein, the authors observed that there was about a 15-fold increase in taurocholate uptake by oocytes expressing OATP1A1 compared with noninjected controls. This finding suggested that OATPs likely plays an important role in hepatic uptake of bile acids.
There are no clear-cut orthologous relationships between OATP1A/1B family of genes in humans and rodents. For example, OATP1A2 is the only human OATP1A isoform, but rats and mice have several OATP1A proteins (OATP1A1, -1A4, -1A5, and -1A6), which probably arose by gene duplication. In humans, OATP1B1 and OATP1B3 are two OATP1B isoforms, whereas rats and mice have only OATP1B2 (Lu et al., 2008). Mouse OATP1A1, -1A4, -1A5, -1A6 and OATP1B2 are all clustered in chromosome 6. Mouse OATP1A1, OATP1A4, and OATP1B2 are expressed in liver, whereas OATP1A5 is expressed in gonads and OATP1A6 is expressed in kidneys (Cheng et al., 2005). Table 10 summarizes some characteristics of rodent and human OATPs important for bile acid uptake.
Rodent and human OATPs important for BA uptake
Observations from OATP-Null Mice
van de Steeg et al. (2010) developed OATP1A/1B-null (Slco1a/1b2–/–) mice. Because of the deletion of the entire Slco1a/1b locus, OATP1A/1B-null mice had all five OATPs deleted— OATP1A1, -1A4, -1A5, -1A6, and OATP1B2. The serum levels of unconjugated bile acids were elevated by about 13-fold in OATP1A/1B-null mice compared with controls, suggesting an important role of mouse OATP transporters in the uptake of unconjugated bile acids. Using OATP1B2-null (Slco1b2–/–) mice, Csanaky et al. (2011) showed that these mice have a 15-fold higher unconjugated serum bile acid levels, thereby clearly identifying OATP1B2 as a key transporter that mediates the basolateral uptake of unconjugated bile acids into the liver.
Further studies (Slijepcevic et al., 2017) revealed an important role of OATP, along with NTCP, in the uptake of conjugated bile acids. When wild-type mice were administered myrcludex B (NTCP inhibitor), there was a small effect on serum taurocholate clearance compared with controls. In contrast, treating OATP1A/1B-null mice with myrcludex B caused a complete abrogation of taurocholate transport, suggesting that NTCP and OATP together mediate hepatic uptake of conjugated bile acids in mice. Next, Slijepcevic et al. (2017) used OATP1A/1B-null mice for expressing the human OATP1B1. The authors found that hepatic expression of human OATP1B1 reversed the conjugated hyperbilirubinemia in OATP1A/1B-null mice but could not block the rise in plasma conjugated bile acids after inhibition of NTCP with myrcludex B. These observations strongly suggest that OATP1B1 can mediate the hepatic uptake of conjugated bile acids, but NTCP is the primary uptake transporter of conjugated bile acids in humans. Using bile acid uptake in U2OS cells transiently overexpressing mouse NTCP, OATP1A1, OATP1B2 and rat OATP1A4, Slijepcevic et al. (2017) demonstrated that mouse OATP1A1 and OATP1B2 were capable of significant taurocholate uptake (10–12 times greater than in untransfected U2OS cells).
Conversion of Secondary Bile Acides to Primary Bile Acids in the Liver
The bile acids transported by enterohepatic circulation to the liver are taken up into hepatocytes by NTCP and OATP, as discussed above, thereby completing the enterohepatic circulatory loop. In the hepatocytes, deconjugated bile acids are again modified by reconjugation with glycine or taurine in the liver. The extent of modification, particularly for rehydroxylation at position 7, is species dependent. For example, DCA does not undergo 7-rehydroxylation in humans, but in mice and rats DCA undergoes 7-rehydroxylation producing CA. In mice, CYP2A12 is responsible for bile acid 7α-rehydroxylation (Honda et al., 2020). In the intestine of rats and mice, unconjugated α-, β-, and ω-MCAs undergo bacterial 7α or 7β-dehydroxylation to yield HDCA or MDCA, but this seems to be a minor pathway compared with 7α-dehydroxylation of CA (Li and Dawson, 2019). Bile acids with a 3β-OH group are epimerized to 3α-OH BAs. Oxo groups are reduced partly or completely to α- and/or β-OH groups in a species dependent manner (Ridlon et al., 2006).
Basolateral Bile Acid Efflux Systems in Hepatocytes
Bile acids can be effluxed across the basolateral (sinusoidal) membrane of hepatocytes back into the sinusoidal blood by MRP3, MRP4, and the OSTα-OSTβ heterodimer (Fig. 11). Under cholestatic conditions, bile acid excretion into the bile canaliculi is limited causing hepatocellular bile acid overload and hepatocyte damage. This condition triggers the alternative clearance path, that is, the transport of bile acids back into the sinusoidal blood by basolateral efflux. Such basolateral efflux transport is a protective mechanism from bile acid-induced damage and is usually coordinated with phase I and II metabolism, often producing substrates that are less toxic (Halilbasic et al., 2013). For example, MRP3 plays a significant role in the basolateral efflux of glucuronidated and sulfated bile acids when canalicular excretion is impaired, as has been shown in obstructive cholestasis in rats (Soroka et al., 2001). Bile acids thus effluxed back into the sinusoidal blood can be eliminated in the urine. The OSTα–OSTβ heterodimer plays two very important functions in bile acid homeostasis; first, to extrude bile acids from the enterocytes into the portal blood to facilitate the enterohepatic circulation of bile acids, and secondly, to perform basolateral efflux of bile acids in the liver to reduce hepatocellular bile acid overload and protect the liver from bile acid-induced damage. The expression of OSTα–OSTβ is induced via FXR. The accumulated bile acids in hepatocytes activates FXR, which in turn upregulates OSTα–OSTβ expression (Dawson et al., 2010).
Bile Acid-Induced Liver Toxicity
Secondary bile acids, such as DCA and LCA, cause cytotoxicity, oxidative stress, and membrane damage, whereas others (e.g., UDCA) have anti-inflammatory properties and can protect enterocytes and colonocytes against oxidative damage. Some of the secondary bile acids produced from primary bile acids are as follows: CA → DCA; CDCA → LCA and UDCA; α-MCA and β-MCA → ω-MCA → HDCA; β-MCA → HCA → MDCA (Winston and Theriot, 2020). Figure 10 shows the metabolic conversion of various primary and secondary bile acids. Song et al. (2011) investigated, among other endpoints, the relative hepatotoxicity of some individual BAs fed to mice and found the order of hepatotoxicity as UDCA < CA < CDCA < DCA < LCA. The lowest concentration of each BA in the feed that produced hepatoxicity was 0.3% for CA and CDCA, 0.1% for DCA, and 0.03% for LCA.
Because an increased hydrophobicity of bile acids is generally known to be associated with an increased tendency to cause cholestasis and cytotoxicity, Heuman (1989) experimentally determined the hydrophobicity index of several primary and secondary bile acids. The higher the value, the greater the hydrophobicity. According to Heuman’s hydrophobicity scale, the following bile acids are ranked from the most hydrophobic (left) to the least hydrophobic (most hydrophilic; right): LCA > DCA > CDCA > CA > HDCA > HCA > UDCA > β-MCA > α-MCA > UCA. Table 11 shows the hydrophobicity indices of these bile acids, and Table 12 shows the sites of hydroxylation on the steroid nucleus for bile acids species found in humans and mice. Figure 10 (Fig. 10) and Figure 11 (Fig. 11) should be placed right after this section.
Heuman’s hydrophobicity indices of some BAs/Bile salts
Hydroxylation sites on the steroid nucleus for BA species found in humans and mice
Regulation of Bile Acid Homeostasis
Cholesterol metabolism to bile acids is regulated through both feed-forward activation by oxysterols and feedback repression by bile acids. The nuclear receptor involved in the feed-forward activation by oxysterols is the liver-X-receptor (LXR), and the nuclear receptor involved in the feedback repression by bile acids is the farnesoid-X-receptor (FXR), which was so named because it was initially thought to be the receptor for farnesol. Later it was found to be a receptor for bile acids, but the name FXR persists.
Role of LXR
In the feed-forward activation pathway, oxysterols activate LXRα, which in conjunction with the liver receptor homolog 1 (LRH-1) induces Cyp7a1 transcription, bile acid synthesis, and cholesterol excretion. The two members of the LXR family are LXRα and LXRβ; LXRα expression is higher in both murine and human liver, and it has a stronger binding affinity to the LXR response element (LXRE) (Peet et al., 1998). The LXRE in the promoter of the mouse Cyp7a1 gene is activated primarily by LXRα.
Observations in LXR-Null Mice
The role of LXR was demonstrated using LXR-null (Lxrα–/–) mice. When LXR-null mice were fed a high cholesterol diet, the mice accumulated massive amounts of cholesterol and had liver abnormalities because these mice could not mount the compensatory increase in bile acid synthesis and elimination due to the lack of activation of the Cyp7a1 gene (Peet et al., 1998). However, the accumulation of more cholesterol in the liver of LXR-null mice may not be solely due to the lack of induction of CYP7A1 but may also be, in part, due to impaired cholesterol elimination pathways that are also regulated by LXR, such as ABCA1, ABCG1, and ABCG5/G8. The effect of LXRα on human CYP7A1 gene transcription is less pronounced. This was determined by the transcriptional regulation of luciferase reporter gene expression under the control of human CYP7A1 promoter in HepG2 cells, a model established for the study of regulation of cholesterol 7α-hydroxylase (Chiang et al., 2001).
Role of FXR/FGF15/19 Pathway
Bile acids are continuously synthesized in the liver. As the bile-acid pool size increases, a feedback mechanism from the intestine is triggered that inhibits de novo bile acid production in the liver. The link here is FXR. There are two FXR systems—one in the liver and the other in the intestine. The intestinal FXR system operates in the FXR/FGF15/19 pathway. FXR-mediated inhibition of hepatic bile acid synthesis uses an endocrine axis that involves communication between the intestine and the liver. In the intestine, bile acid activated intestinal FXR induces the expression of fibroblast growth factor 15 (FGF15 in mouse; FGF19 in humans) in the enterocytes. The FGF15 secreted into the portal blood reaches the liver and binds to its cognate hepatocyte membrane receptor complex—the FGF receptor 4/β-klotho (FGFR4/β-KL) (Figure 9). The binding of FGF15 to FGFR4/β-KL complex activates downstream signaling pathways to mediate postprandial responses, including repression of bile acid synthesis through decreases in Cyp7a1 gene expression. Therefore, ileal FGF15 and hepatic FGFR4/β-KL form an endocrine axis that negatively regulates bile acid synthesis in the liver and maintains bile acid homeostasis (Inagaki et al., 2005; Lin et al., 2007).

The top figure shows the structure of cholic acid after conjugation with glycine to form glycocholate. The middle figure shows the structure of cholic acid after conjugation with taurine to form taurocholate. The lower figure shows glycocholic acid after sulfate conjugation at the 3α-OH. In mammals, the bile acid side chain is primarily conjugated to taurine or glycine, but there exists a remarkable species difference. Bile acid sulfation also shows species difference (see text).

Regulation of bile acid synthesis. FXR–mediated inhibition of hepatic bile acid synthesis involves bile acid, FGF15 (FGF19 in human), and FGFR4 utilizing an endocrine axis that involves communication between the intestine and the liver. Activation of FGFR4 strongly activates the ERK1/2 and to a lesser extent, the JNK1/2 to downregulate both Cyp7a1 and Cyp8b1 gene expression. Bile acid activated FXR induces SHP expression, which also contributes to the downregulation of both Cyp7a1 and Cyp8b1 gene expression.

Metabolic conversion of bile acids to secondary bile acids by the microbiota.
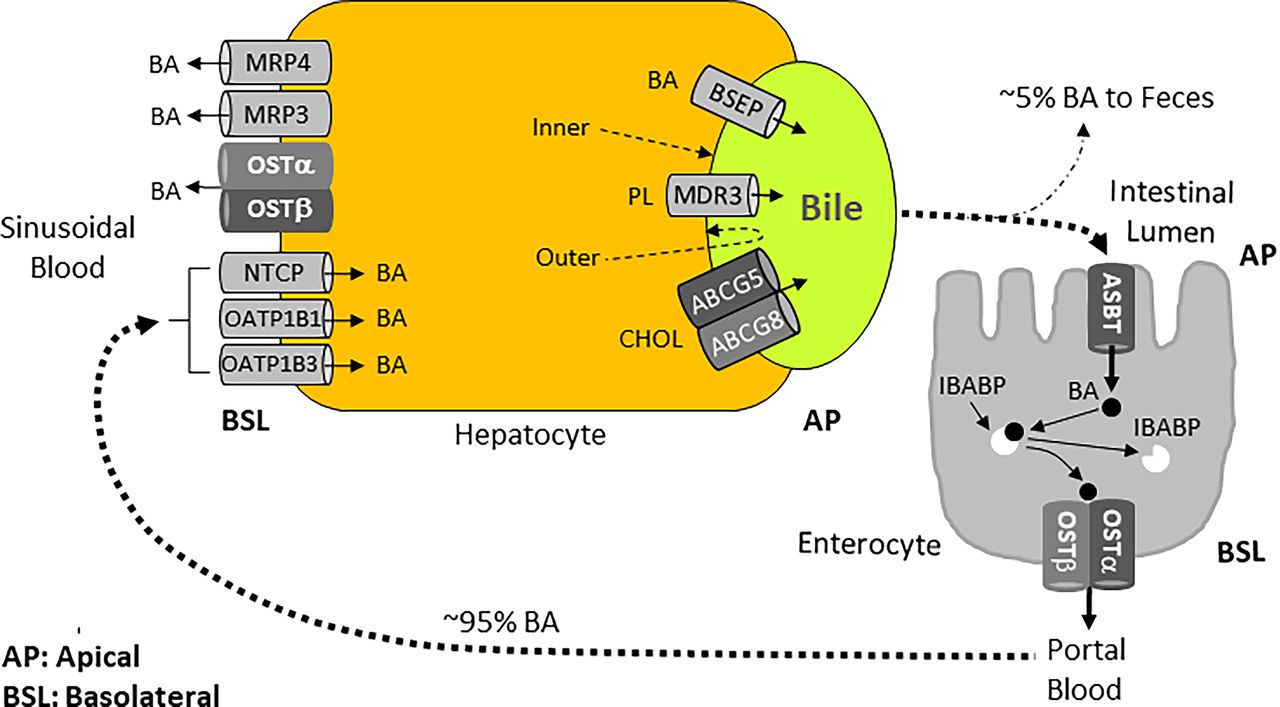
Enterohepatic circulation path and the main bile acid transporters involved. The figure also shows the transporters involved in basolateral efflux of bile acids into sinusoidal blood. This recycling mechanism plays an essential role in maintaining the bile acid pool and normal bile flow, which maintain the bile acid and cholesterol homeostasis.
The human ortholog of FGF15 is FGF19. Like FGF15 in mice, FGF19 in humans represses bile acid synthesis and is induced by FXR. Surprisingly, FGF15 and FGF19 have only 53% amino acid identity (Kliewer and Mangelsdorf, 2015). In humans, serum FGF19 levels peak 1.5 to 2 hours after the postprandial release of bile acids (Lundasen et al., 2006), and the induced FGF19 represses bile acid synthesis. FGF19 level is decreased in subjects administered cholestyramine, a bile acid sequestrant (Lundasen et al., 2006). Overproduction of bile acids causes bile acid diarrhea and lowers circulating FGF19 levels (Walters et al., 2009).
Bile acids were shown to be the cognate ligands of FXR in 1999 independently by three groups (Makishima et al., 1999, Parks et al., 1999, Wang et al., 1999). However, all bile acids and their salts do not bind FXR with the same affinity. Using a cell-free assay that measures ligand-dependent activation of FXR and its interaction with the coactivator SRC-1, Lew et al. (2004) reported the binding affinities of several bile acids and their salts to human FXR. The binding affinity from high (left) to low (right) was found to be as follows:
LCA > CDCA > Tauro CDCA > Glyco CDCA ≫ DCA > UDCA ≫ CA > Tauro CA > Glyco CA
In the liver, bile acids activate the transcription factor FXR, which induces the expression of SHP. Because SHP lacks the DNA-binding domain, elevated SHP protein inactivates LRH-1 by forming a nonfunctional heterodimeric complex leading to promoter-specific repression of the target genes. For example, when SHP binds LRH-1 and/or LXRα, the resulting complex is inhibitory, and it represses the transcription of the Cyp7a1 gene (Brendel, et al., 2002; Goodwin et al., 2000).
The expression of the Cyp8b1 gene is regulated by the fetoprotein transcription factor (FTF) and the hepatocyte nuclear factor 4α (HNF-4α), with FTF probably playing a more important role. SHP-mediated repression of Cyp8b1 transcription is mediated by both FTF and HNF-4α. When SHP binds FTF and/or HNF-4α, the resulting complex is inhibitory, and it represses the transcription of Cyp8b1 (del Castillo-Olivares and Gil, 2001; Zhang and Chiang, 2001).
Observations in FXR-Null Mice
The inhibitory role of bile acid activated FXR on the transcription of Cyp7a1 gene and resulting bile acid synthesis was confirmed using FXR-null (Fxr–/–) mice (Sinal et al., 2000). When challenged with a diet containing CA, FXR-null mice had severe hepatotoxicity and wasting compared with wild-type mice (Sinal et al., 2000). Bile acid activated FXR is now known to decrease the expression of CYP7A1, CYP8B1, CYP27A1, and CYP7B1 (Chiang, 2015). de Aguiar Vallim et al. (2015) reported that FXR activation induces the expression of many transcriptional repressors including the musculoaponeurotic fibrosarcoma oncogene homolog G (MAFG), which directly represses Cyp8b1 gene expression by binding to the MAFG response element (MAREs) in the promoter of the Cyp8b1 gene.
By examining the FGF15 expression in the intestine of wild-type, OSTα-null, FXR-null, and OSTα‒FXR-double null mice, Lan et al. (2012) concluded that the changes in bile acid homeostasis (decreased bile acid content) in OSTα-null mice were due to altered gut-liver bile acid signaling through the FXR‒FGF15‒FGFR4 pathway. The authors found that the expression of CYP7A1 in the liver was decreased in OSTα-null mice, and ileal FGF15 protein was elevated almost 20-fold. These effects were expected because the accumulated bile acids should activate FXR expression in enterocytes, which in turn should upregulate FGF15 expression. FGF15, after being transported to the liver, should ultimately downregulate the expression of CYP7A1 (Figure 9). Conversely, OSTα‒FXR-double null mice have increases in hepatic CYP7A1 expression in the liver due to the loss of FXR and the loss of downstream FXR signaling. Surprisingly, OSTα‒FXR-double null mice had restoration of intestinal cholesterol absorption. The authors concluded that signaling via FXR is required for the paradoxical repression of hepatic bile acid synthesis but not the complex intestinal adaptive changes in OSTα-null mice.
Interestingly, knowledge gained from the whole body FXR-null mice (Sinal et al., 2000), discussed above, was further enriched using tissue-specific FXR-null mice, namely as liver- and intestine-specific FXR-null mice. Using these mice, Kong et al. (2012) showed that the intestinal FXR/FGF15 pathway was critical for suppressing both Cyp7a1 and Cyp8b1 gene expression, but the liver FXR/SHP pathway was important for suppressing Cyp8b1 gene expression and had a minor role in suppressing Cyp7a1 gene expression. Figure 9 illustrates how bile acids negatively regulate their own synthesis. Bile acid activated intestinal FXR induces intestinal FGF15 expression. FGF15 is carried through portal blood to the liver, where it binds to FGFR4 and activates it. Activated FGFR4 in the liver strongly triggers the extracellular signal-regulated kinase 1/2 (ERK1/2), and to a lesser extent c-Jun N-terminal kinase 1/2 (JNK1/2) to downregulate both Cyp7a1 and Cyp8b1 gene expression in the liver (Kong et al., 2012). Bile acid also activates hepatic FXR that induces hepatic SHP expression. Increased SHP expression contributes to the downregulation of both Cyp7a1 and Cyp8b1 gene expression (Kong et al., 2012).
FXR regulates the expression of various bile acid transporters, such as BSEP, NTCP, ASBT, OSTα–OSTβ, and the bile acid conjugation enzymes (e.g., BACS, BAAT) (for an extensive list of the FXR target genes, see Lee et al., 2006). Of these FXR targets, BSEP, OSTα–OSTβ heterodimer, BACS, and BAAT expressions are induced, whereas NTCP and ASBT expressions are repressed. FXR is activated by both free and conjugated bile acids. A highly hydrophilic bile acid, taurine conjugated β-muricholic acid, acts as an FXR antagonist (Sayin et al., 2013). In humans, serum FGF19 peaks about 90–120 minutes after the postprandial release of bile acids, and this peak precedes the repression of bile acid synthesis. FGF19 decreases in subjects administered the bile acid sequestrant, cholestyramine (Lundasen et al., 2006).
Bile Acid Receptor TGR5 and Energy Metabolism
The Takeda G protein-coupled receptor 5 (TGR5) is a G-protein coupled receptor for bile acids. In contrast to FXR that is an intracellular bile acid receptor regulating bile acid homeostasis, TGR5 is a cell-surface bile acid receptor that does not regulate bile acid homeostasis but is important in regulating energy metabolism (Watanabe et al., 2006). TGR5 was identified in 2002 (Maruyama et al., 2002) and was first named the membrane-type receptor for bile acids. TGR5 is also known as the G protein-coupled bile acid receptor 1. Kawamata et al. (2003) reported this as the G-protein coupled receptor for bile acids and named it TGR5, which has become the most widely used name. TGR5 is expressed in many tissues in humans and animals, and it activates various intracellular signaling pathways upon interaction with bile acids. TGR5 exhibits a dose-dependent activation with the following rank order of potency: LCA > DCA > CDCA > CA (Maruyama et al., 2002; Duboc et al., 2014). Interestingly, the secondary bile acids (LCA, DCA) are more potent activators of TGR5 than the primary bile acids (CA, CDCA) (Chaudhari et al., 2021).
TGR5 activation plays an important role in the regulation of basal metabolism and energy expenditure. Glucose homeostasis in the body is maintained by a complex mechanism involving several hormones, receptors, and transporters. An important link between bile acid and glucose homoeostasis is glucagon-like peptide-1 (GLP-1), which is a 30-amino acid peptide hormone derived from the post-translational processing of glucagon and secreted by the intestinal enteroendocrine L cells. GLP-1 promotes glucose-dependent insulin secretion, inhibits glucagon secretion, and reduces gastric emptying and appetite. Bile acids stimulate GLP-1 secretion through TGR5 (Katsuma et al., 2005; Thomas et al., 2009; Klaassen and Cui, 2015).
Bile acids can stimulate adaptive thermogenesis and energy expenditure by signaling via TGR5 (Watanabe et al., 2006). Activation of TGR5 leads to increased intracellular cAMP levels that trigger several downstream events, such as activation of the protein kinase A (PKA) and induction of the cAMP receptor element binding protein (CREB) phosphorylation (Kawamata et al., 2003). The TGR5→cAMP→PKA→CREB activation sequence can regulate several metabolic pathways (Figure 12). Genes containing cAMP response elements can bind CREB and modulate gene expression. One such pathway involves the activation of cAMP-dependent iodothyronine deiodinase type 2 (Dio2) expression; this enzyme converts the biologically inactive thyroxine (T4) to biologically active thyroxine (T3). Formation of T3 increases energy expenditure in all tissues, particularly in the brown adipose tissue and skeletal muscle (Watanabe et al., 2006). Brown adipose tissue is found primarily in newborn infants but is also found in the shoulders and neck in adult humans. Healthy individuals have higher amounts of brown adipose tissue mass/activity, whereas obese individuals seem to have less brown adipose tissue mass/activity. This (re)discovery of the role of brown adipose tissue in adaptive thermogenesis regulation has triggered an interest in using this tissue in enhancing energy expenditure to control body weight and prevent metabolic disorders (Ravussin and Galgani, 2011). The expression of TGR5 and Dio2 in human skeletal muscles suggests a role of TGR5 in a thermogenic mechanism in humans. The accumulating evidence suggests that TGR5 activation may provide a therapeutic approach to improve dysregulation of energy metabolism and reduce obesity by increasing energy expenditure (Taoka et al., 2016).
Schematic diagram of (I) FXR receptor in liver, (II) FXR receptor in intestine, (IIIA) TGR5 receptor in intestine, (IIIB) TGR5 receptor in gallbladder, and (IIIC) TGR5 receptor in fat and muscle (reproduced with permission from Klaassen and Cui. 2015. Drug Metab. Dispos. 43: 1505-1521).
An association between bile acid-TGR5-GLP-1 secretion and improved glucose tolerance has been demonstrated after sleeve gastrectomy, a surgical procedure that removes approximately 75% of the stomach, resulting in the retention of much less food. Using ultra-high-performance liquid chromatography–mass spectrometry-based quantification, Chaudhari et al. (2021) observed an increase in an endogenous bile acid, cholic acid-7-sulfate (CA7S), in the GI tract of both mice and humans after sleeve gastrectomy. They showed that CA7S is a TGR5 agonist that increases TGR5 expression and GLP-1 secretion. Consistent with this finding, administration of CA7S to insulin-resistant mice increases glucose tolerance, which is mediated by TGR5. CA7S remains in the gut and minimizes off-target effects (Chaudhari et al., 2021).
Figure 12 is a schematic diagram of (I) FXR receptor in the liver, (II) FXR receptor in the intestine, (IIIA) TGR5 receptor in the intestine, (IIIB) TGR5 receptor in the gallbladder, and (IIIC) TGR5 receptor in fat and muscle (reproduced with permission from Klaassen and Cui, 2015).
Observations from TGR5-Null and TGR5-Transgenic Mice
Maruyama et al., (2006) developed TGR5-null (Tgr5‒/‒) mice. In these mice, the expression of proteins involved in the hepatic synthesis and transport of bile acids was higher than wild-type controls, but the bile acid pool was not increased; in fact, it was decreased by about 25%, suggesting that TGR5 contributes to bile acid homeostasis. Interestingly, both homozygous and heterozygous TGR5-null females fed a high-fat diet for 2 months showed significant fat accumulation with body weight gain compared with the wild-type mice, suggesting that TGR5 plays an important role in energy homeostasis. TGR5-null mice fed a lithogenic diet are protected against cholesterol gallstone formation (Maruyama et al., 2006; Duboc et al., 2014).
Thomas et al. (2009) showed that glucose tolerance was improved in transgenic mice overexpressing TGR5, and it was accompanied by increased GLP-1 and insulin secretion. This observation strongly suggests a role of bile acids and the bile acid receptor TGR5 in glucose metabolism through the Bile Acid–TGR5–GLP-1 pathway. When the authors added a TGR5 agonist (6-alpha-ethyl-23(S)-methyl-cholic acid) to a high-fat diet, glucose tolerance, insulin and GLP-1 secretion, and insulin sensitivity in both liver and muscle of TGR5-overexpressing mice improved, but not in TGR5-null (Tgr5–/–) mice. TGR5-overexpressing mice have enhanced GLP-1 secretion and insulin release in response to a glucose load, and TGR5-null mice have impaired glucose tolerance (Thomas et al., 2009). Therefore, the data from TGR5-overexpressing and TGR5-null mice collectively suggest that TGR5’s role in glucose metabolism is bile acid dependent (Pols et al., 2011).
Lessons Learned from Experiments on the Activation and Inactivation of Certain Transcription Factors
Several receptors present in the liver and intestine act as ligand-activated transcription factors, the ligands being various endogenous and exogenous substances. These ligand-activated transcription factor receptors help enhance the transport and biotransformation of chemicals. Examples include aryl hydrocarbon receptor (AhR), constitutive androstane receptor (CAR), pregnane X receptor (PXR), peroxisome proliferator activated receptor (PPAR), and nuclear factor erythroid 2-related factor 2 (NRF2). Table 13 illustrates the effect of activating these receptors by a corresponding ligand and/or the knockout of the receptor on bile acid homeostasis. The data include bile acid concentrations and mRNA expression of various enzymes, transporters, and transcription factors from 18 experiments with the data expressed as treated/nontreated or the knockout (null)/wild-type mouse.
Effect of agonists (ag) and knockout (null) of receptors on bile acid concentrations and gene expression in mice in comparison with control wild-type (WT) mice. Arylhydrocarbon Receptor (AhR) data are from Csanaky et al. (2018), Constitutive Androsterone Receptor (CAR) data are from Lickteig et al. (2016, 2019), Pregnane X Receptor (PXR) data are from Csanaky et al. (unpublished data), Peroxisome Proliferator Activated Receptor alpha (PPARα) data are from Zhang et al. (2017, 2018), and Nrf2 data are from Zhang et al. (2020) and Liu et al. (2021). Data are presented as percent of control. Values not statistically different from controls are noted as 100%.
The Value of Serum Bile Acid Concentrations
The serum bile acid concentrations have often been thought to reflect the concentration of bile acids in the liver. However, as shown in Table 13 this is not always true. While there was a similar percent change in bile acid concentrations in the serum as in the liver for a few of the experimental protocols, this was not true for most of the experiments. For example, the bile acid concentrations in the liver of the male and female CAR-null mice and the CAR agonist treated mice decreased 35%–50%, but the concentration in serum did not change. An even more extreme example is the female mice administered the AhR agonist that resulted in a decrease in the concentrations of bile acids in the liver but an increase in the serum. It is not surprising that the bile acid concentration in the serum does not reflect the concentration in the liver as the concentration in the serum is dependent on their absorption from the intestine and the first-pass extraction by the bile acid transporters in the liver. In addition, less than 0.05% of the bile acids in the body is in the serum.
Maintenance of Bile Acid Concentrations in the Liver
The maintenance of bile acid concentrations in the liver is thought to be important to provide sufficient bile acids for the intestinal absorption of lipids and lipid-soluble vitamins, and yet not so high that they will produce liver injury. Bile acid concentrations in the liver result from the balance of uptake of bile acids from the portal circulation and synthesis of new bile acids in the liver, and that excreted into the bile. Under the protocols of these 18 experiments, surprisingly 16 protocols resulted in a decrease in bile acid concentrations in the liver (Table 13). Thus, bile acid concentrations in the liver are not tightly regulated as usually thought.
Biliary Excretion of Bile Acids
Biliary excretion of bile acids is essential for their transfer to the intestine. However, most experimentalists do not cannulate the bile duct and thus there is minimal data available on biliary excretion. As noted in Table 13, the biliary excretion of bile acids was not altered by 10 of the 18 experimental protocols. Biliary excretion of the bile acids remained constant or even increased when the bile acid concentrations in the liver decreased about 50%. This might suggest that the primary factor the body is attempting to regulate is to maintain the correct concentration of bile acids in the gut. This can be achieved by regulating bile acid secretion.
The Bile Acid/FXR/FGF15/CYP7a1 Paradigm after Activation or Knockout of Various Transcription Factors
As noted previously, the homeostasis of bile acids in mice is thought to involve the following steps in sequence: (I) bile acid-mediated activation of the intestinal FXR, (II) increases in FGF15 expression, (III) secretion of bile acids and FGF15 into the portal blood, (IV) interaction of FGF15 with the FGFR4/β-KL in the liver, and (V) decreased expression of CYP7A1, resulting in decreased synthesis of bile acids.
Bile acid-mediated expression of FXR not only decreases CYP7A1 expression in the liver, but also (I) decreases NTCP expression in the liver (hence reduces uptake of bile acids into the liver) and (II) increases BSEP expression in the liver (hence increases efflux of bile acids out of the liver). The combined effects of such differential expression of these transporters in the liver should decrease the concentration of bile acids in the liver, resulting in prevention/reduction of bile acid-induced hepatotoxicity.
In the intestine, in addition to the bile acid-FXR induction of FGF15, bile acids (I) decrease the expression of ASBT (hence reduced uptake of bile acids into the enterocytes) and (II) increase the expression OSTα/OSTβ (hence increased efflux of bile acids out of the enterocytes into the blood). The combined effects of such differential expression of these transporters in the intestine is a decrease in the amount of bile acids in the enterocytes and prevention/reduction of bile acid-induced enterocyte damage.
Of the 18 groups of mice shown in Table 13, 10 groups had altered expression of FXR in the intestine. Five of the groups had a decrease in FGF15 expression (AhR agonist in female mice, PXR agonist in female mice, PPARα agonist in male mice, PPARα null in female mice, and NRF2 null in male and female mice). According to the intestinal FGF15-CYP7A1 paradigm, one would expect corresponding increases in CYP7A1 in these same groups of mice. However, only in two of these five groups was there an increase in CYP7A1. In six groups of mice, an increase in FGF15 expression was observed in the intestine (AhR agonist in male mice, PXR null in male and female mice, PXR agonist in male mice, PPARα null in female mice, and NRF2 agonist in female mice). Again, according to the FGF-CYP7A1 paradigm, one would anticipate that the CYP7A1 would decrease in these mice; however, only in three of the six groups was there a decrease in CYPA1. Thus, in many of these groups of mice, there is a poor inverse expression pattern between FGF15 and CYP7A1.
The intestinal FXR-FGF15 pathway not only is thought to have a downregulating effect on CYP7A1 expression and a corresponding decrease in bile acid synthesis, but also a downregulating effect on NTCP expression to decrease the uptake of bile acids into the liver. However, of the 18 groups of mice, only three groups showed an inverse expression pattern between FGF15 and NTCP. It is also thought that the FXR-FGF15 pathway increases BSEP to enhance the transport of bile acids into bile. However, only four groups showed a direct association between the expression of FGF15 and BSEP whereas two groups showed an inverse association. Thus, overall, there is a poor association between FGF15 and the expression of bile acid transporters in the liver.
Bile acids in the enterocytes not only activate FXR to increase the expression of FGF15, but also decrease ASBT expression to reduce the uptake of bile acids into enterocytes and increase the expression of OSTα/OSTβ to increase the efflux of bile acids out of the enterocytes into the portal circulation. The opposite expression of these two transport systems is thought to help decrease the concentration of bile acids in the enterocytes and protect them from bile acid toxicity. However, the data in Table 13 indicate that the observed expression of both ASBT and OSTα/OSTβ do not correlate, inversely and directly respectively, with the expression of FGF15 in most groups of mice to regulate bile acid concentrations in the enterocytes and hepatocytes.
Lack of Association between Intestinal FXR Expression and Bile Acid Concentrations in the Liver
Of the 18 protocols, only six exhibited an inverse association between the FXR mRNA in the intestine and BA concentrations in the liver.
Lack of Association between the Expression of CYP7A1, CYP7B1, and CYP8B1
As noted previously, CYP7A1 is the enzyme that hydroxylates the C-7 position in the classic route of bile acid synthesis, whereas CYP7B1 does the same reaction for the alternative route of bile acid synthesis. CYP8B1 is the 12-hydroxylase that is responsible for the formation of cholic acid. There was no association between CYP7A1 and CYP7B1 expression as seven of the protocols increased and three decreased CYP7A1, whereas only two resulted in an increase of CYP8B1 and 12 decreased it. CYP8B1 was the most stably expressed of these three enzymes as only four protocols increased it and three decreased CYP8B1 (Table 13).
Lessons Learned from Other Experimental Protocols
Experiments were conducted to assess bile acid homeostasis in mice under various physiologic and environmental conditions: fed 2% resin; bile-duct ligation (BDL); 40% caloric restriction; CYP7A1-transgenic (TG); 6 PM/6 AM circadian rhythm, CYP450-reductase-null; germ-free, fed 0.10% CA or CDCA, gender (female/male), or fed an HMG-CoA reductase inhibitor (Table 14).
Effect of various experimental protocols on bile acid concentrations and gene expression in mice in comparison with control mice. Data for the mice fed 2% resin in their diet is from Zhang and Klaassen (2010) and doctorate dissertation of Peizhen Song (2010), data for the 12hr bile duct ligation is from Zhang et al. (2012), data for 40% diet restriction is from Fu and Klaassen (2013), data for the Cyp7a-Transgenic (TG) mice is from Li et al. (2011), data for the circadian rhythm experiments at 6pm/6am sampling are from Zhang et al. (2011), data for the Cyp450-reductase null mice is from Cheng et al. (2014), data for the germ-free mice is from Selwyn et al. (2015), data for the mice fed 0.1% cholic (CA) or chenodeoxycholic (CDCA) are from Song et al. (2011, 2015) and Zhang and Klaassen (2010), data from female (F) and male (M) mice are from Lickteig et al. (2019), and the data for the HMG-CoA reductase inhibitor is from Fu et al. (2014). Data are presented as percent of control. Values not statistically different from controls are noted as 100%.
Changes in Serum and Liver Bile Acid Concentrations
A poor association was observed between changes in the concentrations of bile acids in the serum and liver after various protocols (Table 14). At 12 hours of bile duct ligation (BDL), the bile acid concentration in the liver increased 18-fold and in serum 576-fold. The bile acid resin decreased liver bile acid concentrations 85%, but serum concentrations remained constant. Germ-free mice have twice the bile acid concentration in the liver and a fourfold higher concentration in the serum.
The Bile Acid/FXR/FGF15/CYP7A1 Paradigm
The bile acid/FXR/FGF15/CYP7A1 paradigm exhibited a poor inverse association between FGF15 and CYP7A1, seen after the alteration of various receptors described in the previous section (Fig. 13); however, a clear inverse association was observed in some of these experimental models (Fig. 14). For example, mice fed the 2% resin diet and the CYP450-reductase-null mice both had more than an 85% reduction in FGF15 mRNA in the ileum and a 3- to 4-fold increase in CYP7A1 mRNA in the liver. Most markedly, the HMG-CoA reductase inhibitor decreased FGF15 by 86% in the intestine and increased CYP7A1 11-fold in the liver. A 2- to 4-fold increase in FGF15 in the intestine in CYP7A1-TG mice and in mice fed 0.10% CA decreased CYP7A1 by more than 95% in the liver. However, there was no inverse association between intestinal FGF15 and CYP7A1 in the liver in the caloric-restriction experiments, the circadian rhythm experiments, and female/male mice.
An increase in FGF15 is thought not only to decrease CYP7A1 but also increase BSEP and decrease NTCP expressions in the liver, collectively resulting in less bile acid accumulation in hepatocytes. But this association of expressions was seldom observed in these various experimental models. Likewise, a decrease in FGF15 in the intestine is thought to decrease ASBT and increase OSTα and OSTβ expressions in the intestine, collectively resulting in less bile acid accumulation in the enterocytes, but this too was seldom observed in the various experimental models.
The Bile Acid-FXR-FGF15-CYP7A1 Paradigm in CYP2C70 Null-Mice
As noted earlier, CYP2C70-null mice were engineered to produce mice that have a pattern of bile acids similar to that in humans. These “humanized” mice have 2.4- to 4.8-fold higher concentrations of bile acids in their livers than do wild-type mice. The mice engineered by Honda et al. (2020) have 8%–25% lower FGF-15 in the intestine without an increase in CYP7A1 in the liver, rather a 70%–90% decrease in CYP7A1. The CYP2C70 mice engineered by Straniero et al. (2020) have a 68% increase in FGF15 in the intestine and a 60% decrease in CYP7A1 in the liver. Most remarkable in both engineered mice, there was a marked decrease in CYP8B1, which is essential in the synthesis of CA (Table 15).
Bile acid concentrations and gene expression of humanized CYP2C70-null mice. The table has been created based on the data from Honda et al. (2020) for the first two columns and from de Boer et al. (2020) for the third column. Data are presented as percent of control.
The above discussion underscores the complexity and the paradoxical nature of FXR activation and the regulation of FXR target genes. In other words, FXR upregulation or downregulation may regulate FXR-target genes that may not be consistent with expectation.
Dysregulation of Bile Acids and Liver Diseases
Some chronic liver diseases associated with the dysregulation of bile acids are (1) primary sclerosing cholangitis (PSC), (2) primary biliary cholangitis (PBC), previously known as primary biliary cirrhosis, (3) nonalcoholic fatty liver (NAFL), and (4) nonalcoholic steatohepatitis (NASH). Both NAFL and NASH are nonalcoholic fatty liver disease (NAFLD). NAFL is a condition in which fat builds up in the liver; whereas in NASH, there is inflammation and liver damage along with fat in the liver (https://www.niddk.nih.gov/health-information/liver-disease/nafld-nash/definition-facts). Since the first demonstration in 1972 that UDCA (3α,7β) could dissolve gallstones, UDCA has been used to treat gallstone diseases and other hepatobiliary dysfunctions (Makino and Tanaka, 1998). However, the results in humans are mixed. Table 16 lists some of the clinical trials reported in the public domain and some associated publications.
Clinical studies conducted to address the four types of liver diseases associated with the dysregulation of bile acids (PBC, PSC, NAFL, NASH)
PSC
PSC is a chronic male-predominant inflammatory and cholestatic condition that advances very slowly. In PSC. inflammation damages intrahepatic and/or extrahepatic bile ducts, making them hard, narrow, and eventually blocked. Impairment of bile duct function reduces bile flow, resulting in bile accumulation in the liver and causing liver damage. Many patients with PSC end up requiring a liver transplant, typically about 10 years after being diagnosed with the disease. Most cases of PSC occur in association with inflammatory bowel disease. There is no effective medical therapy for halting the disease progression (Rabiee and Silveira, 2021). UDCA treatment can improve liver histology (confirmed by liver biopsy) and serum ALT/ASP levels in patients with PSC, but data supporting any long-term efficacy or long-term survival are lacking (Stiehl, 1994).
Moreover, high dose UDCA increased the medical complications and mortality in patients with PSC (Stiehl, 1994; Meadows et al., 2020). Patients with PSC have decreased expression of hepatic bile acid receptors—FXR and TGR5. Interestingly, de Boer et al. (2021) reported that female CYP2C70-null mice develop considerable pathologic features with age, and treatment with UDCA reverses the liver pathology in these mice.
PBC
PBC is a chronic female-predominant inflammatory and cholestatic liver disease resulting from progressive destruction of the intrahepatic (smaller) bile ducts. With the impairment of bile duct function, bile builds up in the liver, causing liver damage. In up to 73% of patients, PBC can coexist with other autoimmune diseases. Sjogren’s syndrome is the most common condition associated with PBC; both are characterized by chronic autoimmune inflammation (Mago and Wu, 2020).
Patients are benefiting from pharmacological agonists of FXR and peroxisome proliferator-activated receptors. UDCA was the first FDA-approved therapy for patients with PBC exhibiting altered serum liver enzyme levels. The recommended adult dosage is 13–15 mg/kg/d administered in two to four divided doses with food (https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/020675s017lbl.pdf). Obeticholic acid (6α-ethyl-CDCA) is a synthetic derivative of CDCA and is a high affinity ligand and agonist for FXR (Bowlus, 2016). It has been suggested that obeticholic acid treatment can be delivered in conjunction with UDCA in PBC or as a monotherapy in patients who do not tolerate UDCA (Hirschfield et al., 2015). Obeticholic acid is about 100 times more potent as an FXR ligand than CDCA and does not interact with TGR5. It activates FXR in the ileum, resulting in the downregulation of ASBT, which decreases bile acid reabsorption from the distal ileum. It also increases the expression of FGF19, which decreases bile acid synthesis in liver via CYP7A1 (Bowlus, 2016). Obeticholic acid was approved by the FDA for treatment of PBC in 2016 (https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/207999s003lbl.pdf). However, in May 2021, the FDA issued a warning that obeticholic acid should not be used in patients with PBC with advanced cirrhosis because it can cause serious harm (https://www.fda.gov/safety/medical-product-safety-information/ocaliva-obeticholic- acid-intercept-pharmaceuticals-drug-safety-communication-due-risk-serious- liver).
Although PSC and PBC have primarily autoimmune etiologies resulting in cholestasis and progressive biliary ductal destruction due to damage of biliary epithelial cells, they are well-defined individual disease states with distinct features and specific diagnostic criteria based on clinical symptoms, serologic, immunologic, and histologic findings. In very rare cases, there may be overlapping features (Mago and Wu, 2020).
NAFL and NASH
Both NAFL and NASH are different types of NAFLD. NAFL is a condition in which fat builds up in the liver, the occurrence being the highest in populations with obesity and type II diabetes (Vernon et al., 2011). NASH is a type of NAFLD and is characterized by inflammation and liver cell damage. NAFL often progresses to NASH, then to advanced fibrosis and hepatocellular carcinoma (Vernon et al., 2011). Thus, patients with NASH also have fatty liver. NASH affects about 3%–6% of the US population. Patients with NAFLD show a moderate elevation of total bile acid levels in the serum and an altered bile acid composition. Moreover, compared with healthy controls, patients with NAFLD have increased CYP7B1 mRNA levels and decreased CYP8B1 mRNA levels in the liver, and elevated serum levels of the conjugated bile acids. It is thought that the protective effects of bile acids against NAFLD progression is mediated through FXR activation (Gottlieb and Canbay, 2019). Clinically, most patients with NAFLD are asymptomatic, have mild to moderate elevations of serum aminotransferase levels, clinical hepatomegaly, and features of fatty liver on imaging. Liver biopsy is the only accepted method for diagnosis. Currently, there are no specific approved therapies. Lifestyle modification, including weight loss, is the mainstay of treatment. Approximately 20% of patients with NASH develop cirrhosis, a potentially life-threatening condition (Vernon et al., 2011; Sheka et al., 2020). Several clinical trials have been conducted in which obeticholic acid and an FGF-19 analog have been used separately (Table 16).
Conclusion and Future Prospect
Although the pathophysiology of bile acid metabolism in hamsters appears to mimic that of humans (van Golen et al., 2018), much has been learned about the biology of bile acids from studies in mice. It is generally thought that we know all the enzymes for the synthesis of bile acids, as well as the transporters for establishing their enterohepatic circulation, that is, the uptake and efflux of bile acids into and out of the liver and intestine. The discovery of FXR as a bile acid receptor, and especially the overall pathway of the bile acid-FXR-FGF15-CYP7A1 pathway in regulating bile acid homeostasis was considered as the culmination of knowledge to understand bile acid synthesis and homeostasis. In experimental protocols where there are major increases or decreases in the body burden of bile acids, the intestinal bile acid-FXR-FGF15-CYP7A1 explained most of the obtained data and aided our understanding of bile acid homeostasis. However, the data described in this review using over 30 animal models (Tables 13, 14, 15) indicate that there are many situations when there is not an inverse expression of FGF15 in the intestine and CYP7A1 in the liver. This suggests there must be additional pathways that regulate CYP7A1. Not much knowledge has been sought on what regulates bile acid concentrations in the liver, but the present review indicates that many experimental protocols decrease bile acid concentrations in the liver, indicating that bile acid concentrations in the liver are not tightly regulated. Unfortunately, the concentration of bile acids in the blood is not a biomarker for the concentration in the liver. In contrast, biliary excretion, which is seldom quantified by experimentalists, appears to be more consistent between the various experimental protocols. This might be because the primary purpose of bile acids is to aid in the absorption of lipids and lipid soluble vitamins. The question remains, what other factors are important in the regulation of bile acids in the various compartments of the body, and how does the structure of the various bile acids play into the regulation. Thus, while much has been learned about bile acid homeostasis, much appears yet to be learned.
Acknowledgments
The authors would like to thank Drs. Paul Dawson, John Chiang, and Youcai Zhang for critically reading the manuscript and providing constructive comments.
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Choudhuri, Klaassen.
Footnotes
- Received August 19, 2021.
- Accepted October 20, 2021.
This study received no external funding.
The opinions expressed in this article are the authors’ own and do not necessarily reflect that of the U.S. Food and Drug Administration, U.S. Department of Health and Human Services, or the Federal Government. The authors have no conflict of interest.
↵1 Current affiliation: Retired.
ABBREVIATIONS
- ABC
- ATP-binding cassette
- Ah
- R, aryl hydrocarbon receptor
- ASBT
- apical sodium-dependent bile acid transporter
- BAAT
- bile acid-CoA‒amino acid N-acyltransferase
- BACO-SCPA
- bile acid-coenzyme A thioester
- BACS
- bile acid-CoA synthase
- BRIC2
- benign recurrent intrahepatic cholestasis type 2
- BSEP
- bile salt export pump
- CA
- cholic acid
- CAR
- constitutive androstane receptor
- CDCA
- chenodeoxycholic acid
- CMC
- critical micellar concentration
- CTX
- cerebrotendinous xanthomatosis
- CYP
- cytochrome P450
- DCA
- deoxycholic acid
- –FGF
- fibroblast growth factor
- FGFR4
- fibroblast growth factor receptor 4
- FXR
- farnesoid-X-receptor
- GLP-1
- glucagon-like peptide-1
- HCA
- hyocholic acid
- HDCA
- hyodeoxycholic acid
- IBABP
- ileal bile acid-binding protein
- β-KL
- β-klotho;
- LCA
- lithocholic acid
- LXR
- liver-X-receptor
- α-MCA
- α-muricholic acid
- β-MCA
- β-muricholic acid
- ω-MCA
- ω-muricholic acid
- MDR
- multidrug resistance
- MRP2
- multidrug resistance-associated protein
- NAFL
- nonalcoholic fatty liver
- NAFLD
- nonalcoholic fatty liver disease
- NASH
- nonalcoholic steatohepatitis
- NRF2
- nuclear factor erythroid 2-related factor 2
- NTCP
- sodium (Na+)-taurocholate cotransporting polypeptide
- OATP
- organic anion transporting polypeptides
- OH
- Hydroxyl group
- OST
- heterodimeric organic solute transporter
- PBC
- primary biliary cholangitis
- PE
- phosphatidylethanolamine
- PFIC2
- progressive familial intrahepatic cholestasis type 2
- P-gp
- p-glycoprotein
- PPAR
- peroxisome proliferator activated receptors
- PS
- phosphatidylserine
- PSC
- primary sclerosing cholangitis
- PXR
- pregnane X receptor
- SHP
- small heterodimer partner
- SULTS
- sulfotransferases
- T-BA
- taurine-conjugated bile acid
- TCA
- taurine-conjugated cholic acid
- TGR5
- Takeda G protein-coupled receptor 5
- UCA
- ursocholic acid
- UDCA
- ursodeoxycholic acid
- U.S. Government work not protected by U.S. copyright.





{kind=link}
{kind=link}
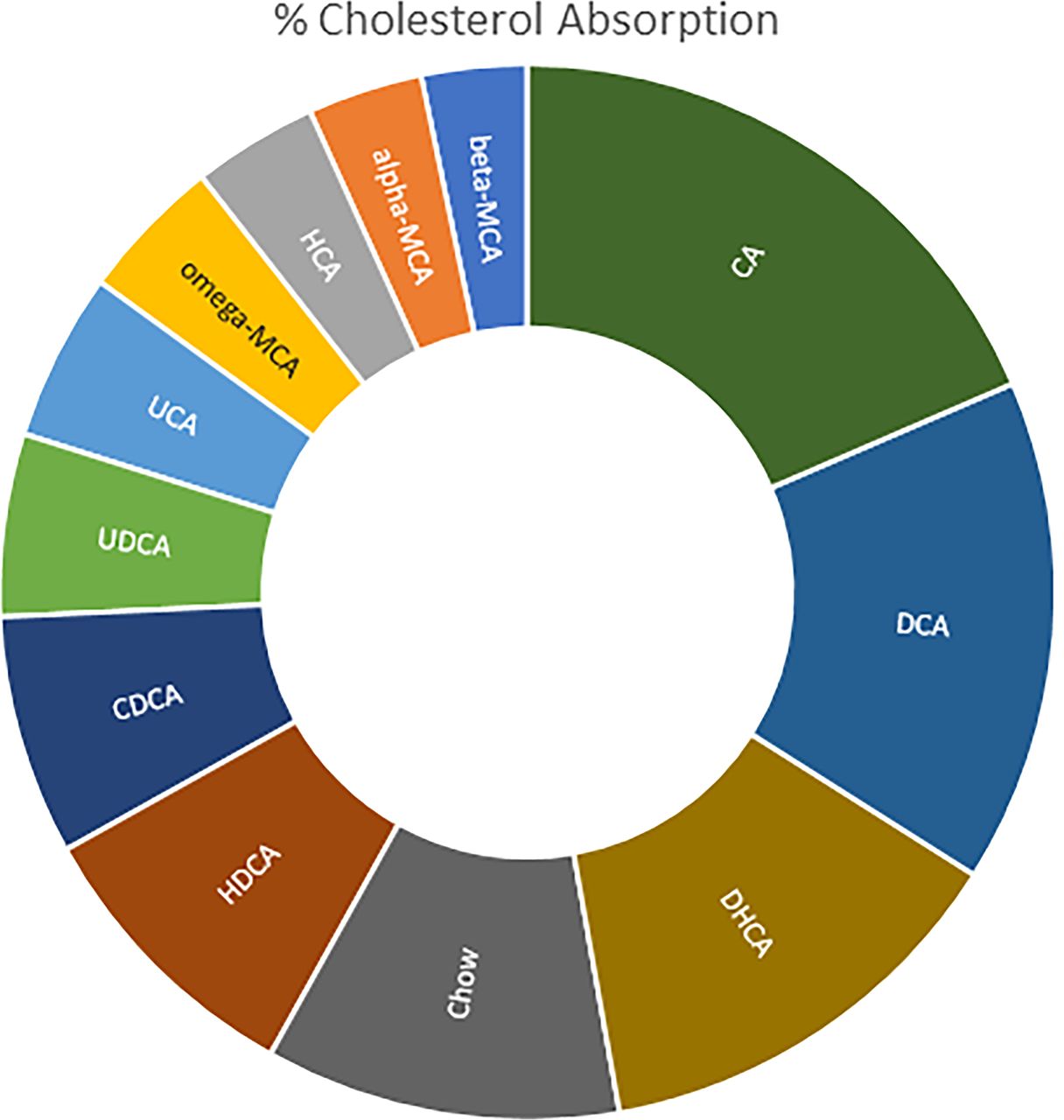{kind=link}
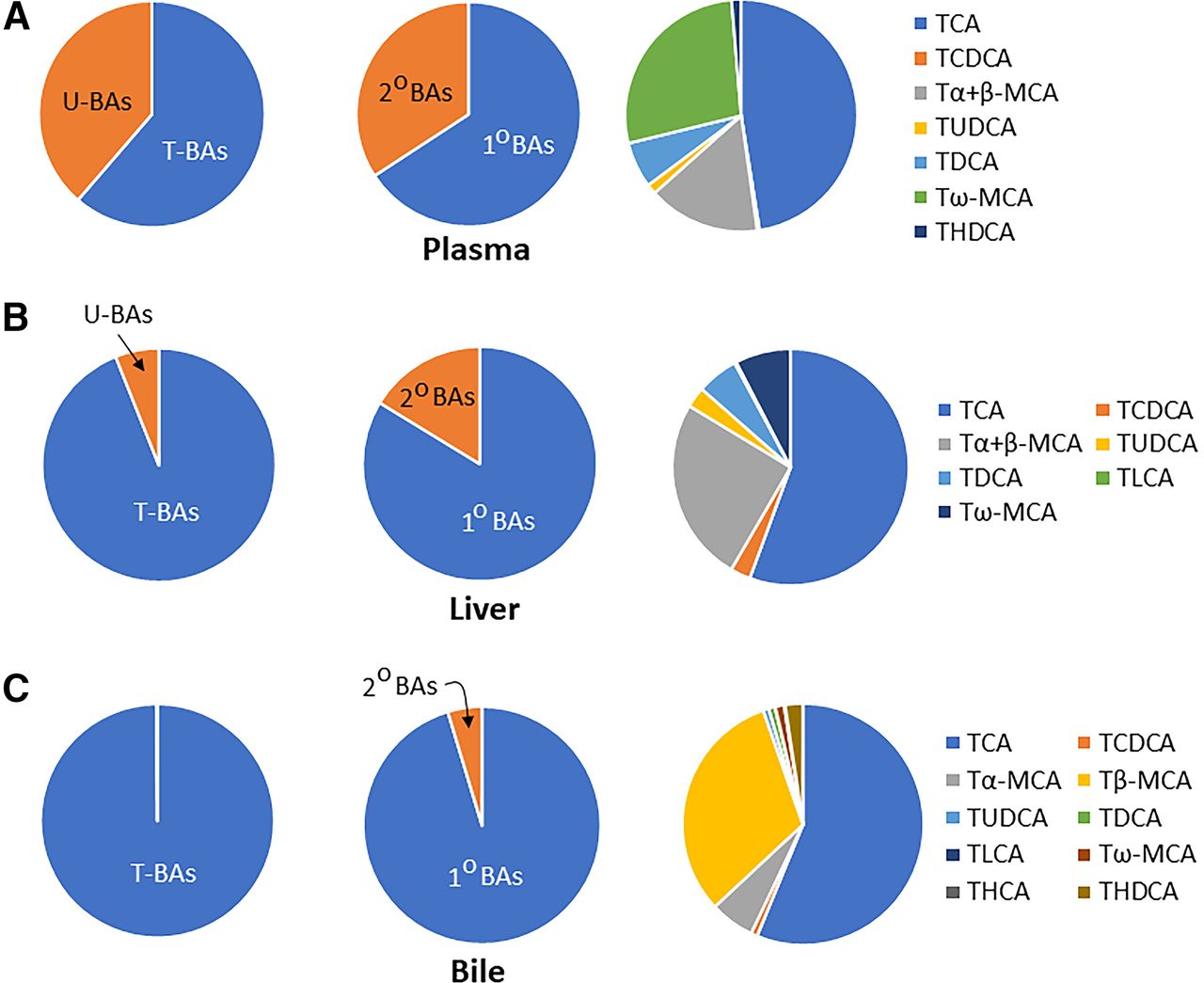{kind=link}
{kind=link}
{kind=link}
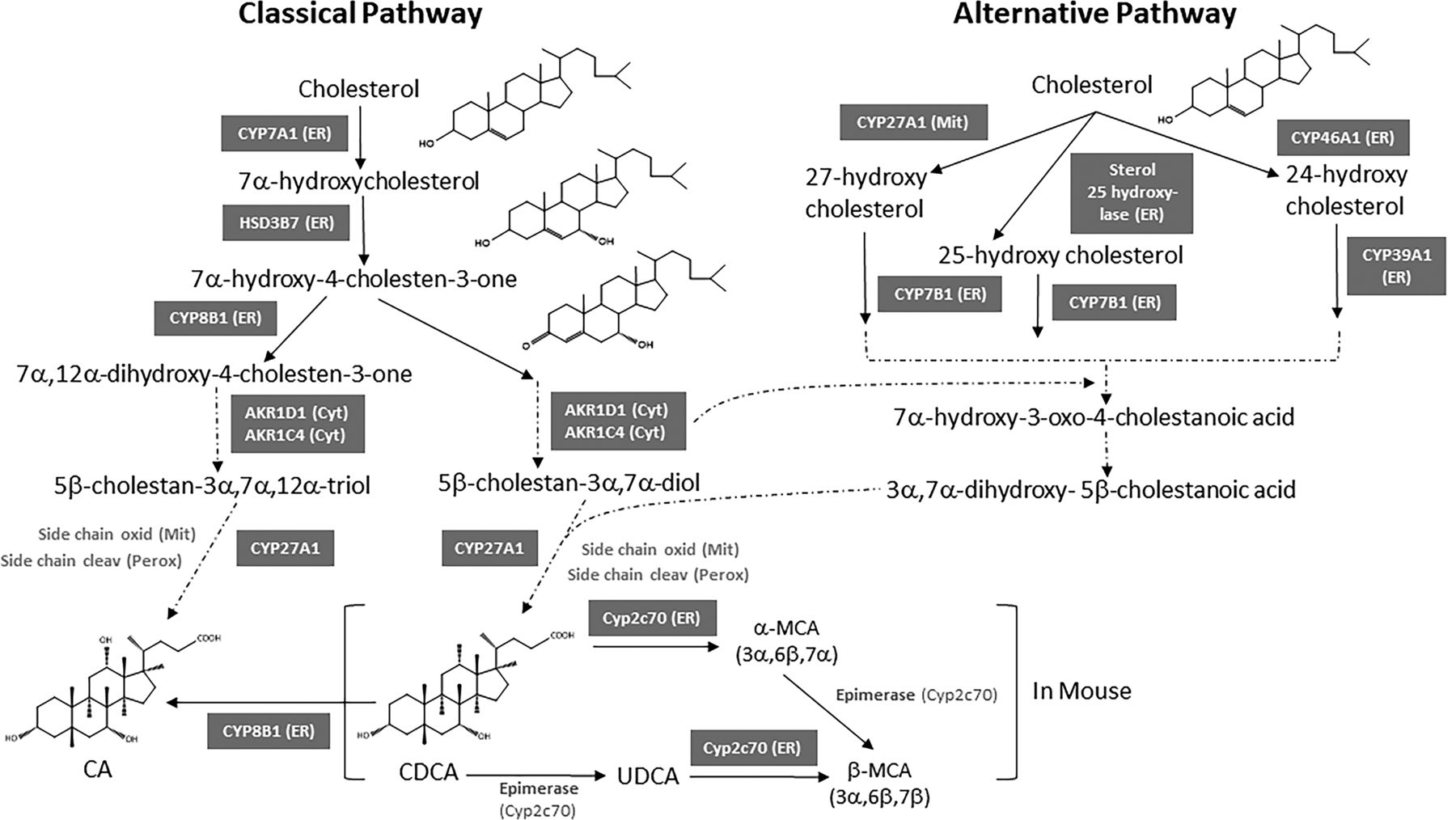{kind=link}
{kind=link}
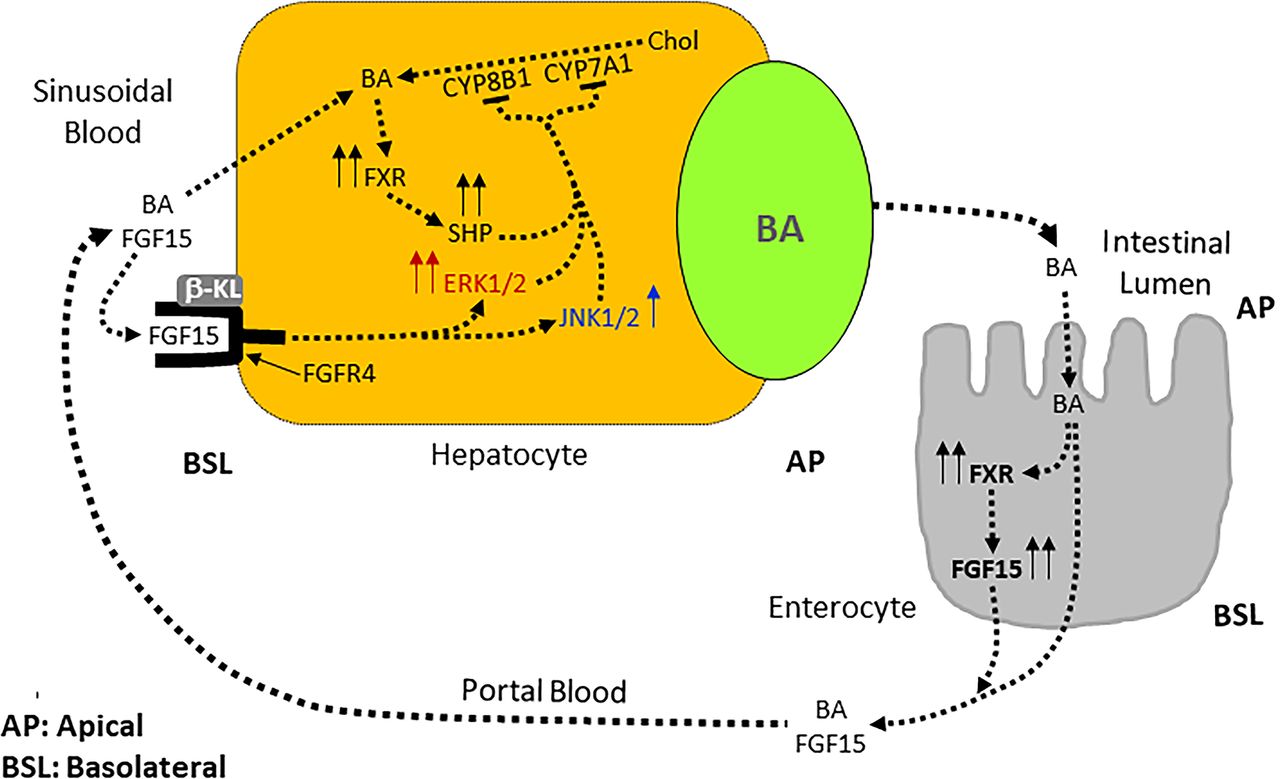{kind=link}
{kind=link}
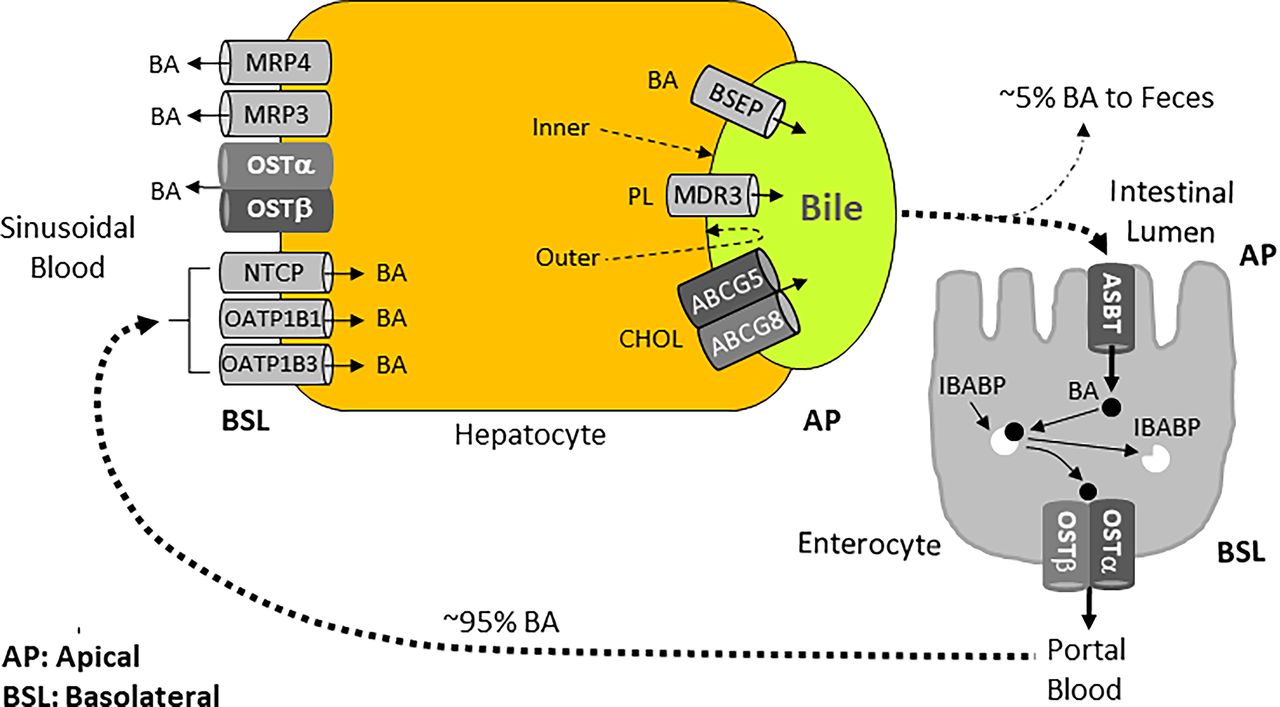{kind=link}
{kind=link}