


Abstract
Characterization of the pharmacokinetics and biodistribution of therapeutic proteins (TPs) is a hot topic within the pharmaceutical industry, particularly with an ever-increasing catalog of novel modality TPs. Here, we review the current practices, and provide a summary of extensive cross-company discussions as well as a survey completed by International Consortium for Innovation and Quality members on this theme. A wide variety of in vitro, in vivo and in silico techniques are currently used to assess pharmacokinetics and biodistribution of TPs, and we discuss the relevance of these from an industry perspective, focusing on pharmacokinetic/pharmacodynamic understanding at the preclinical stage of development, and translation to human. We consider that the ‘traditional in vivo biodistribution study’ is becoming insufficient as a standalone tool, and thorough characterization of the interaction of the TP with its target(s), target biology, and off-target interactions at a microscopic scale are key to understand the overall biodistribution on a full-body scale. Our summary of the current challenges and our recommendations to address these issues could provide insight into the implementation of best practices in this area of drug development, and continued cross-company collaboration will be of tremendous value.
SIGNIFICANCE STATEMENT The Innovation and Quality Consortium Translational and ADME Sciences Leadership Group working group for the absorption, distribution, metabolism, and excretion of therapeutic proteins evaluates the current practices and challenges in characterizing the pharmacokinetics and biodistribution of therapeutic proteins during drug development, and proposes recommendations to address these issues. Incorporating the in vitro, in vivo and in silico approaches discussed herein may provide a pragmatic framework to increase early understanding of pharmacokinetic/pharmacodynamic relationships, and aid translational modeling for first-in-human dose predictions.
Introduction
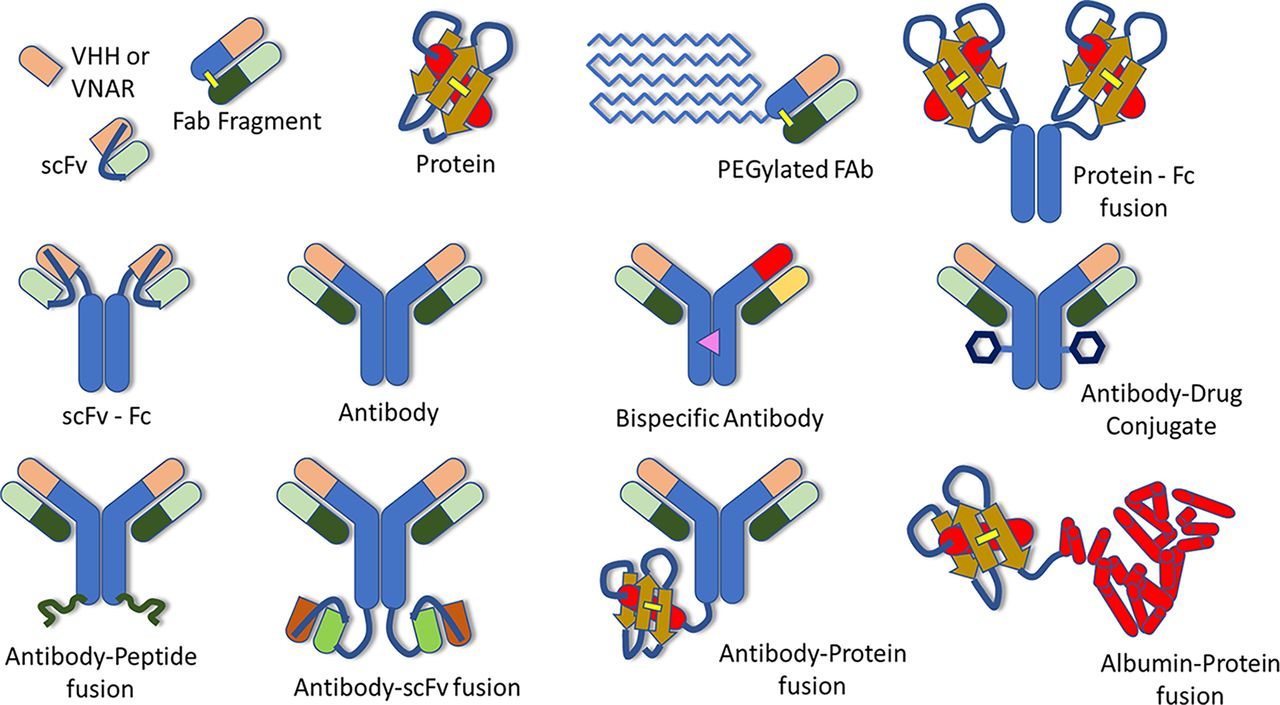
Understanding the absorption, distribution, metabolism, and excretion (ADME) of therapeutic proteins (TPs) is an important step in their development as therapeutic entities. In 2019, the Translational and ADME Sciences Leadership Group (TALG) within the International Consortium for Innovation and Quality (IQ consortium) launched a cross-company working group to identify the current practices for characterizing ADME properties of TPs within the pharmaceutical industry. To accomplish this, an industry-wide survey was conducted within IQ member companies, and the working group also collected data submitted to the US Food and Drug Administration by reviewing regulatory biologics license application (BLA) submission packages of TPs approved by the US Food and Drug Administration between 2011 and 2020. Results of the survey and an evaluation of ADME data submitted within BLAs for TPs within this time period are summarized in a separate paper, along with future perspectives and recommendations for conducting ADME studies for internal decision making and regulatory submissions (Bolleddula et al., 2022). Another paper from this working group focuses on the biotransformation of therapeutic proteins along with its impact on pharmacokinetics (PK), pharmacodynamics (PD) and immunogenicity (Walles et al., 2022). The objectives of our paper are to summarize the current practices within the pharmaceutical industry for the characterization of the PK and biodistribution of TPs, as well as the associated challenges and limitations, and to provide our recommendations and future perspectives. We will cover a broad range of TPs, focusing on modalities currently being developed or considered as potential future drug candidates by the companies contributing to this paper, although inevitably, the list will not be exhaustive. This will include IgG-like molecules, such as monoclonal antibodies (mAbs) and crystallizable fragment (Fc)-containing polyspecific molecules, as well as antibody fragments, proteins, and protein-fusion or peptide-fusion molecules (Fig. 1). Therapeutic modalities that are considered out of scope for this paper are small molecules, nanoparticles and microspheres, gene and cell therapies, peptides, and antibody drug conjugates, since many of these have been addressed in other IQ TALG working groups.
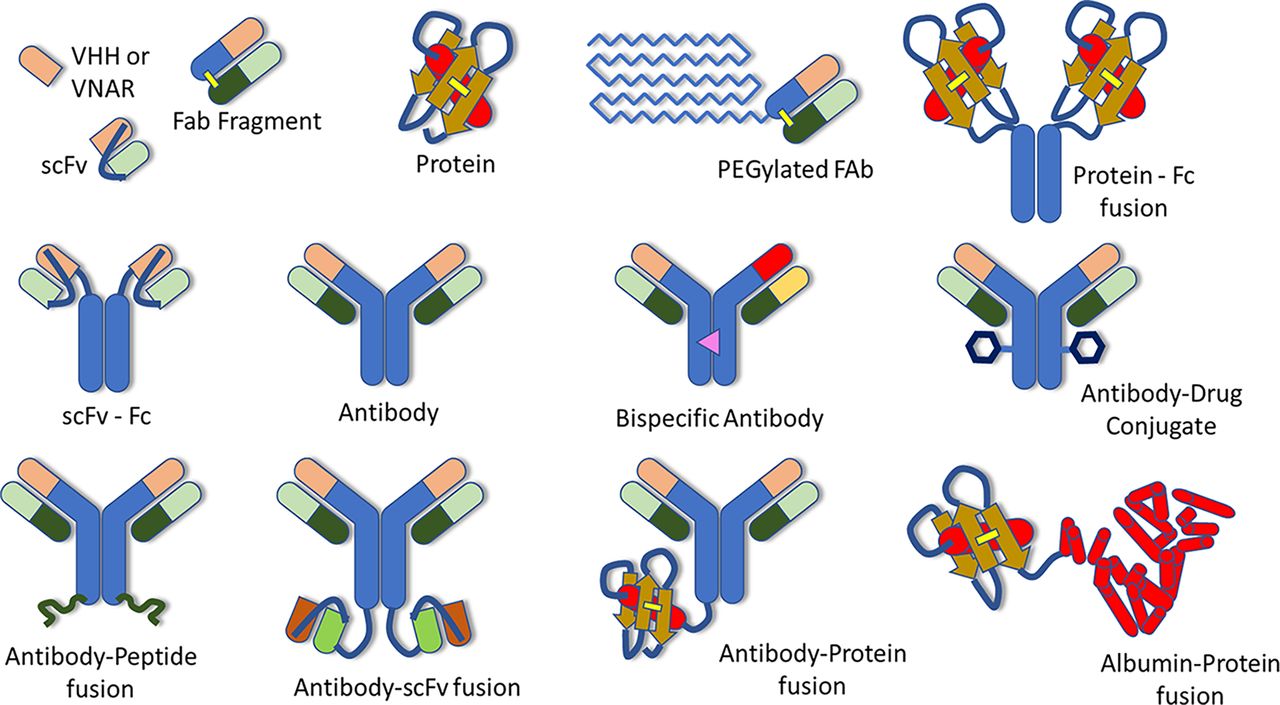
Examples of therapeutic proteins. VHH – camelid antibody; VNAR – variable new antigen receptors; PEG – polyethylene glycol; scFv – single chain variable fragment; Fab – antigen binding fragment; Fc – crystallizable fragment. Reproduced with permission from Bolleddula et al. (2022) and Walles et al. (2022).
While much is already known about the ADME of mAbs, the increasing variety of non-mAb TPs under development has yielded molecules possessing a wide range of PK and biodistribution characteristics. Smaller-sized TPs generally penetrate more efficiently into tissues, but at the expense of more rapid elimination and shorter duration in the systemic circulation. The latter aspect has led to the development of TPs which have been engineered to improve their circulating half-life. Common modifications include increasing the molecular weight and hydrodynamic radius and adding or modulating the interaction with the neonatal Fc receptor (FcRn) via amino acid engineering or via fusion to protein- or peptide-based molecules, such as the Fc domain of IgG, human serum albumin, unstructured hydrophilic, biodegradable protein polymers (Podust et al., 2016), or to different types of peptides (Strohl, 2015). Conjugation to synthetic polymers, such as polyethylene glycol is also a well-known approach to modulate systemic half-life (Veronese and Pasut, 2005). Other modifications to the native structure of TPs, such as glycoengineering or point mutations of amino acids to modulate effector function can also have an impact on the PK and biodistribution properties of their native TPs (Saunders, 2019).
Since our paper is written from an industry point of view, its main focus is on the understanding of PK and biodistribution characteristics essential for early go/no-go decisions within a project during preclinical development of TPs, including drug design, lead selection and dose projection for first-in-human studies. Measurement and modeling of clinical PK and biodistribution data are out of scope, although we emphasize that learning and confirming via feedback from clinical data are an important part of model-informed drug development. The first part of this paper will briefly describe the key factors affecting the PK and biodistribution of TPs, and the prior best practices and guidance available for how these processes can be characterized. The second part will discuss the relevant in vitro and in vivo studies and modeling approaches that are vital to understand PK and biodistribution of TPs during drug development, as well as their PK/PD relationships for efficacy and toxicity. We place particular emphasis on TPs that may present challenges to the standard paradigms, i.e., behave differently from mAb-like molecules with soluble targets, and may thus require more ‘bespoke’ approaches. The third part of this paper will describe modeling approaches for preclinical to clinical translation of systemic PK and biodistribution data for TPs to provide predictions of dose-exposure-response relationships in humans. Finally, we discuss the key challenges and current limitations of the above aspects, and provide our recommendations for how these challenges may be addressed within an industry setting. A summary of challenges and recommendations as discussed throughout this paper can be found in Table 1.
Current challenges and limitations for the characterization of biodistribution of therapeutic protein, and recommendations and perspectives from the absorption, distribution, metabolism, and excretion of therapeutic proteins Innovation and Quality consortium Translational and ADME Sciences Leadership Group working group
Part 1: PK and Biodistribution of Therapeutic Proteins
Key Factors Affecting PK and Biodistribution of TPs
The concentration and kinetics of drug exposure at the site of action define the pharmacological activity of therapeutic molecules (Rizk et al., 2017). The mechanisms governing the PK and biodistribution of TPs depend on the combination of their intrinsic physicochemical and structural attributes and their kinetic interactions with the host’s physiology. Structural properties of TPs include molecular weight, hydrodynamic radius, charge, glycosylation pattern, hydrophobicity, and overall systemic stability (self-aggregation, non-specific interactions, biotransformation, and catabolism). Kinetic interactions with on-target or off-target antigens, FcRn, and with members of the scavenger receptor family expressed on liver resident macrophages (Kupffer cells), hepatocytes, and sinusoidal endothelial cells of liver also play an important role in biodistribution of TPs. Passive movement of molecules from the bloodstream to tissues is determined by blood flow, lymphatic drainage, vascular leakiness, tissue permeability, membrane surface area, and concentration and pressure gradients. Generally, TPs below the renal clearance molecular weight cut-off can extravasate and penetrate tissues faster, but are also more likely to be eliminated via glomerular filtration, which significantly shortens their systemic exposure and half-life. Elimination of larger TPs occurs mainly via intracellular catabolism following uptake via non-specific cellular pinocytosis. Upon internalization and trafficking, they interact with FcRn receptors in early endosomes, where the acidic microenvironment (pH∼6) promotes IgG and albumin-FcRn ligation. FcRn-bound molecules are then recycled to the cell membrane, where the neutral pH results in their release into the bloodstream or interstitial space. This ingenious process prevents lysosomal degradation and is the main factor conferring the long half-life to these molecules (Roopenian and Akilesh, 2007; Sockolosky and Szoka, 2015). In preclinical models, it has been shown that FcRn-containing endothelial and hematopoietic cells are the primary sites for the maintenance of IgG concentrations in vivo (Montoyo et al., 2009). Antigen binding and immune complex formation leads to interactions with members of the Fc-gamma receptor family, which contribute to the elimination of Fc-containing immune complexes or aggregates (Hepburn et al., 2006). The attributes of the target, i.e., soluble or membrane-bound, physiologic location, valency, density, and internalization, recycling, and turnover rates, may influence the target-mediated drug disposition characteristics.
Understanding the specific molecular attributes and processes responsible for desirable PK and ADME properties of TPs, combined with advances in protein-engineering, has provided a rational experimental path for the improved design, optimization, and selection of successful lead candidate drugs (Lagasse et al., 2017). For example, half-life extension of small-sized TPs can be achieved via polyethylene glycol conjugation (Swierczewska et al., 2015), albumin binding (Hoefman et al., 2015), or by recombinant engineering as part of a longer unstructured polypeptide chain (Wunder et al., 2003; Podust et al., 2013). Fc-fusion is also an effective approach to increase systemic exposure of TPs with otherwise undesirable PK properties, as demonstrated for etanercept (Duivelshof et al., 2021). Fc-engineering is another means of modulating the half-life of IgG-like molecules. An Fc-modified anti-respiratory syncytial virus mAb with amino acid substitutions M252Y/S254T/T256E (YTE) to increase binding to FcRn (Oganesyan et al., 2014) was shown to extend the half-life up to 100 days in humans (Robbie et al., 2013).
Measurement of PK and Biodistribution of TPs
Typically, the systemic concentration of TPs is determined in blood, plasma, or serum sampled from the relevant animal species, quantified using ligand-binding assays, such as ELISA, electrochemiluminescence (Woodbury et al., 2019; Eangoor, 2020), or mass spectrometry assays, such as liquid chromatography–tandem mass spectrometry or liquid chromatography–high-resolution mass spectrometry (Chang et al., 2021; Khaowroongrueng et al., 2021). For TPs with targets in non-systemic locations, while systemic exposure can sometimes be considered a surrogate for exposure at the target site, it may be essential to characterize the biodistribution of the TP to the site of action and/or other tissues or fluids to aid translational PK/PD modeling or proof of mechanism of action. Biodistribution of TPs can be characterized by collecting tissue samples and measuring concentration of TP via bioanalytical methods, such as ELISA, liquid chromatography–tandem mass spectrometry, liquid chromatography–high-resolution mass spectrometry, matrix-assisted laser desorption ionization, or radiometric analysis in tissue lysates via quantitative whole body autoradiography. Alternatively, in vivo biodistribution may be measured in the intact animal via radiolabeling or fluorescence conjugation and then measured by positron emission tomography, immunofluorescence, or other imaging techniques (Williams, 2012). More recently, large-pore microdialysis or open flow microperfusion have also emerged as minimally invasive techniques to measure the time course of tissue or tumor interstitial fluid concentrations of TPs (Hummer et al., 2021). It is important to thoroughly understand the advantages and limitations of each analytical technique, e.g., is residual blood contamination an issue? What is the spatial resolution of the measurement? Is the intact TP measured or are catabolites, antigen-TP complexes or anti-drug antibody-TP immune complexes included? Does the imaging probe confer non-native biodistribution properties to the original TP? Furthermore, when multiple mechanisms are involved in the PK and biodistribution of a TP, in vivo biodistribution studies may only give an overall view of the net effect of the various processes. Thus, combining biodistribution data with additional in vitro analyses or within a mechanistic modeling framework could elucidate the relative contributions of each mechanism to the overall PK and biodistribution (see Parts 2 and 3, and Table 1).
Prior Guidance and Best Practices for Characterizing PK and Biodistribution of TPs within the Pharmaceutical Industry
Although a review of regulatory guidance and submissions for PK and biodistribution of TPs is out of scope here, it is noteworthy that our survey of IQ member companies found that 77% of respondents analyze tissue biodistribution of TPs in preclinical species, whereas a review of BLAs submitted from 2011 to 2020 found that only 22% contained preclinical biodistribution data (Bolleddula et al., 2022). In the absence of specific regulatory guidance, it is valuable to evaluate scientific publications from industry, which can provide a view of the pragmatic approaches carried out by pharmaceutical companies. However, there are relatively few cross-industry white papers or consensus articles on this topic. A white paper on the ADME characterization of antibody drug conjugates was published by another IQ TALG working group (Kraynov et al., 2016), as well as some review articles in a special issue of AAPS Journal “ADME of therapeutic proteins” (Prueksaritanont and Tang, 2012; Vugmeyster et al., 2012; Xu and Vugmeyster, 2012). The remaining available ‘industry’ literature are mainly publications by individual pharmaceutical or biotechnology companies. Many of these focus on describing the mechanisms behind biodistribution and potential ways to improve the design of TPs to encourage favorable biodistribution and PK properties (Tabrizi et al., 2010; Datta-Mannan, 2019). Other industry publications have broadened the focus to include reviews of the various experimental and mathematical modeling approaches available to characterize the biodistribution of TPs, albeit mainly focusing on mAbs and Fc-containing molecules (Lee, 2013; Tibbitts et al., 2016; Conner et al., 2020). We consider that experimental and modeling approaches should be combined as a crucial element of the drug development strategy for TPs from late discovery up to the clinic (Marshall et al., 2019; Wang et al., 2019; Elmeliegy and Ghobrial, 2021). In addition to reducing cost, these approaches will also align with the ‘3R’ principles ( replacement, reduction, and refinement), which are fundamental to the ethical use of animals in drug development (Fenwick et al., 2009). In the remainder of this white paper, we will summarize the current and emerging industry practices on the experimental and translational modeling approaches to characterize the biodistribution of TPs, with particular focus on novel modalities, and cite noteworthy examples from industry.
Part 2: Characterizing and Understanding Preclinical Biodistribution and PK/PD Relationships for Efficacy and Toxicity
Several considerations underpin the preclinical development of TPs, including efficacy, toxicology, and ADME properties, as well as various other developability aspects. During early discovery and development, in vitro and in vivo models are often used to screen multiple drug candidates, and in vitro-in vivo correlations (IVIVCs) may be generated to aid in designing TPs with desirable PK and biodistribution properties, such as systemic half-life, or specific biodistribution into one or more tissue types. Although IVIVCs for clearance of TPs are less well established than for small molecules, some progress has been described enabling internal decision making on candidate molecules (Avery et al., 2018). However, significant gaps remain due to the limited number of molecules analyzed in such studies, and the development of large-scale correlations for PK and/or biodistribution of TPs would likely require wider collaboration across industry and academia.
In Vitro Methods to Assess Molecular Properties
The molecular size, hydrophobicity, surface charge cluster location, and nonspecific or off-target interactions of a TP with the physiologic environment can influence its PK and biodistribution characteristics, and can vary both between and within different classes of TPs. The presence of certain native structural components can give an indication of hydrophobicity and charge characteristics, e.g., the Fc domain of a given IgG subtype is often similar, although alteration through Fc-engineering of amino acids or posttranslational modifications, such as glycosylation can confer differences in physicochemical properties which ultimately affect PK and biodistribution (Zhou and Qiu, 2019). For antibody-derived TPs, the variable domain(s) can have very different hydrophobicity and charge properties determined by the amino acid sequence diversity within these domains. Variable region charge, charge patchiness and hydrophobicity influence the rate of pinocytosis due to electrostatic interactions with negatively charged components of the glycocalyx of cells. Furthermore, changes in variable region charge have also been reported to alter FcRn-mediated recycling via changes in the interaction with the FcRn receptor (Schoch et al., 2015). Within industry, these intrinsic properties are typically evaluated in a variety of early-stage developability assays, including specific or nonspecific binding to cell-membrane proteins, FcRn interaction assessments, self- and cross-interactions, and polyspecificity (Jain et al., 2017).
Quantitative Structure-Pharmacokinetic Relationships (QSPKR)
QSPKR has been described for the characterization and optimization of small molecules (Xu and Mager, 2011), with the aim to link molecular descriptors to PK properties via traditional regression methods, nonlinear statistical techniques, and machine learning. Although challenging due to the multifactorial mechanisms underlying the PK of TPs, such in silico approaches will become more important with increasing capabilities to correlate structure and properties of large molecules to in vivo data. They could also help to reduce the number of animal PK studies for PK characterization of TPs, a critical aspect since non-human primates (NHPs) are often used for this purpose, and target expression and (non-)responder species have to be accounted for. Recently, in silico methods were investigated as a way to predict the PK of therapeutic antibodies based on their sequences, and it was shown that a combination of in vitro and in silico descriptors can enrich for antibodies with desirable PK properties (Grinshpun et al., 2021). A recent example of QSPKR for non-mAb TPs demonstrated the influence of size and charge of unstructured polypeptides within targeted fusion proteins on their PK and biodistribution in a preclinical tumor xenograft mouse model (Brandl et al., 2019). Another recent example presented an in silico tool for the prediction of half-life for peptides conjugated to serum albumin (Hijazi, 2021). These types of QSPKR could be used to streamline the design of synthetic or biologically-based fusion proteins developed using ‘plug-and-play’ approaches, and potentially replace some of the target-independent in vitro studies currently used to screen for PK properties in cases in which in vitro to in vivo translation is inherently challenging. Given the ability of QSPKR to predict in vivo plasma clearance based on, for example, molecular size, the plasma exposure of TPs may be predicted across a range of doses and potentially scaled across species via allometry (Li et al., 2017). Furthermore, when combined with biodistribution coefficients for TPs of the relevant size (Li et al., 2016), the tissue exposure may also be predicted to better anticipate target engagement or toxicity.
Binding Affinity and Internalization via Target Antigen(s)
In addition to driving pharmacological activity, binding of the TP to target antigen(s) can also influence its biodistribution. The nature of the antigen, whether soluble or membrane associated/internalizing is key for the fate of the TP (Tabrizi et al., 2006; Tibbitts et al., 2016). In vitro assays are often carried out within the industry to measure the equilibrium rate constant for binding (KD) and the kinetics of binding: the association rate (ka) and dissociation rate (kd). These can be measured in a variety of cell-free in vitro systems, including surface plasmon resonance (SPR) and biolayer interferometry (BLI) assessing the binding of ligands to immobilized targets. To understand and quantify the activity of increasingly complex molecules (bispecifics, TP-conjugates, etc.) and to account for the multiple factors involved in the disposition of TPs, other assays such as internalization assays using flow cytometry or imaging techniques, such as confocal microscopy (Vainshtein et al., 2015), cell binding assays to investigate effects of avidity or different target densities in case of bispecifics (Register et al., 2021) and patient-derived cells or tissues (Bondza et al., 2017), are becoming established in vitro or ex vivo systems within industry. Emerging technologies may also enable the direct monitoring of antibody–target binding dynamics in living systems and tumors, e.g., bioluminescence resonance energy transfer (BRET) imaging system (Tang and Cao, 2020). Although sometimes used as ‘standalone’ information within a drug project, binding affinity and internalization data can also be implemented in quantitative PK/PD models (see Part 3).
Binding Affinity and Kinetics to ‘Off-Target’ Molecules
This is sometimes called ‘secondary pharmacology’ and has recently been reviewed in detail with emphasis on drug discovery within the industry (Jenkinson et al., 2020). Binding to ‘off-target’ receptors, such as Fc-gamma receptors and scavenger receptors, can trigger potentially undesirable side effects and may also influence the PK and biodistribution of TPs. Binding of the TP to other ‘off-target’ receptors can trigger receptor-mediated endocytosis, which can be exploited for organ-targeting via receptors such as the transferrin receptor (Daniels et al., 2012) or insulin receptor (Xiao and Gan, 2013). ‘Off-target’ cross-reactivity can be assessed via binding of the TP to a wide range of receptors and cell-surface proteins expressed in an array, and quantified by immunohistochemical methodologies (Dostalek et al., 2017). More detailed data on binding affinity and uptake kinetics can be assessed in cell-based assays using cells expressing endogenous receptors, or in receptor-knockout cell systems. Measurements made using cell-based assays allow the assessment of the net effect of the physicochemical properties of the TP and its binding kinetics to the components present in the in vitro system.
For Fc-containing TPs, recycling via FcRn plays an important role in prolonged systemic exposure (Qi and Cao, 2021). FcRn binding properties of five human IgG1 mAbs, determined by biolayer interferometry with the immobilized FcRn receptor, were found to reasonably correlate with their human half-lives (Souders et al., 2015). These studies further showed that even modifications distant to Fc, e.g., in the complementarity-determining region, could impact FcRn binding and thereby PK properties. Several cell-based functional assays have been developed in the past years to rank Fc mutant mAbs with respect to their expected pharmacokinetic properties (Jaramillo et al., 2017). For correlation to in vivo PK, transgenic FcRn mice are typically used as they have been reported to give a better prediction of non-target-mediated clearance in NHPs and humans for mAbs versus wild-type rodents (Avery et al., 2016).
Following cellular uptake by non-specific fluid-phase pinocytosis or receptor-mediated endocytosis, endosomal trafficking and sorting leads to either recycling, transcytosis or lysosomal degradation of the TP (Gurbaxani et al., 2013). The kinetic parameters of some of these processes can be estimated via in silico models based on in vitro and/or in vivo data, and can allow molecule ranking and potentially predictions for future molecules with respect to PK and biodistribution properties, particularly if a good IVIVC has been established for a particular modality or scaffold. However, as TP scaffolds and formats of increasing complexity are developed that deviate from typical mAbs, existing IVIVC need to be re-established, albeit with limited in vivo data, since animal PK studies are often only performed for promising drug candidates within industry.
In Vivo Methods to Assess the Impact of Molecular Properties on PK and Biodistribution
Two key approaches are often employed when studying the PK and biodistribution of TPs in vivo: one is to characterize the underlying, non-target-mediated PK and disposition, while the second, subsequent approach is to assess target-mediated effects. The first approach allows the evaluation of the relationship between physicochemical and in vitro properties of the TP with PK and biodistribution independent of the target, assessing the effects of aspects, such as FcRn affinity, charge, size, route of administration, etc. Evaluation of systemic PK, along with tissue concentrations, commonly performed with radio- or fluorescently-labeled TPs, can be achieved, for example, in species in which target is not present. Dose dependency is not necessarily investigated in these cases, as clearance is often dose-proportional. This approach can be used to compare modalities, e.g., mAbs versus scFv, by assessing differences in tissue penetration or residency (Li et al., 2016). The second approach evaluates the impact of target-mediated drug disposition (TMDD), essentially repeating the above experiment(s) in target-expressing, cross-reactive species, e.g., human transgenic mouse or NHP. By exploring different dose levels, the magnitude of TMDD can be assessed, along with the exposure in various physiologic locations. Comparing in vivo biodistribution data with in vitro binding affinity data may be essential for more complex TP modalities. For example, the relative binding affinity of each arm of an anti-human epidermal growth factor receptor 2/cluster of differentiation 3 bispecific antibody was shown to highly affect both tumor and tissue distribution in a transgenic mouse model (Mandikian et al., 2018). Furthermore, in vivo studies may elucidate the major influencing moiety on the biodistribution of TP-conjugates or fusion proteins; for example, it was shown for an anti-N-(5'-phosphoribosyl)anthranilate isomerase-interleukin-2 antibody-cytokine fusion protein that the interleukin-2 moiety entirely governed its biodistribution in a syngeneic mouse model, rather than the antibody (Tzeng et al., 2015). When considering biodistribution data or modeling for translational purposes, it is important to employ the appropriate labeling and measurement approaches, as discussed in Part 1, and to recognize the assumptions involved: namely, that physiologic distribution of TPs in animals is translatable across species, and that target expression in transgenic animals or NHPs is similar to that in humans. If these assumptions do not hold, it may be necessary to generate additional experimental target biology data in each relevant species to perform scaling appropriately.
Since our IQ survey results showed that 50% of responding companies analyzed tissue biodistribution of TPs in humans (Bolleddula et al., 2022), it will be important to harness this data to assess the predictivity of preclinical biodistribution data and/or translational models. Target protein expression and turnover measurements have been used to develop more robust quantitative models of PK and biodistribution processes in humans to guide the design of improved TPs against the same target during early development phases (Farrokhi et al., 2018). By incorporating a variety of in vitro and in vivo data into quantitative in silico models, the effect of adjusting certain drug-specific properties (e.g., molecular size, target binding affinity), or the effect of physiologic variability (e.g., target expression and turnover) on the predicted plasma or tissue exposure can be explored. The various types of quantitative models used to accomplish this, as well as their associated approaches for translation to humans, are further discussed in Part 3.
Overall, it is clear that a wide range of in silico, in vitro and in vivo approaches are currently used within the pharmaceutical industry to characterize the PK and biodistribution of TPs (Fig. 2, and Table 1), and this list is ever-increasing with the emergence of new modality TPs. ‘Standalone’ in vitro and in vivo data are no longer sufficient to understand the complex interplay behind the various mechanisms behind PK and biodistribution; IVIVC, QSPKR, and quantitative, predictive models which integrate multiple parameters are increasingly becoming the norm within drug development of TPs.

Key in vitro and in vivo data to characterize the pharmacokinetic and biodistribution of therapeutic proteins during drug development.
Part 3: Modeling Approaches for Translating PK, Biodistribution, and PK/PD to the Clinical Setting
While predicting the active dose range in humans is a regulatory requirement prior to first-in-human studies, it is also desirable to predict dose-exposure-response relationships in humans to ensure that a clinical study is feasible and has a reasonable probability of providing therapeutic benefit to patients while limiting toxicity. In addition to providing evidence of drug exposure at the target site, preclinical biodistribution data could also help to anticipate in which physiologic locations toxicities may be observed in patients. Encouragingly, our survey results showed that 82% of responding companies used data from biodistribution studies in translational PK/PD modeling for efficacy and safety predictions (Bolleddula et al., 2022). Since biodistribution of TPs cannot be separated from target biology, a ‘totality of evidence’ approach must be employed during the translation of preclinical biodistribution data to humans, combining data from in vitro assays, in vivo studies, and in silico approaches. Nevertheless, the complexity of the quantitative model(s) used for this purpose depends greatly on factors such as the TP structure, mechanism of action, target antigen(s), and human patient characteristics.
Translation of Models Built Using Systemic PK/PD Data
Compartmental PK models assuming first-order distribution and clearance rate processes may be appropriate when target receptor occupancy is high, i.e., high concentrations of TP relative to target abundance. However, for TPs being dosed below the target saturation range, a TMDD model may be required (Mager and Jusko, 2001). Translational TMDD models can be built based on systemic PK measurements and/or in vitro data for the target-mediated component of the model. A good understanding of target biology is particularly relevant in cases in which the target is expressed to a different extent or in different tissues in the preclinical species compared with humans. Usually, the nonspecific distribution and clearance parameters are scaled using allometry, often from NHP studies, if the mechanisms behind these processes are expected to be similar between animals and humans (Li et al., 2019). Allometry for predicting linear clearance processes (Betts et al., 2018) using transgenic FcRn mice could alternatively be used at an earlier stage. It is worth noting that the net systemic clearance of some TPs may be confounded by additional biotransformation processes, such as deconjugation for TP-conjugates (Mahmood, 2021), so the particularities of each TP modality need to be considered when using allometry. Interspecies translation of target-related model parameters in TMDD models may be carried out in different ways. Experimental data measured in human-derived in vitro or ex vivo systems can be used directly as input parameters for target abundance and kinetics, as well as binding affinity and complex degradation or internalization rate, with sufficient confidence in the IVIVC of these parameters. Alternatively, a quantitative analysis exploring translational rules for TMDD models with target-related parameters estimated using in vivo monkey data for mAbs could be used as a guide (Singh et al., 2015). This approach assumes a high sequence homology of the target, similar target expression, and absence of other substantial differences in PK between monkeys and humans, which is not always the case, particularly in human disease.
TMDD models predicting systemic PK and/or receptor occupancy are more appropriate for some TPs than others. For example, for TPs with blood-based targets, a good relationship between systemic PK and pharmacological activity is expected. In practice, TMDD models are also often used as a pragmatic solution within drug development, even when biodistribution processes may lead to a different time course of TP exposure at the target tissue, e.g., solid tumors, poorly perfused tissues, or those with limited extravasation. An alternative would be to integrate TMDD into site-of-action models to identify the limiting factors for distribution and receptor occupancy at the target site (Tiwari et al., 2016). TMDD models may also inform how the interplay between nonspecific and target-mediated clearance, as well as target abundance, can influence the PK of a TP. As target abundance increases, TPs with low nonspecific clearance are increasingly rapidly eliminated via target-mediated clearance (Fig. 3A), whereas if nonspecific clearance is high, this route of elimination will take precedence over target-mediated clearance of unbound TP, and binding of the TP to the target will actually result in a greater retention of TP (as TP-target complex) in the systemic circulation (Fig. 3B). An example of a small-sized TP whose distribution and elimination are strongly influenced by its target is the first approved nanobody, caplacizumab. Free caplacizumab is rapidly cleared by renal elimination, but target-bound caplacizumab remains longer in the circulation (Sargentini-Maier et al., 2019), and its slower elimination is likely via catabolism in the liver, as demonstrated by a mouse biodistribution study reported in the BLA (Ulrichts et al., 2011). Although empirical TMDD models can be pragmatic for predictions of human PK, they do not provide information about the predicted exposure of the TP in other tissues, which may be of interest for PK/PD relationships in the clinical setting. Additional complexity is encountered when more than one target antigen is present, such as for bispecific molecules (Rhoden et al., 2016). In such cases, more mechanistic models may be required (see Table 1).

Simulations showing the effect of changing target abundance on the time course of total therapeutic protein (TP) concentrations in the systemic circulation (where total TP = free TP + TP bound to target), for (A) an ‘IgG-like’ TP with a slow elimination rate (0.046 day−1); (B) a ‘small-sized’ TP with a fast elimination rate (4.6 day−1). Target abundance was varied between 0.001 nM and 10 nM, and binding affinity of TP to target was fixed to 0.1 nM. Arrows indicate the direction of change in the concentrations of TP as target abundance increases. The simulations were performed in Berkeley Madonna version 8.3.18 using the two-compartment full target-mediated drug disposition model structure and parameter values from (Dua et al., 2015), unless otherwise stated. See Dua et al., 2015 for model equations and code.
Translational Quantitative Systems Pharmacology (QSP) and Physiologically Based Pharmacokinetic (PBPK)/PD Models
Aside from anticipating the influence of PK on efficacy or toxicity, predicting PK/PD in human is particularly important when the PD of the target also has an influence on the PK of the TP. These complex exposure-response relationships for novel modality TPs may be captured in translational QSP models, as was recently demonstrated for PF-06671008, a P-Cadherin/cluster of differentiation 3 dual-affinity retargeting antibody molecule containing a human IgG1 Fc domain to extend its half-life (Betts et al., 2019). The model incorporated both biodistribution and T cell kinetics, including T cell proliferation and contraction, and was used to translate preclinical efficacy data to the clinic. In another example, translational PK/PD modeling of the upregulation of the interleukin-2 receptor by the pharmacological action of the novel therapeutic antibody-cytokine conjugate cergutuzumab amunaleukin, and thus the subsequent impact on the target-mediated PK of this TP, was used to guide dose selection and scheduling of assessments in the first-in-human study (Grimm et al., 2016).
PBPK models haven been developed for mAbs, ranging in complexity from ‘minimal PBPK’ to full-body models, with some success in predicting human PK and biodistribution (Glassman and Balthasar, 2019). A key factor in the use of PBPK models for mAbs and newer TP modalities is the inclusion of FcRn, and thus the TP-FcRn binding affinity is a key parameter for TPs with modified FcRn binding (Qi and Cao, 2021). The extension of PBPK models to non-mAb TPs, such as antibody fragments, has been explored within academia (Li et al., 2021), and if it proves promising, industry may increasingly employ this approach to aid drug design. One current challenge with PBPK models for TPs is obtaining the drug-specific data that are used as input parameters within the model, for which standardized experimental conditions and IVIVC do not yet exist, in contrast to the well-established in vitro methodologies and scaling strategies for small molecules. Nevertheless, some success has recently been reported using in vitro input data in PBPK models for mAbs (Jones et al., 2019). PBPK models may be most valuable when used together with pharmacodynamic modeling approaches to interrogate target engagement in tissues that may be less accessible to large molecules, or in diseased tissues (Vugmeyster et al., 2012). This may be challenging for newer modality TPs, but is beginning to be addressed, e.g., for polyvalent molecules, such as bispecifics (Gibbs et al., 2020), and dual-targeting fusion proteins (Nguyen et al., 2020). As discussed above, TP-target interactions can affect PK of TPs, so PBPK models must also take into account any complex target dynamics, such as target turnover and shedding (Li et al., 2014). A key challenge for PBPK/PD and QSP approaches is how to translate PD model parameters from preclinical species to humans. Using experimental in vitro or animal values either unmodified or scaled to humans using allometry or IVIVC, or using literature values for similar TPs and targets may be considered. These approaches for translating PD parameters may require robust experimental data and hypothesis testing in preclinical species, prior to human translation. Retrospective evaluation of the success of predictions made using translational PK/PD models for TPs, via clinical PK and PD data, will be required to evaluate their suitability during drug development, as was demonstrated for the anti-FcRn therapy rozanolixizumab (Li and Balthasar, 2019a; Li and Balthasar, 2019b). Continued cross-industry collaboration and sharing of experiences will be vital in providing confidence in these approaches.
Conclusions and Perspectives
Due to rapid developments in sampling and detection technologies that can be used in preclinical and clinical studies, combined with the need to better understand biodistribution of TPs, increased efforts can be observed in this area of drug development for TPs. With an increasing number of non-mAb-like new TP modalities, we can expect that more question-based, fit-for-purpose in vitro and in vivo studies will be conducted within industry. The emphasis will certainly be on incorporating various types of biodistribution data in translational PK/PD models for the opportunity to better understand the exposure-response relationships driving efficacy and safety.
A crucial aspect of drug development for TPs is the prediction of the optimal dose range in patients, based on the dose-exposure-response relationship. This is highly influenced by PK and biodistribution properties, which can be characterized by a combination of experimental studies along with modeling and simulation approaches. Modeling and simulation for TPs ranges from simple compartmental models that do not account for target binding kinetics, to more mechanistic models that may consider tissue distribution and kinetic interactions with the target and other endogenous receptors as key components in the disposition of the TP. Consequently, PBPK and QSP models are becoming more commonly used within the industry for TPs; however, the challenge remains of translating the in vitro and in vivo data used in these models to the clinical setting. An additional consideration is how to streamline experimental and modeling efforts, distinguishing ‘nice to have’ from ‘need to have’ information to enable key decision-making within drug development. Table 1 provides a summary of the major challenges and limitations that we have identified with the current paradigms for characterizing TP biodistribution, as well as our recommendations for addressing these issues.
The wide range of modalities encompassed in the term ‘therapeutic proteins’ also presents a challenge to the standardization of approaches. Currently, measuring and modeling systemic PK and target engagement can be sufficient for ‘simpler’ TP modalities such as mAbs, without the need for tissue biodistribution studies which may add little information for dose-related decision-making within industry. However, more complex TP modalities may require different approaches and rely more heavily on additional experimental data and mathematical modeling of the interconnected mechanistic pathways. At the design stage of drug development, developing QSPKR for particular TP types will be useful to anticipate what effect modifications to the native protein structure will have on the individual kinetic processes that govern in vivo PK and biodistribution. Since several design factors may simultaneously affect in vivo PK, multiple QSPKR may need to be incorporated within a PBPK model framework to predict their net effect on overall PK and biodistribution, rather than ‘standalone’ QSPKR models. QSPKR could also ultimately replace several of the in vitro-derived parameters of PBPK models, particularly where IVIVC is poor due to differences in the in vitro versus in vivo environments, or is dependent on individual companies’ assay conditions. Nevertheless, experimental data remains an important part of the drug development process for TPs, so we recommend that modeling and simulation can also be used to guide the design of in vitro and in vivo studies, for example by predicting the concentration-time course of the TP within tissues of interest to select the relevant drug concentration for in vitro assays, or drug dose for in vivo preclinical studies. Ultimately, it will be desirable to ‘work backward’ from human PK/PD models to fine-tune the relevant PK- and biodistribution-influencing properties of the TP, with the caveat that this approach requires availability of clinical data to establish predictivity. In the absence of clinical data, preclinical biodistribution data may be used to inform or verify quantitative models, but we recommend that the role of the ‘traditional’ in vivo biodistribution study is carefully considered before being carried out for TPs. Preferential localization or accumulation of the TP within a given tissue should not be confused with proof of target engagement, since TPs or their catabolites can be non-specifically bound or sequestered within tissues without binding the target and/or eliciting their pharmacological effect.
As novel TP structures emerge with a greater extent of engineering and modification compared with endogenous molecules, additional challenges will arise. The ever-expanding range of experimental systems, animal models, and bioanalytical techniques adapted for different TP types will necessitate a clear understanding of their advantages and limitations, to allow for correct interpretation of the data generated. Additionally, a key consideration when developing quantitative correlations and models for highly engineered TPs will be to account for any immunogenicity that develops against the TP in vivo, as this may affect the PK profile, as well as the efficacy and safety profile. As the technological development of new TP modalities continues to accelerate, characterization of the biodistribution of TPs will be a constantly moving target, thus a close collaboration between industry, academia, and regulatory bodies will be vital to keep pace with the field.
Acknowledgments
The authors would like to thank Brooke Rock, Kevin Brady, Rob Webster, Cong Wei, Bo Wen, Jeffrey Kurz, Anthony Lee, and Dian Su, for participating in helpful discussions. The authors would like to thank IQ TALG for support of this work.
Authorship Contribution
Wrote or contributed to the writing of the manuscript: Ball, Bruin, Escandón, Funk, Pereira, Yang, Yu.
Footnotes
- Received March 9, 2021.
- Accepted January 6, 2022.
This work received no external funding.
No author has an actual or perceived conflict of interest with the contents of this article.
Abbreviations
- ADME
- absorption, distribution, metabolism, and excretion
- Fc
- crystallizable fragment
- FcRn
- neonatal Fc receptor
- IQ
- Innovation and Quality consortium
- IVIVC
- in vitro–in vivo correlation
- mAb
- monoclonal antibody
- NHP
- non-human primate
- PBPK
- physiologically-based pharmacokinetic
- PD
- pharmacodynamic
- PK
- pharmacokinetic
- QSP
- quantitative systems pharmacology
- QSPKR
- quantitative structure PK relationship
- TALG
- Translational and ADME Sciences Leadership Group
- TMDD
- target-mediated drug disposition
- TP
- therapeutic protein
- Copyright © 2022 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}