


Visual Overview
Abstract
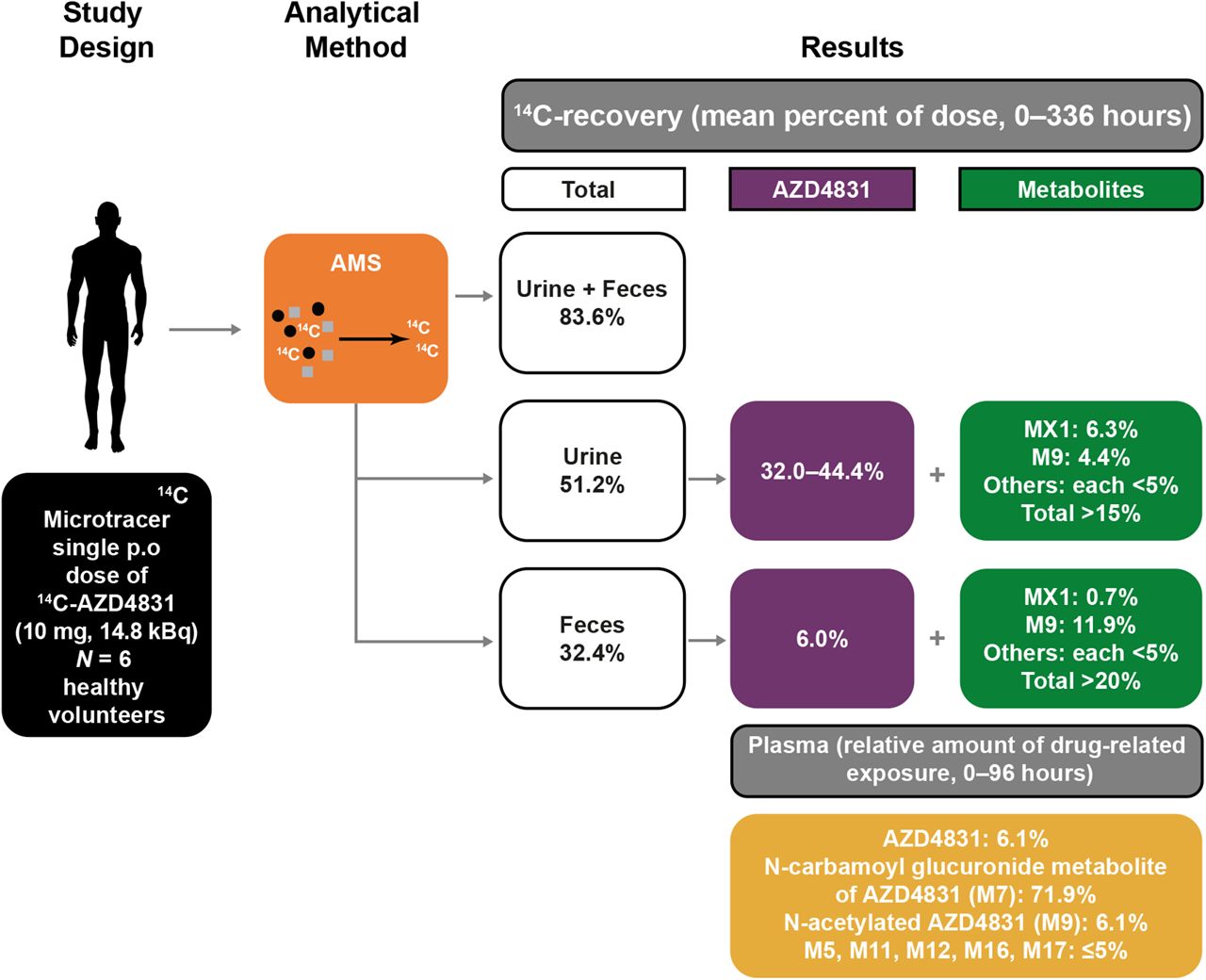
This study evaluated the mass balance and disposition of AZD4831, a novel myeloperoxidase inhibitor, in six healthy participants using a 14C-labeled microtracer coupled with analysis by accelerator mass spectrometry (AMS). A single oral dose of 10 mg 14C-AZD4831 (14.8 kBq) was administered as a solution, and 14C levels were quantified by AMS in blood, urine, and feces over 336 hours postdose. AZD4831 was rapidly absorbed, and AZD4831 plasma concentrations declined in a biphasic manner, with a long half-life of 52 hours. AZD4831 was eliminated via metabolism and renal excretion. An N-carbamoyl glucuronide metabolite of AZD4831 (M7), formed primarily via UGT1A1, was the predominant circulating metabolite. Presumably, M7 contributed to the long half-life of AZD4831 via biliary elimination and hydrolysis/enterohepatic recirculation of AZD4831. On average, ∼84% of administered 14C-AZD4831 was recovered by 336 hours postdose (urine, 51.2%; feces, 32.4%). Between 32%–44% of the dose was excreted as unchanged AZD4831 in urine, indicating renal elimination as the major excretory route. Only 9.7% of overall fecal recovery was recorded in the first 48 hours, with the remainder excreted over 48%–336 hours, suggesting that most fecal recovery was due to biliary elimination. Furthermore, only 6% of unchanged AZD4831 was recovered in feces. Overall, the fraction of the administered AZD4831 dose absorbed was high. 14C-AZD4831 was well tolerated. These findings contribute to increasing evidence that human absorption, distribution, metabolism, and excretion studies can be performed with acceptable mass balance recovery at therapeutically relevant doses and low radiolabel-specific activity using an AMS-14C microtracer approach.
SIGNIFICANCE STATEMENT In this study, the human absorption, distribution, metabolism, and excretion (hADME) of the novel myeloperoxidase inhibitor AZD4831 was assessed following oral administration. This included investigation of the disposition of M7, the N-carbamoyl glucuronide metabolite. Resolution of challenges highlighted in this study contributes to increasing evidence that hADME objectives can be achieved in a single study for compounds with therapeutically relevant doses and low radiolabel-specific activity by using an AMS-14C microtracer approach, thus reducing the need for preclinical radiolabeled studies.
Introduction
Heart failure (HF) with preserved ejection fraction (HFpEF) occurs when the heart is unable to sufficiently fill with blood due to stiffness of the left ventricle and inability of the muscle to relax (Dunlay et al., 2017). HFpEF represents about half of all HF cases and is highly prevalent in patients with hypertension, diabetes, obesity, metabolic syndrome, and chronic kidney disease (Oktay and Shah, 2015). Chronic HF is a leading cause of hospitalization in the United States and represents a significant clinical and economic burden (McCullough et al., 2002; Husaini et al., 2011; Azad and Lemay, 2014; Agarwal et al., 2021).
Myeloperoxidase (MPO) is an abundant enzyme stored within primary granules of neutrophils and monocytes and is not released until leukocyte activation and degranulation (Zhang et al., 2001). MPO is unique in its ability to generate reactive chlorinating species and promotes oxidative damage to host tissues at sites of inflammation, including atherosclerotic lesions (Podrez et al., 2000). Evidence supports that MPO acts as a modulator of vascular tone in vivo and identifies MPO as a systemic regulator of vasomotion and, thus, a potential therapeutic target for HF treatment (Zhang et al., 2001; Baldus et al., 2003; Brennan et al., 2003; Tang et al., 2007; Nelander et al., 2021).
AZD4831—(R)-1-(2-(1-aminoethyl)-4-chlorobenzyl)-2-thioxo-2,3-dihydro-1H-pyrrolo[3,2-day]pyrimidin-4(5H)-one—is a novel, covalent MPO inhibitor with high selectivity for MPO at low oral doses (Inghardt et al., 2022). In phase I studies in healthy volunteers, AZD4831 was rapidly absorbed with a long plasma half-life, dose-dependently inhibited MPO in whole blood, and was generally well tolerated (Gan et al., 2019; Nelander et al., 2021). In the phase IIa SATELLITE study in HFpEF patients (NCT03756285), target engagement (MPO inhibition) was confirmed, and AZD4831 was well tolerated (Lam et al., 2021).
A microtracer approach combined with accelerator mass spectrometry (AMS) has several strengths over conventional liquid scintillation counting–based methodologies for human absorption, distribution, metabolism, and excretion (hADME) studies (Beaumont et al., 2014; Flach et al., 2016; Spracklin et al., 2020; Young et al., 2022). AMS allows detection of 14C administered at 3.7–37 kBq (0.1–1 µCi), compared with conventional doses typically ranging from 1850–7400 kBq (50–200 µCi). As individuals receive minimal radiation exposure, the need for preclinical studies to support dosimetry calculations is spared, and performance of the clinical trial is consequently expedited (Combes et al., 2003; Lappin and Garner, 2003; Spracklin et al., 2020), an important factor in the development of AZD4831. Furthermore, initial studies with high specific activity 14C-AZD4831 highlighted a potential radiochemical stability issue that may have impacted shelf life and potential utility to support clinical studies (unpublished observations). Such radiochemical stability risks can be mitigated using a microtracer approach where materials with low specific activity can be used (Beumer et al., 2007). Finally, AMS allows for detection of microtracer levels over a long follow-up (Lappin and Garner, 2003; Beumer et al., 2007; Gupta et al., 2018), radiocarbon usage is reduced, and problems with radioactive waste disposal are reduced.
Roffel et al. (2016) reviewed >40 oral hADME studies involving administration of regular or low doses of 14C combined with either liquid scintillation counting or AMS analysis. Among the 12 studies conducted with low doses of 14C, six showed recoveries of ≥90%. Low‐dose studies were more often conducted on drugs with a plasma elimination half-life of the parent drug of >100 hours (5 of 12 studies) than regular‐dose studies (1 of 26 studies) to avoid exposure to radiation burden. However, the authors concluded that both low‐ and regular‐dose hADME studies provide adequate data to support decision-making for further drug development (Roffel et al., 2016).
We describe herein the human disposition of 10 mg 14C-AZD4831 (14.8 kBq; 0.4 µCi) as a solution following a single oral dose, i.e., the intended therapeutic administration route. The study used a 14C-labeled microtracer coupled with AMS to evaluate mass balance, pharmacokinetics, and extent of distribution of total 14C into the blood, as well as routes and rates of elimination of AZD4831 in healthy volunteers. To support the pharmacokinetic objective, we separately investigated the potential for racemization of AZD4831, itself an R-enantiomer. During the clinical mass balance study, samples were collected for metabolite profiling and identification. To further understand the disposition of M7, we complemented the mass balance studies by investigating the enzymes involved in the metabolic pathway of AZD4831 to form M7, the predominant circulating N-carbamoyl glucuronide in vitro, as recommended by health authorities (https://www.fda.gov/regulatory-information/search-fda-guidance-documents/in-vitro-drug-interactionstudies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions), and also the absorption and elimination of M7 in rats.
Materials and Methods
Chemicals, Reagents, and Equipment
AstraZeneca supplied unlabeled AZD4831, which was manufactured by SynTheAll Pharmaceutical Co. (Shanghai, China). Radiolabeled 14C-AZD4831 (Fig. 1) was manufactured by Pharmaron (Rushden, UK) following the procedure described previously (Inghardt et al., 2022). The solid material was isolated following crystallization from ethanol and water. Quotient Sciences (Nottingham, UK) prepared 14C-AZD4831 oral solution 10 mg (14.8 kBq; 0.4 µCi) as a clear, colorless, pH-adjusted solution (pH 3) at the clinical site. Metabolite M7 was synthesized as described previously (Jurva et al., manuscript submitted). Acetanilide and paracetamol were purchased from Sigma-Aldrich (St. Louis, MO). ANU (Austrailian National University) sucrose was purchased from the National Institute of Standards and Technology (Gaithersburg, MD). Uridine 5′-diphosphoglucuronic acid, magnesium chloride, alamethicin, D-saccharolactone, β-estradiol, 4-methyl umbelliferone, lamotrigine, 1-naphthol, propofol, zidovudine, and oxazepam were purchased from Sigma-Aldrich (Stockholm, Sweden). 5,5-Diethyl-2-imino-1,3-diphenyl-1,3-diazinane-4,6-dione was supplied by Compound Management AstraZeneca. BD Supersomes Enzymes Baculovirus-Insect Cell-Expressed Human uridine diphosphate glucuronosyltransferase (UGT) was purchased from Corning Life Sciences B.V. (Amsterdam, The Netherlands). Isoflurane (Forene) was purchased from Abbott GmbH & Co. (KG, Wiesbaden, Germany). A list of the analytical equipment used in the hADME study is shown in Supplemental Table 1. Analytical details relating to the supporting studies (e.g., configurational stability, in vitro phenotyping, and rat studies) are provided in the Supplemental Information.
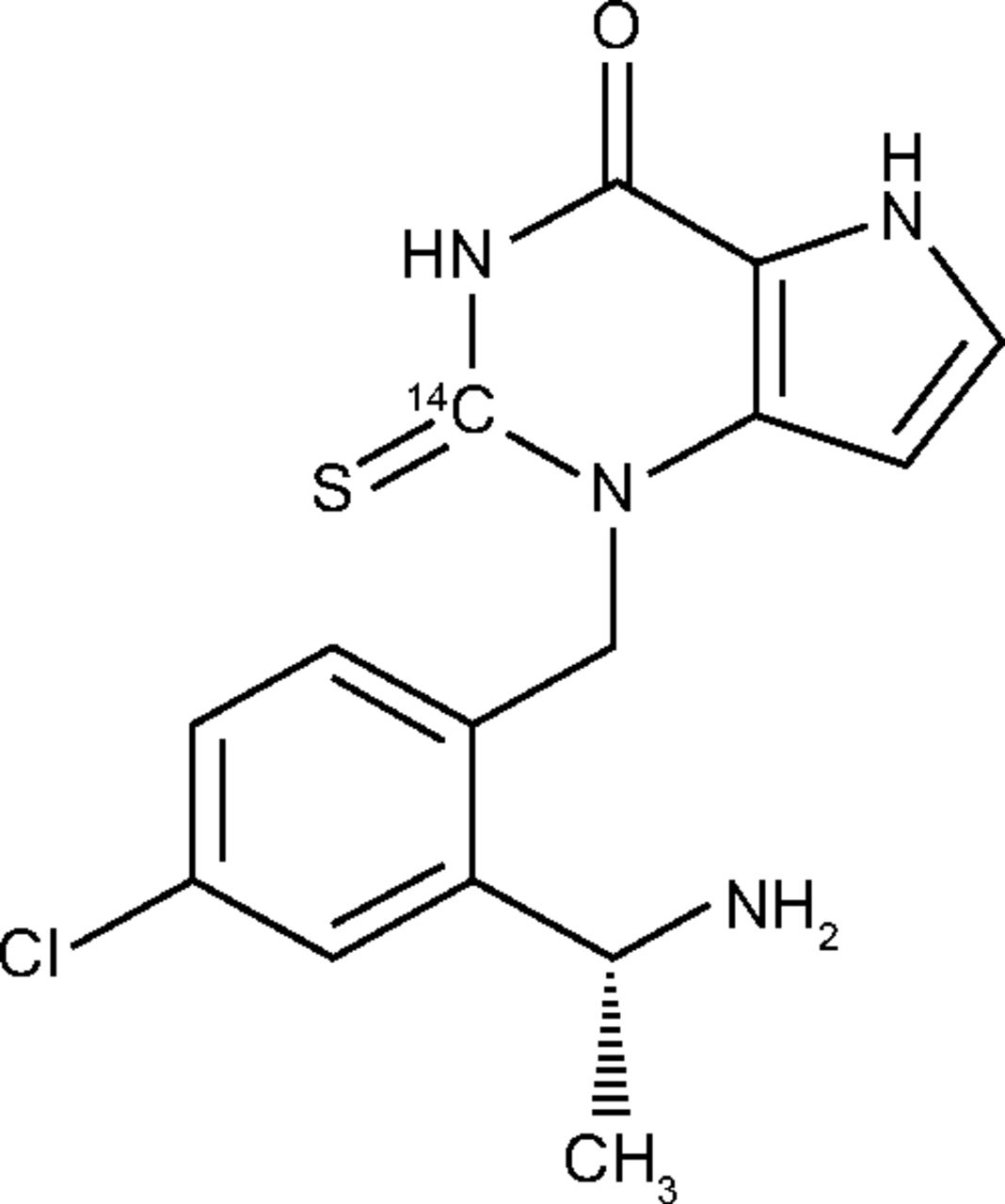
Structure of 14C-AZD4831 (an R-enantiomer) showing the position of the 14C label.
Mass Balance Clinical Study Design
A phase I, open-label, nonrandomized, single-dose, single-center, single-period mass balance study was conducted in healthy male volunteers at Quotient Sciences (Nottingham, UK; NCT04407091). The primary objective was to determine cumulative (urine and feces) mass balance recovery after a single oral dose of 14C-AZD4831 from predose until 336 hours postdose. Key secondary objectives over the same period were to determine routes and rates of elimination of 14C-AZD4831, the pharmacokinetic profile of AZD4831 in plasma and urine, and the metabolic profiles of AZD4831 in plasma, urine, and feces. The total duration of the study (from screening through follow-up) was ∼8 weeks. Participants were screened up to 28 days before dosing, were admitted to the study center prior to drug administration (day −1), and remained resident until 336 hours postdose (day 15). Participants returned for a follow-up visit 7–10 days after discharge.
The study adhered to the principles of the Declaration of Helsinki and International Council for Harmonization Good Clinical Practice Guidelines and was approved by the Office for Research Ethics Committees Northern Ireland, Lisburn. The administration of radioactive substances was conducted under the Department of Health Administration of Radioactive Substances Advisory Committee. All participants provided written informed consent prior to any study procedures.
Participants
Participants were enrolled from a panel of volunteers recruited by Quotient Sciences. Healthy male volunteers aged 18–65 years, with a body mass index of 18.0–30.0 kg/m2 and without clinically relevant disease, were eligible for the study. Key inclusion and exclusion criteria are provided in Supplemental Table 2.
Rationale for Dose Selection
Single oral doses of AZD4831 from 5 mg to 405 mg and multiple doses from 5 mg to 15 mg were previously studied in healthy participants (Gan et al., 2019; Nelander et al., 2021). Target engagement studies have revealed potentially beneficial clinical effects after an MPO inhibition of 50%, which is anticipated to be achieved or exceeded at steady state after once-daily dosing of 5 mg AZD4831 (Lam et al., 2021; Nelander et al., 2021). Due to the long half-life of AZD4831 in humans, there is a two- to threefold accumulation in exposure following once-daily dosing to steady state compared with single-dose administration (Nelander et al., 2021). Therefore, a single oral dose of 10 mg was selected for this study to cover the proposed therapeutic exposure range.
The 14C dose was determined to be adequate to allow the study objectives to be met using AMS methodology but below the upper limit of a microtracer dose. The associated radiation exposure complied with the guidance from the International Commission on Radiologic Protection (1992) Guidelines for Category I studies (<0.1 mSv), and a dose ≤16.4 kBq (0.44 µCi) was selected, resulting in a total effective dose of 9.2 μSv.
Collection of Blood Samples and Excreta
Following dosing, whole blood, plasma, urine, and fecal samples were collected up to 336 hours postdose for study assessments. For AZD4831 and total 14C analysis in plasma, samples were collected before dosing and at 0, 0.25, 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 10, 12, 24, 36, 48, 72, 96, 120, 144, 168, 192, 216, 240, 264, 288, 312, and 336 hours postdose. Whole blood samples for total 14C were collected before dosing and at 1, 3, 5, 8, 12, 24, 36, 48, 72, 96, 120, 168, and 336 hours postdose. Blood samples used for metabolite profiling and identification in plasma were collected before dosing and at 0.5, 1, 2, 3, 4, 6, 8, 12, 24, 36, 48, 72, and 96 hours postdose. Blood samples for quantification of AZD4831, total 14C, and metabolite profiling and identification were collected into tubes containing potassium ethylenediaminetetraacetic acid as anticoagulant. After mixing and centrifugation at 1500g for 10 minutes at 4°C, plasma was harvested and transferred to 5 mL (predose) or 3.5 mL (postdose) polypropylene tubes. Plasma samples were frozen within 70 minutes of collection and were stored at −80°C or below until analysis.
Urine samples for the analysis of AZD4831, total 14C, and metabolite profiling and identification were collected predose (the first void of the day), at 0–6, 6–12, 12–24, 24–36, and 36–48 hours postdose, and then daily (at 24-hour intervals) until discharge. Samples were collected into individual 500-mL polypropylene containers, weighed, pooled for the specific intervals (into 2-L and 4-L containers), and stored at 2–8°C. Samples were separated into aliquots of 2–5 mL for total 14C analysis, metabolite profiling and identification, and analysis of AZD4831.
Fecal samples for total 14C analysis, metabolite profiling and identification, and analysis of AZD4831 were collected predose (between admission and before dosing) and then daily (all fecal samples at 24-hour intervals) until discharge. Samples were collected into individual propylene containers, weighed, pooled for the specific intervals, and stored at −20°C until analysis.
Determination of Total 14C-Concentrations by AMS Analysis
Combustion AMS combines an elemental analyzer for sample combustion and total carbon measurement, interfaced for CO2 transfer using a zeolite concentration step to the AMS, which determines the 14C/12C ratio (van Duijn et al., 2014). The combination of total carbon in a defined sample volume with the 14C/12C ratio provides the 14C concentration. Urine and fecal samples (after homogenization) were analyzed individually to determine total 14C for the mass balance analysis using combustion AMS. Plasma and whole blood samples were also analyzed for total 14C to assess the pharmacokinetic profile of total 14C and to determine blood-plasma partitioning. In short, an aliquot of 5 µL of each blood or plasma sample, 15 µL of each urine sample, and 30 mg of each fecal homogenate sample (diluted or undiluted with blank matrix) were dried and placed in the elemental analyzer, which acted as an autosampler for combustion AMS analysis (van Duijn et al., 2014). Fecal homogenates were analyzed in triplicate; the lower limit of quantification (LLOQ) in fecal homogenates was 2.78 mBq/g homogenate, which equals an LLOQ of AZD4831 of 27.6 pmol Eq. per gram of feces. Plasma LLOQ was 1.51 mBq/mL, which equals an LLOQ of AZD4831 of 3.00 nmol Eq/L. Whole blood LLOQ was 5.28 mBq/mL, which equals an LLOQ of AZD4831 of 10.5 nmol Eq./L. Urine LLOQ was 1.08 mBq/mL, which equals an LLOQ of AZD4831 of 2.15 nmol Eq./L. All measured 14C levels were corrected for individual background values from respective predose or blank samples.
Preliminary recovery analysis showed low overall excretion of 14C for urine samples. Since AZD4831 and its metabolites were potentially bound to the plastic of the collection containers, an extended sample processing was performed for the urine samples to release drug material from the surface of plastic sample tubes and improve recovery (details given in Supplemental Information). Extended processing of plasma and fecal samples was not pursued.
Sample Pooling for Metabolite Profiling and Identification
The methodology for metabolite profiling and identification was different to that used to determine total 14C. Based on the total 14C results, a pooling strategy for metabolite profiling activities was determined. For plasma samples, a Hamilton pool was prepared for each participant, representing the area under the plasma concentration-time curve (AUC) from 0–96 hours (AUC0–96h) by combining volumes in proportion to the time interval between individual samples (Hamilton et al., 1981). These six pooled samples were combined into a single overall AUC0–96h pooled sample by equivolumetric mixing of each participant’s pool. Urine and fecal samples were pooled over a 0–336-hour interval for each participant (including all timepoints assessed). Urine pools were prepared by using equal percentages per timepoint based on volume, and a single overall urine pool was then prepared by equivolumetric mixing of each participant’s pool. To prepare a pool of fecal homogenates, a 0–336-hour pool of homogenates was made for each subject using an equal percentage of homogenate per timepoint (based on the total weight of fecal homogenates); individual fecal pools were then mixed using an equal weight of pool from each subject.
To generate liquid chromatography (LC)-AMS with high-resolution mass spectrometry (HRMS) metabolite profiles in all matrices, the pooled samples were extracted (plasma and fecal homogenates) by protein precipitation with cold MeOH/MeCN (1/1, v/v). Following centrifugation, the supernatant was removed and mixed with 5 µL of dimethyl sulfoxide, evaporated to dryness under nitrogen, and reconstituted in MeOH/MeCN/H2O (12.5/12.5/75, v/v/v). An aliquot of the reconstituted solution was subsequently injected onto an ultraperformance LC (UPLC; Waters Corp., Milford, MA) system coupled to a HRMS (Q Exactive, Thermo Fisher Scientific, Waltham, MA). The pooled urine sample was centrifuged prior to injection onto the LC-AMS-HRMS. The eluate was split between the fraction manager (approximately 58%) to provide the fractions used offline to generate the metabolite profiles by AMS analysis and the Q Exactive (approximately 42%) to provide online HRMS data for metabolite identification. The split ratios were determined by collecting the entire flow from the split arm to the fraction collector, weighing the collected material, and determining its density. Knowing the total flow coming out of the column, the ratios were then determined. Extraction efficiencies for plasma and fecal homogenate (93% and 69%) and column recoveries for plasma, urine, and fecal homogenate (101%, 94%, and 97%, respectively) were deemed adequate. Consequently, LC-AMS metabolite profiles were generated quantitatively together with the corresponding LC–mass spectrometry profiles. Data-dependent tandem mass spectrometry (MS/MS) was applied to obtain fragmentation spectra for both known and unknown metabolites. The analytical details for metabolite characterization are described extensively and reported separately (Jurva et al., manuscript submitted). Analytical details relating to the chromatographic and mass spectrometric conditions used are summarized in Supplemental Table 3, the latter being based on those described previously (Holmberg et al., 2022).
Analysis of Pharmacokinetics of Nonradiolabeled AZD4831
Plasma concentrations of AZD4831 were determined using liquid-liquid extraction followed by LC-MS/MS detection. After addition of internal standard and 2% ammonium hydroxide in water to the plasma aliquot, samples were extracted with ethyl acetate. The organic layer was removed, and the extract was evaporated to dryness under nitrogen at 40°C, reconstituted in acetonitrile:water:formic acid (20/80/0.2, v/v/v), and an aliquot analyzed by LC-MS/MS. Urine concentrations of AZD4831 were determined using sample dilution followed by LC-MS/MS analysis. The LLOQ of AZD4831 in plasma and urine was 2 nmol/L and 20 nmol/L, respectively. Both methods were validated prior to sample analysis and published (Gan et al., 2019; Nelander et al., 2021), and all study samples were analyzed within the confirmed storage periods. The results from the in-study quality control samples, calibration standards, and incurred sample reanalysis samples were evaluated, and it was concluded that both methods performed acceptably for this study (data not shown). The fraction of 14C-AZD4831 in the 10 mg dose was insignificant (∼0.2% based on mass) and so was not accounted for in the pharmacokinetic calculations.
Pharmacokinetic Analysis
The pharmacokinetic parameters for AZD4831 and total 14C were estimated by noncompartmental analysis methods using Phoenix WinNonlin software (v8.0, Certara Inc.). Pharmacokinetic parameters measured included the following the Cmax, AUC from dosing extrapolated to infinity, AUC from dosing to last measurable concentration, the time from dosing at which Cmax was apparent (Tmax), elimination half-life (t1/2), half-life associated with the terminal slope of a semilogarithmic concentration-time-curve, apparent total body clearance of drug from plasma, apparent volume of distribution, and renal clearance of drug from plasma.
Safety Analysis
During each study visit, participants were questioned regarding the occurrence of any adverse events (AEs; serious and nonserious), and events were documented. Other safety measures included clinical laboratory parameters, vital signs (blood pressure, heart rate, oral temperature, respiratory rate), electrocardiogram, and physical examinations.
Configurational Stability of AZD4831 in Humans
The potential for chemical or metabolic racemization of AZD4831 to its corresponding S-enantiomer was investigated using chiral LC-MS/MS. Although the determination of the potential for racemization of AZD4831 was not a prespecified objective of the hADME study, it was included in this article to rationalize the use of nonchiral LC analysis in the hADME study of AZD4831, itself an R-enantiomer. Human plasma samples from healthy volunteers dosed with AZD4831 during the multiple ascending dose (MAD) study were pooled (Nelander et al., 2021; Jurva et al., manuscript submitted). Calibration standards, quality control samples, and plasma samples, including blanks, were processed using protein precipitation with MeCN and analyzed by LC-MS/MS to measure the levels of AZD4831 and its S-enantiomer separately. Additional detail is provided in the Supplemental Information.
In Vitro Phenotyping Studies
Phenotyping of UGTs Responsible for the Formation of Metabolite M7
Based on the recommendations from the US Food and Drug Administration (U.S. Food & Drug Administration, 2012) for compounds with glucuronidation ≥ 25% of total metabolism, seven recombinant UGT isoforms were selected for this phenotyping study. Recombinant UGT enzymes 1A1, 1A3, 1A4, 1A6, 1A9, 2B7, and 2B15 were preincubated with the pore-forming agent alamethicin (25 μg/mL) for 30 minutes on ice before the addition of 4.0 mM magnesium chloride, 0.12 mM D-saccharolactone, and 2.5–200 µM of control substrates (Supplemental Table 4) or 10 µM of AZD4831. After 10 minutes of preincubation with shaking at 37°C in 95% air, 5% CO2, and saturated humidity in a HeraCell 150i incubator (Thermo Fisher Scientific, Waltham, MA), the reaction was initiated by the addition of the cofactor uridine-5′-diphosphoglucuronic acid (5.0 mM). The reaction was conducted in potassium phosphate buffer 0.1 M, pH 7.4 (control substrates), or sodium bicarbonate buffer 0.1 M, pH 7.4 (AZD4831), and H2O for a final incubation volume of 250 µL. Aliquots were taken at 0.5, 60, and 180 minutes, and the reaction was stopped with MeCN containing 5% formic acid and 100 nM of the internal standard 5,5-diethyl-1,3-diphenyl-2-iminobarbituric acid. All reactions were run in triplicates. The pH of the reaction was monitored over the 3-hour incubation and remained neutral during the course of the experiment.
Metabolite analysis.
Formation of M7, the N-carbamoyl glucuronide of AZD4831, and glucuronide metabolites of control substrates for the individual recombinant UGT enzymes was analyzed by UPLC quadrupole time-of-flight MS. The percent contribution from each of the seven tested isoforms to the metabolism of AZD4831 to M7 was estimated using relative activity factors previously determined by Busse et al. (2020) (Supplemental Table 5). The percent contribution for all of the individual isoforms was obtained for the carbamoyl glucuronide formation pathway by dividing the scaled activity by total activity as follows:



The analytical details for the study are described in the Supplemental Information.
In Vivo Pharmacokinetics of M7 in Rats
Determination of the in vivo pharmacokinetics of M7 in rats was not a prespecified objective of the hADME study, but it was performed to further understand the disposition of M7 from a preclinical perspective and strengthen the interpretation of the hADME data. Two days prior to dosing, male Han Wistar rats were prepared by cannulation of the left carotid artery for blood sampling and by cannulation of the right jugular vein for intravenous administration. The catheters were filled with heparin (100 IU mL−1), exteriorized at the nape of the neck, and sealed. Surgery was performed under anesthesia with isoflurane. After surgery, the rats were housed individually with free access to food and water. Approximately 16 hours prior to dosing, the animals were deprived of food and fasted until 4 hours after dosing; animals had free access to drinking water throughout. On the experiment day, metabolite M7, the N-carbamoyl glucuronide of AZD4831, was administered intravenously at a dose of 5 mg/kg in 100% saline into the jugular vein and orally at a dose of 30 mg/kg (0.5% hydroxypropyl methyl cellulose and 0.1% Tween in water) by oral gavage. At predefined timepoints (before dosing and at 1, 2, 4, 8, and 24 hours after dosing), blood samples ∼0.150 mL were withdrawn from the carotid artery. Blood samples were collected in heparinized plastic tubes and centrifuged, within 30 minutes, for 5 minutes at 10,000g and at 4°C. Plasma was transferred to a 96-well plate and stored at −20°C until analysis by LC-MS/MS.
Results
Configurational Stability of AZD4831
Using chiral LC-MS/MS for human plasma samples from the MAD study, levels of the S-enantiomer of AZD4831 were below the LLOQ (3.89 nmol/L) in the pooled plasma samples at individual timepoints, the AUC0–24h plasma pooled samples, or each participant’s plasma samples (Table 1). As shown in Fig. 2, representative overlaid chiral chromatograms of extracts of plasma samples from participants receiving AZD4831 and plasma samples spiked with the S-enantiomer at 3.1 nM and 6.2 nM, indicating that racemization of AZD4831 was not observed in humans. Therefore, this confirmed that analysis of AZD4831 conducted by nonchiral LC-MS/MS in the clinical mass balance study was justified.

Representative overlaid chromatograms showing the separation of AZD4831 from its S-enantiomer (3.1 nM and 6.2 nM) and metabolite M7 following chiral LC-MS/MS analysis of pooled plasma samples from the MAD clinical study (Nelander et al., 2021). The high peak at RT 13.8 minutes between AZD4831 and the S-enantiomer is the metabolite M7. RT, retention time.
LC-MS/MS plasma concentrations of AZD4831 and the S-enantiomer in human pooled plasma samples from the MAD study (Nelander et al., 2021; Jurva et al., manuscript submitted)
Clinical Mass Balance Study
Participants
Six healthy males aged 22–39 years were enrolled in the study. Four participants were White (66.7%), and two were Black or African American (33.3%); mean (standard deviation) body mass index was 27.5 (1.3) kg/m2, no participants were current smokers, and all had an alcohol consumption of 0–10 units per week. Demographics and baseline characteristics of participants are summarized in Supplemental Table 6.
Pharmacokinetics of AZD4831
Plasma.
Following a single oral dose of 14C-AZD4831 10 mg (as a solution), AZD4831 was detected in plasma from the first sampling timepoint at 0.25 hours onwards in all participants (Fig. 3A). Pharmacokinetic analysis was calculated from concentration profiles generated by LC-MS/MS. Pharmacokinetic parameters of 14C-AZD4831 are presented in Table 2. Tmax was reached between 0.5 and 6.1 hours postdose, and concentrations then declined in a biphasic manner and remained quantifiable from 96 to 168 hours postdose. Terminal slopes were reliably determined for all participants, and resulting values of t1/2 ranged from 36.3 to 73.4 hours, with a geometric mean t1/2 of 52.3 hours (Table 2).
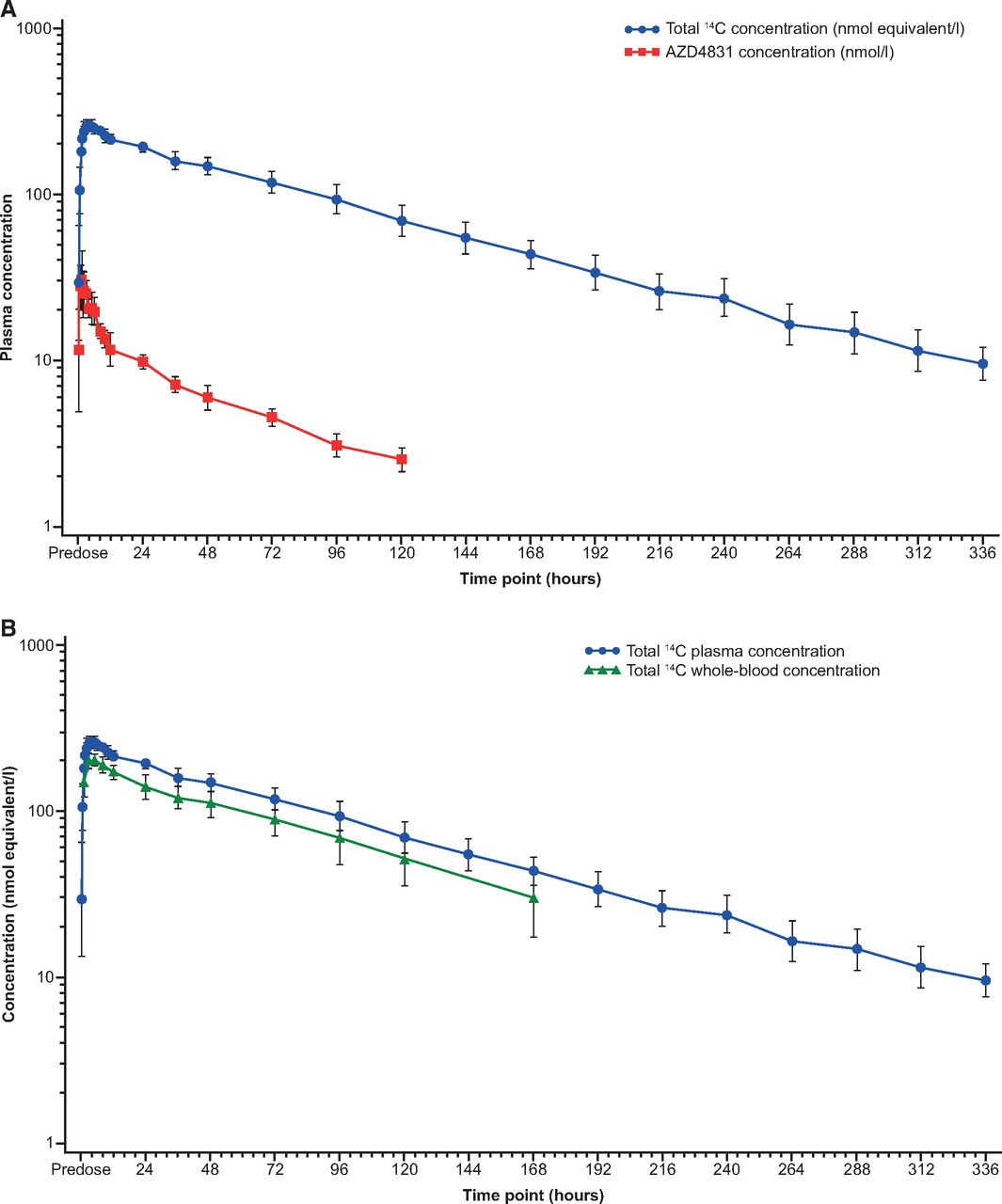
Geometric mean (geometric standard deviation) concentration versus time profiles of AZD4831 and total 14C in (A) plasma and (B) whole blood following administration of a single oral dose of 10 mg of 14C-AZD4831 (14.8 kBq, in solution) in healthy male volunteers.
Geometric mean (geometric %CV) plasma and whole blood pharmacokinetic parameters following a single dose of 10 mg of 14C-AZD4831 (14.8 kBq, as a solution) in healthy male volunteers
Urine.
An average of 44.4% (based on unlabeled compound analysis of urine samples by LC-MS/MS) of the dose administered was recovered as unchanged AZD4831 in urine by the end of the sampling period (336 hours), with about 15.3% being recovered within the first 24 hours postdose.
Pharmacokinetics of Total 14C in Plasma
Following a single oral dose of 14C-AZD4831 10 mg (as a solution), total 14C was detected in plasma from the first sampling timepoint at 0.25 hours onwards in all participants (Fig. 3A). Pharmacokinetic parameters of total 14C are presented in Table 2. Tmax occurred between 3 and 8 hours postdose. Following Cmax, plasma total 14C declined in a monophasic or biphasic manner and remained quantifiable for up to 336 hours postdose. Terminal slopes were reliably determined for all participants, and resultant values of t1/2 ranged from 62.2 to 83.5 hours, with a geometric mean t1/2 of 73.5 hours (Table 2). The concentration profile of total 14C following administration of a single oral dose of 14C-AZD4831 is shown in Fig. 3A.
Blood-Plasma Partitioning of 14C-AZD4831–Derived Radioactivity
Total 14C was quantifiable in whole blood in all participants from the first sampling timepoint at 1 hour postdose and remained quantifiable over 120–336 hours postdose. The geometric mean (%CV) whole blood-to-plasma total 14C concentration ratios ranged from 0.72 (14.9%) to 0.84 (21.6%), which indicated nonpreferential distribution of total 14C to the cellular components of whole blood. There were no notable time-dependent differences in the geometric mean whole blood-to-plasma total 14C concentration ratios (Fig. 3B).
Mass Balance and Excretion
Following a single oral dose of 14C-AZD4831 10 mg (as a solution), an average of 83.6% of the 14C administered was recovered by the end of the sampling period (336 hours) (Table 3). On average, 51.2% of the total 14C was recovered from urine, and 32.4% was recovered from feces. An overview of total 14C concentrations in urine samples measured before and after extended extraction, involving additional volumes of MeCN before sample dilution (MeCN/urine 1/1, v/v) followed by extended extraction of the sample tubes (Supplemental Information), improved recovery in the urine from approximately 37% to 51%, indicating that some binding to plastics had occurred. Within the first 24 hours postdose, on average 17.9% and 5.0% of total 14C was recovered in urine and feces, respectively (Supplemental Table 7). The cumulative fractions of 14C dose excreted as total 14C in urine, feces, and total (urine and feces combined) are shown in Fig. 4 and Supplemental Table 7. A summary of total 14C in urine and feces combined, expressed as amount of 14C (nmol equivalent) and fraction of dose, is shown in Table 3. The excreted 14C accounted for 71.8% of the dose excreted in urine and feces between 0–168 hours, and the residual 14C excretion continued throughout the 336-hour collection period (Table 3; Supplemental Table 7).

Arithmetic mean (standard deviation) cumulative fraction of 14C dose (%) excreted as total 14C in urine, feces, and total (urine plus feces) following administration of a single oral dose of 10 mg of 14C-AZD4831 (14.8 kBq, as a solution) in healthy male volunteers. Fe, fraction of total 14C excreted expressed as a percentage of the dose administered.
Summary of total 14C in urine and feces combined (nmol equivalent), and fraction of dose excreted as total 14C in urine and feces combined, following administration of a single oral dose of 10 mg of 14C-AZD4831 (14.8 kBq, as a solution) in healthy male volunteers
Data are arithmetic. Unit for Ae is nmol equivalent
Metabolite Profiling and Identification
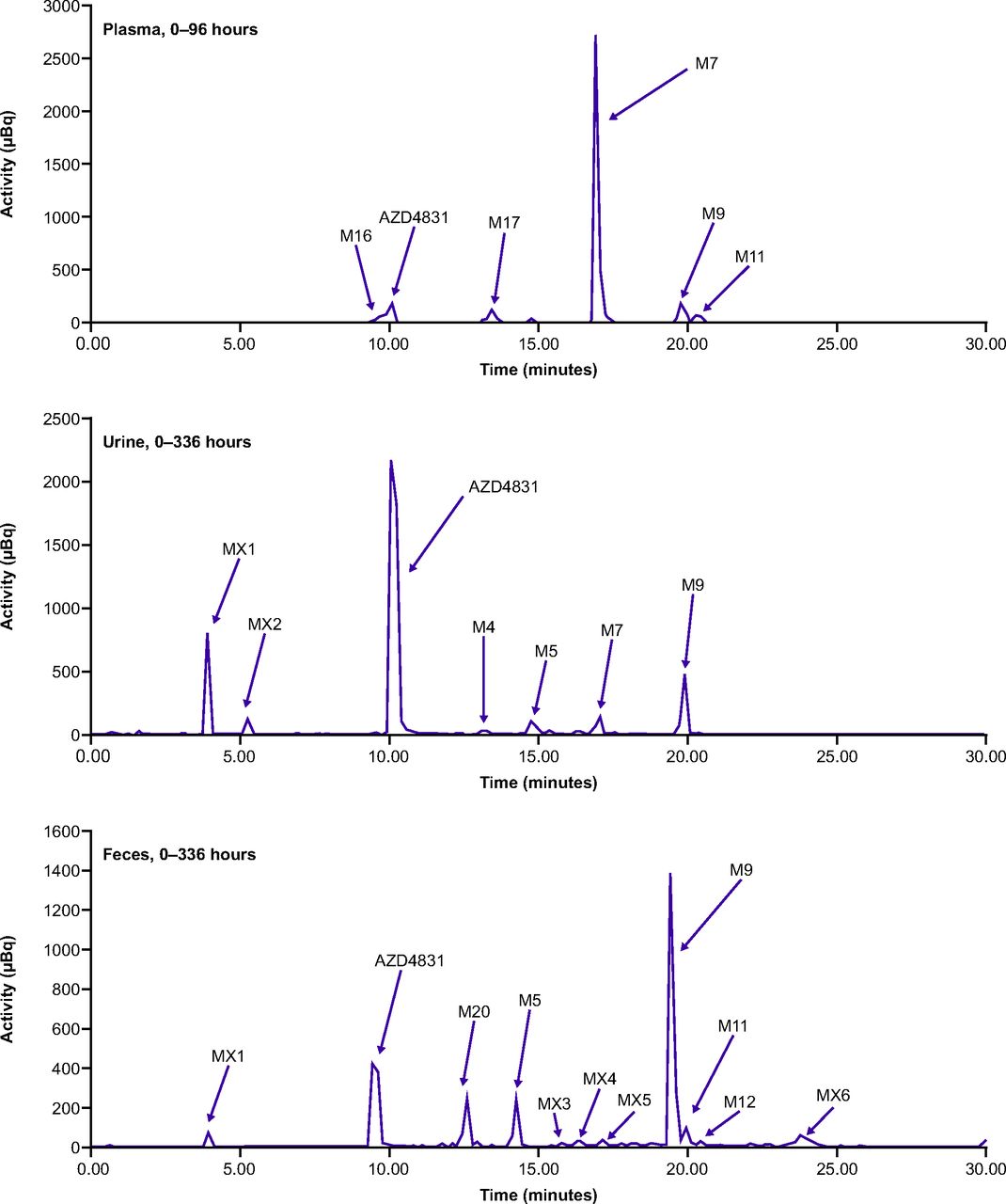
Representative UPLC chromatograms of pooled plasma (0–96 hours), pooled urine (0–336 hour), and pooled fecal extracts (0–336 hours) are presented in Fig. 5. Following oral administration of 14C-AZD4831 10 mg (as a solution), structures of nine metabolites (M4, M5, M7, M9, M11, M12, M16, M17, and M20) were identified, whereas six metabolites or metabolite fractions (MX1–MX6) were detected but not characterized (Jurva et al., manuscript submitted).

Representative UPLC chromatograms showing metabolite profiles in plasma (0–96-hour pool), urine (0–336-hour pool), and feces (0–336-hour pool) following administration of a single oral dose of 10 mg of 14C-AZD4831 (14.8 kBq, as a solution) in healthy male volunteers.
Quantitative percent estimates of 14C and dose are presented, and metabolite profiles in plasma, urine, and feces are shown in .Tables 4 and 5 and in Fig. 5. Unchanged AZD4831 accounted for 32.0% and 6.0% of the dose excreted in urine and feces, respectively, whereas in a pooled plasma sample representing AUC0–96h, which represents more than 90% of the total drug-related exposure, AZD4831 accounted for 6.1% of the total drug-related material (DRM), corresponding to 11.9 nmol Eq./L based on the total drug-related exposure of 197 nmol Eq./L.
Exposure to AZD4831 and metabolites identified in plasma (AUC pool 0–96 hours) following administration of a single oral dose of 10 mg of 14C-AZD4831 (14.8 kBq, as a solution) in healthy male volunteers
Quantitative estimates of AZD4831 and identified metabolites in urine and feces (both pooled 0–336 h) (% of dose) following administration of a single oral dose of 10 mg of 14C-AZD4831 (14.8 kBq, as a solution) in healthy male volunteers
The major metabolite of AZD4831 in human plasma was the N-carbamoyl glucuronide M7, accounting for 71.9% of total exposure of DRM in plasma. AZD4831 and six metabolites were all present at <7% of DRM each. AZD4831 was the major component in urine (32.0% of the dose based on single AMS chromatograms of a 0–336-hour pool). The N-acetyl metabolite (M9) was the most abundant characterized metabolite in urine and accounted for 4.4% of the dose; the uncharacterized metabolite MX1 accounted for 6.3% of the dose. The amount of AZD4831 recovered in feces was 6.0% of the dose, and metabolite M9 was the most abundant metabolite, accounting for 11.9% of the dose.
The structural characterization of metabolites of AZD4831 has been thoroughly studied and reported separately (Jurva et al., manuscript submitted). The structures of M5, M7, M9, M11, and M12 were confirmed by comparison with authentic standards. A proposed metabolite scheme for AZD4831 showing the characterized metabolites in humans only is presented in Fig. 6.

Proposed metabolite scheme for 14C-AZD4831 following administration of a single oral dose of 10 mg of 14C-AZD4831 (14.8 kBq, as a solution) in healthy male volunteers. Shaded atoms indicate different possible sites of metabolism. The asterisk marks the position of the 14C label in AZD4831.
Safety
A total of 13 AEs were reported by six participants who received a single oral dose of 14C-AZD4831 10 mg (as a solution). All AEs were mild, none were considered related to 14C-AZD4831, and there were no serious AEs. Diarrhea was the most commonly reported AE (five times by three participants). There were no clinically relevant findings in clinical laboratory evaluations, vital signs, electrocardiograms, or physical examinations.
In Vitro Phenotyping of UGTs Responsible for the Formation of Metabolite M7
The results of an in vitro study, using seven UGT isoforms scaled to predict the relative contribution of each isoform in human liver microsomes, indicated that AZD4831 is a substrate of the following UGT isoforms: 1A1, 1A3, 1A9, and 2B7, where 1A1 is the major enzyme contributing toward 96% of the total AZD4831 carbamoyl glucuronidation among the isoforms tested. AZD4831 was not a substrate of UGT isoforms 1A4, 1A6, and 2B15 (Fig. 7; Table 6). Glucuronide metabolites for the control substrates could be detected in all UGT isoforms, indicating that the test system was performing to expected standard. No other UGTs were examined as a large proportion of glucuronidation is explained through formation via UGT1A1 and to a lesser percent by UGT isoforms 1A3, 1A9, and 2B7. However, contribution from other UGT isoforms cannot be excluded.
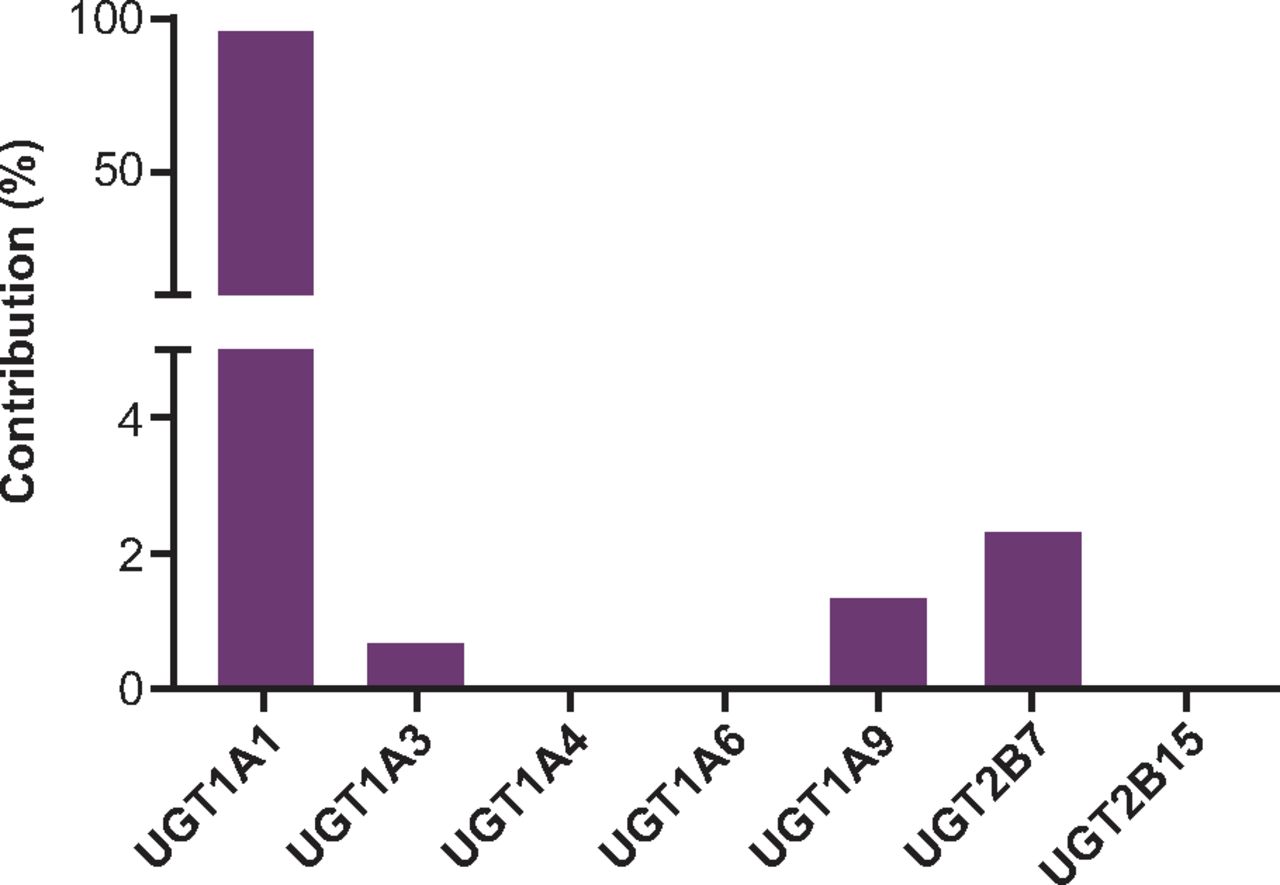
Summary of the percent contribution of individual UGT isoforms responsible for the formation of M7 following incubation with AZD4831 in recombinantly expressed UGTs.
Summary of the percent contribution of individual UGT isoforms responsible for the formation of M7 following incubation with AZD4831 in recombinantly expressed UGTs
In Vivo Pharmacokinetics of M7 in Rats
Metabolite M7 displayed moderate clearance and low volume of distribution, a relatively short t1/2 of 0.5 hours, and only 0.6% oral bioavailability. Monitoring of AZD4831 in the same pharmacokinetic samples revealed that AZD4831 was detectable in the circulation following both intravenous and oral doses of M7. Exposure (AUC) of AZD4831 in rats was approximately threefold higher following oral versus intravenous dosing when dose adjusted (AUC, 2.31 versus 0.73 hour·µmol/L, respectively). For the intravenous dose, the Tmax for AZD4831 (∼5 hours) was substantially delayed compared with that for M7 (0.03 hours). For the oral dose (30 mg/kg), the observed AUC for AZD4831 was 66-times greater than that for M7 (13.9 versus 0.21 hour·µmol/L, respectively) (Supplemental Fig. 1).
Discussion
The hADME study investigated the mass balance recovery, pharmacokinetics, and metabolism of AZD4831. In this small population of healthy volunteers, a single oral dose of 10 mg 14C-AZD4831 given as a solution (14.8 kBq; 0.4 µCi) was well tolerated, consistent with previous studies (Gan et al., 2019; Nelander et al., 2021). The clinical study design allowed for sample collection over 336 hours to ensure complete recovery of the 14C dose via excretion. Routes and rates of elimination and the pharmacokinetics of AZD4831 were characterized, as well as the extent of distribution of total 14C into the blood. Samples were also collected for the purpose of metabolite profiling, and the enzyme responsible for the formation of M7 was studied in vitro. Additionally, chiral analysis of human plasma samples from the MAD study (Nelander et al., 2021; Jurva et al., manuscript submitted) confirmed that conversion of AZD4831 to its S-enantiomer was not quantifiable in plasma samples as levels were below the LLOQ. Therefore, the use of traditional nonchiral analysis to generate concentrations of AZD4831 for the modeling of pharmacokinetic profiles was justified.
The absorption of AZD4831 was relatively fast, and concentrations declined in a biphasic manner, as previously reported (Nelander et al., 2021). Results from both the plasma metabolite profiling and the plasma pharmacokinetics of unlabeled AZD4831 indicate that exposure due to AZD4831 is low compared with total DRM in circulation, accounting for only 4.4% of circulating plasma total radioactivity based on area under the plasma concentration-time curve from dosing extrapolated to infinity and confirm that M7, the N-carbamoyl glucuronide metabolite of AZD4831, is the predominant circulating drug-related component in plasma, accounting for >70% of total 14C over the 0–96 hour plasma profile. Furthermore, the geometric mean whole blood-to-plasma total 14C concentration ratios indicated nonpreferential distribution of total 14C to the cellular components of whole blood. However, it should be noted that in a separate in vitro study of parent drug and metabolites M7, M9, M11, and M12, the blood-to-plasma ratios ranged from 0.57 to 1.2 (unpublished observations).
The recovery of total 14C following a single oral dose of 14C-AZD4831 was 84% in excreta (urine and feces) over the 336-hour sampling period. Although a total recovery of >90% is “preferable” for human mass balance studies (https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-pharmacology-considerations-human-radiolabeled-mass-balance-studies), a recovery of >80% of 14C in humans is deemed acceptable, according to the literature standard (Roffey et al., 2007; Penner et al., 2009; Coppola et al., 2019; Singh et al., 2022) for relevant clinical dose with low radiocarbon if explained by biologic factors. In our study, nonspecific adsorption to the plastic collection containers and subsample tubes was clearly a factor that influenced the recovery of 14C in the urine samples (as demonstrated by increased recovery after exhaustive extraction of the urine sample tubes) and at least partially contributed to the lower than 100% of total 14C recovery. The improved recovery did not include any unaccounted 14C lost prior to the exhaustive extraction of the urine sample tubes, and so recovery in urine can be assumed to be higher than 51.2%. Extended processing of plasma and fecal samples was not required due to, in our experience, the proteins and lipids in plasma and feces that prevent adsorption to plastic. Notably, it has been reported that compounds with a shorter total 14C half-life (<50 hours) tend to achieve greater total recovery than compounds with a longer half-life (Roffey et al., 2007). Indeed, the recovery rate of total 14C was slow, with 13% of the dose being excreted during the second week of sample collection. These findings are consistent with the relatively high oral absorption and long plasma half-life reported for AZD4831 (Gan et al., 2019; Nelander et al., 2021).
The majority of total 14C was recovered via urinary excretion (51.2%), and 32%–44% of the dose was unchanged AZD4831 in urine; thus, the majority of total 14C recovered in the urine was AZD4831. The range of 32%–44% for the dose recovered as unchanged AZD4831 reflects the different modes of detection and limits of quantification from the two analytical methods used (i.e., a single AMS chromatogram of a 0–336-hour pool for metabolite profiling compared with unlabeled compound analysis of all individual urine samples by LC-MS/MS). The cumulative fraction of dose excreted in feces as total radioactivity was 32.4% during the total collection period of 336 hours postdose. Only 9.7% of the dose recovered (as total radioactivity) was excreted in the first 48 hours postdose, with the remainder excreted over 48–336 hours. As per the literature, mean overall transit time throughout the entire gastrointestinal tract was reported to be 30–40 hours, with upper and lower limits of normal of 70 and 14 hours, respectively (Rao et al., 2011; Castellino et al., 2013). Therefore, together with the low fecal recovery during the initial 48 hours and the small amount of unchanged AZD4831, this suggests that the majority of fecal recovery over the 336-hour interval is due to biliary elimination and not poor absorption of AZD4831. Therefore, based on excretion data, it can be concluded that renal clearance is the main route of elimination for AZD4831 and that orally administered AZD4831 is well absorbed. In addition, renal clearance of AZD4831 was estimated to be 12.8 L/h, which is higher than the product of the unbound fraction in plasma (0.349) (Jurva et al., manuscript submitted) and the glomerular filtration rate (7.5 L/h: 7.5 L/h × 0.349 = 2.6 L/h) (Davies and Morris, 1993). Thus, this indicates that urinary excretion is not solely by glomerular filtration but also involves active tubular secretion of AZD4831.
Metabolism also contributes substantially to the elimination of AZD4831. In the clinical study, the biotransformation of AZD4831 involved modification primarily at the thiourea and primary amine moieties of the xanthine derivative, as observed for preclinical species (Jurva et al., manuscript submitted) with no human-specific metabolites observed. Metabolite profiling shows that although AZD4831 was the primary drug-related component in excreta over 336 hours, N-acetylated AZD4831 (M9) was the most abundant metabolite, accounting for 4.36% and 11.9% of the dose in urine and feces, respectively. Intramolecular cyclization with sulfur elimination of AZD4831 is the most prominent phase I biotransformation reaction leading to metabolites M4 and M5, whereas M11 and M12 were relatively minor metabolites in human samples. N-carbamoyl acyl glucuronide conjugation (M7) and N-acetylation (M9) were the major phase II biotransformation reactions observed for AZD4831 in this clinical study, whereas M16, M17, and M20 (desulfurized N-acetyl conjugate) were minor metabolites in human samples.
The finding that M7 is the predominant circulating metabolite of AZD4831 in human plasma contrasts with observations in mouse, rat, and dog. Exposure to M7 in preclinical toxicology studies nevertheless exceeded that observed in humans, ensuring adequate toxicological coverage of the metabolite as reported by Jurva et al. (manuscript submitted). High plasma binding of M7 (>99.8%) in humans is likely to contribute to the low clearance of M7. Support for this hypothesis is found in the reported preclinical species with higher free fractions of M7, and the resulting half-lives of M7 and AZD4831 in these species are shorter. In addition, the circulation of M7 at high exposure with only low levels of M7 found in excreta in humans suggests hydrolysis of M7 to the parent compound and/or enterohepatic recycling into the systemic circulation before excretion, thereby contributing to the long half-life of AZD4831. When M7 was administered by oral and intravenous dosing to rats, circulating AZD4831 was observed, suggesting that M7 is hydrolyzed followed by reabsorption of AZD4831 and supporting the involvement of enterohepatic recycling in the disposition of AZD4831.
Results from the in vitro studies presented in this article indicate that among the isoforms tested, UGT1A1 is the primary enzyme involved in the formation of M7, although UGT1A3, UGT1A9, and UGT2B7 are also involved to a lesser extent, consistent with observations reported previously for a primary amine molecule (Gunduz et al., 2010). Given that UGTs are typically high-capacity enzymes with high Km, drug-drug interactions with AZD4831 are expected to be low (Mackenzie et al., 2010). Additionally, with multiple enzymes involved in this metabolic pathway, even if coadministered drugs inhibit one UGT isoform, the pharmacokinetics of AZD4831 are unlikely to be affected, as described for other drugs, e.g., febuxostat (Mukoyoshi et al., 2008).
In conclusion, a 14C-labeled microtracer approach coupled with analysis by sensitive AMS was successfully applied to study the disposition and mass balance recovery of AZD4831 in humans and to expedite the clinical development of this new molecular entity. The elimination of AZD4831 is via metabolism and renal excretion. The recovery of 14C-AZD4831 and metabolites showed that a high fraction of AZD4831 is absorbed and that renal elimination is the principal route of excretion. M7, the N-carbamoyl glucuronide metabolite of AZD4831, is the predominant circulating metabolite of AZD4831 in plasma. M7 is formed primarily via UGT1A1, and preclinical evidence shows that once eliminated in bile, M7 can undergo hydrolysis before being recirculated in the system. Alternatively, it can also be excreted as AZD4831 following hydrolysis. These findings support the further clinical development of AZD4831 as a potential treatment of HF and possibly other indications.
Acknowledgments
The authors thank the participants in this study, and the personnel at Pharmaron, Quotient Sciences, and TNO who were involved in the conduct of this study. Medical writing support was provided by Shaun W. Foley (Hons), ISMPP CMPP, and editorial support was provided by Sharmin Saleque of Core Medica, London, UK, supported by AstraZeneca.
Authorship Contributions
Participated in research design: Gränfors, Amilon, Menakuru, Garkaviy, Weidolf, Gopaul.
Conducted experiments: Menakuru, Sandinge, Yan, Andersson, Pelay-Gimeno, Vaes, de Ligt.
Performed data analysis: Sandinge, Amilon, Yan, Heijer, Andersson, Jurva, Weidolf, Pelay-Gimeno, Vaes, de Ligt, Gopaul.
Wrote or contributed to the writing of the manuscript: Bhattacharya, Sandinge, Bragg, Heijer, Yan, Andersson, Jurva, Pelay-Gimeno, Vaes, de Ligt, Gränfors, Amilon, Lindstedt, Menakuru, Garkaviy, Weidolf, Gopaul.
Footnotes
- Received August 30, 2022.
- Accepted December 15, 2022.
This study was funded by AstraZeneca.
C.B., A.-S.S., J.Y., U.J., V.S.G., M.H., R.A.B., M.G., L.C.A., and P.G. are employees of AstraZeneca and may hold stock in the company. L.W., C.A., and E.-L.L. are former employees of AstraZeneca and may hold stock in the company. M.P.-G., W.H.J.V., and R.A.F.d.L. are employees of TNO. S.R.M. is an employee of Quotient Sciences.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AE
- adverse event
- AMS
- accelerator mass spectrometry
- AUC
- area under the plasma concentration-time curve
- AUC0–96h
- area under the plasma concentration-time curve from 0–96 hours
- DRM
- drug-related material
- hADME
- human absorption, distribution, metabolism, and excretion
- HF
- heart failure
- HFpEF
- heart failure with preserved ejection fraction
- HRMS
- high-resolution mass spectrometry
- LC
- liquid chromatography
- LLOQ
- lower limit of quantification
- MAD
- multiple ascending dose
- MPO
- myeloperoxidase
- MS/MS
- tandem mass spectrometry
- t1/2
- elimination half-life
- Tmax
- time at maximum plasma concentration
- UGT
- uridine diphosphate glucuronosyltransferase
- UPLC
- ultraperformance liquid chromatography
- Copyright © 2023 by The Author(s)
This is an open access article distributed under the CC BY Attribution 4.0 International license.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}