
Abstract
Various members of the cytochrome P450 (CYP) superfamily have the capacity of metabolizing omega-6 and omega-3 polyunsaturated fatty acids (n-6 and n-3 PUFAs). In most mammalian tissues, CYP2C and CYP2J enzymes are the major PUFA epoxygenases, whereas CYP4A and CYP4F subfamily members function as PUFA hydroxylases. The individual CYP enzymes differ in their substrate specificities as well as regio- and stereoselectivities and thus produce distinct sets of epoxy and/or hydroxy metabolites, collectively termed CYP eicosanoids. Nutrition has a major impact on the endogenous CYP-eicosanoid profile. “Western diets” rich in n-6 PUFAs result in a predominance of arachidonic acid-derived metabolites, whereas marine foodstuffs rich in n-3 PUFAs shift the profile to eicosapentaenoic and docosahexaenoic acid-derived metabolites. In general, CYP eicosanoids are formed as second messengers of numerous hormones, growth factors and cytokines regulating cardiovascular and renal function, and a variety of other physiological processes. Imbalances in the formation of individual CYP eicosanoids are linked to the development of hypertension, myocardial infarction, maladaptive cardiac hypertrophy, acute kidney injury, stroke and inflammatory disorders. The underlying mechanisms are increasingly understood and may provide novel targets for the prevention and treatment of these disease states. Suitable pharmacological agents are under development and first proofs of concept have been obtained in animal models.
Abbreviations: AA arachidonic acid, ALA alpha-linolenic acid, COX cyclooxygenase, CYP cytochrome P450, DHA docosahexaenoic acid, EDHF endothelium-derived hyperpolarizing factors, EDP epoxydocosapentaenoic acid, EEQ epoxyeicosatetraenoic acid, EET epoxyeicosatrienoic acid, EPA ecosapentaenoic acid, HDoHE hydroxydocosahexaenoic acid, HEPE hydroxyeicosapentaenoic acid, HETE hydroxyeicosatetraenoic acid, HETrE hydroxyeicosatrienoic acid, I/R ischemia-reperfusion, KO knockout, LA linoleic acid, LOX lipoxygenase, PLA2 phospholipase A2, PUFA polyunsaturated fatty acid, ROS reactive oxygen species, sEH soluble epoxide hydrolase, SHR spontaneously hypertensive rat, TAC transverse aortic constriction, TG transgene, WT wild-type.
Similar content being viewed by others

Keywords
- Arachidonic acid
- Eicosapentaenoic acid
- Docosahexaenoic acid
- Hydroxylases
- Epoxygenases
- Hypertension
- Ischemia/reperfusion injury
- Cardiac hypertrophy
6.1 Introduction: Discovery of the Third Branch of the Arachidonic Acid Cascade
The discovery chain leading to our current understanding of the pivotal role of cytochrome P450 (CYP) enzymes in the generation of biologically active metabolites of polyunsaturated fatty acids (PUFAs) was initiated with a series of seminal findings in the 1980s. In 1981, three laboratories demonstrated that liver and kidney microsomal as well as purified CYP enzymes catalyzed the oxygenation of arachidonic acid (AA; 20:4 n-6) [1–4]. Structural characterization of the metabolites indicated that CYP enzymes can metabolize AA via three reaction types [5, 6] (Fig. 6.1): (1) allylic oxidation to form cis,trans-conjugated “mid-chain” hydroxyeicosatetraenoic acids (5-, 8-, 9-, 11-, 12- and 15-HETE); (2) hydroxylation at or near the terminal methyl group (ω-/(ω−1)-hydroxylase reaction) yielding 20-, 19-, 18-, 17- and 16-HETE; and (3) olefin epoxidation (epoxygenase reaction) generating four regioisomeric epoxyeicosatrienoic acids (5,6-, 8,9-, 11,12- and 14,15-EET), each of which can be formed as either the R,S or the S,R enantiomer.

Bioactivation of arachidonic acid (AA). Cyclooxygenases (COX), lipoxygenases (LOX), and CYP enzymes initiate the production of biologically active AA metabolites. CYP enzymes are able to metabolize AA by three different reaction types. Olefin epoxidation results in formation of regioisomeric epoxyeicosatrienoic acids (EETs). Hydroxylation at or near the terminal methyl group generates hydroxyeicosatetraenoic acids (HETEs) and allylic oxidation produces cis,trans-conjugated “mid-chain” HETEs (compare Sect. 6.1). The individual CYP enzymes involved in AA metabolism differ in their reaction specificities as well as regio- and stereoselectivities and thus produce enzyme specific sets of hydroxy- and epoxy metabolites (compare Sect. 6.2)
Subsequent studies revealed the presence of EETs as endogenous constituents in rat liver, rabbit kidney, and human urine providing the first proof for an active role of CYP enzymes in AA metabolism under in vivo conditions [7–10]. The biological tissues contained unique sets of regio- and stereoisomeric EETs substantiating the enzymatic origin of these metabolites and stimulating the search for the individual CYP enzymes involved in the regio- and enantioselective epoxidation of endogenous AA pools [11] (compare Sect. 6.2). Moreover, it became clear that CYP enzymes require free AA as a substrate suggesting that phospholipase-mediated AA release from membrane phospholipids provides the starting point for the formation and action of CYP-dependent AA metabolites under in vivo conditions (compare Sect. 6.3). Phospholipase activation is a common feature of the receptor-mediated actions of numerous vasoactive hormones, growth factors and cytokines. Accordingly, CYP-dependent AA metabolites were increasingly recognized to function as “second messengers”. This concept became the key for our present understanding of how PUFA-metabolizing CYP enzymes are integrated into the regulation of a wide variety of physiological and pathophysiological processes.
Early studies on the potential physiological roles of CYP-dependent AA metabolites revealed effects on renal salt reabsorption and vascular tone and led to the hypothesis that alterations in the formation of 20-HETE and EETs contribute to the pathophysiology of hypertension [12]. Actually, it was then that the results obtained with two animal models of genetic hypertension until now have shaped our thinking and research about the renal and cardiovascular functions of 20-HETE and EETs (compare Sect. 6.4). A study published in 1989 demonstrated that increased renal 20-HETE production contributes to the elevation of blood pressure in spontaneously hypertensive rats (SHR) [13]. Later on, the development of hypertension in salt-sensitive Dahl rats was attributed to the inability of this strain to upregulate renal EET biosynthesis in response to salt loading [14]. In conclusion, it appeared that hypertension can be caused by an imbalance of pro-(20-HETE) and antihypertensive (EETs) AA metabolites produced by CYP hydroxylases and CYP epoxygenases, respectively. Challenging this simplified view, a series of further studies showed that Dahl salt-sensitive rats exhibit not only a deficiency in EET formation but also in renal CYP hydroxylase expression and 20-HETE production [15]. The apparent paradox was solved after recognizing that the prohypertensive role of 20-HETE is related to its action as a potent vasoconstrictor in the vascular system of the kidney, whereas the antihypertensive role of 20-HETE is based on its capacity to inhibit sodium reabsorption in different segments of the nephron [16]. In contrast to the dual and site-specific role of 20-HETE, the vascular and tubular actions of EETs are apparently unidirectional and antihypertensive because they promote both vasodilation and salt excretion [16, 17].
Taken together, these early biochemical and pathophysiological studies established the CYP-dependent formation of biologically active hydroxy- and epoxymetabolites of AA as the so-called “third branch of the AA cascade” complementary to the previously discovered cyclooxygenase (COX) and lipoxygenase (LOX) initiated pathways of prostanoid and leukotriene formation [18] (Fig. 6.1). Collectively, the AA metabolites produced via all three pathways as well as nonenzymatic reactions are termed eicosanoids (from Greek eicosa = twenty, reflecting that these metabolites are derivatives of a 20 carbon fatty acid). In general and also in the present review, the term eicosanoid is used more broadly to also include related metabolites derived from other PUFAs. Currently, over a hundred different eicosanoids have been identified and the analysis of their specific biological functions has remained a highly active area of research [19].
Noteworthy in the historical context, the CYP branch of eicosanoid formation was discovered almost 50 years after recognizing the essentiality of PUFAs in the mammalian diet, 20 years after elucidating the enzymatic formation and structure of prostaglandins [20], 10 years after aspirin-like antiinflammatory and analgesic drugs were shown to act by inhibiting prostaglandin formation [21], and shortly after understanding the biosynthetic pathway of leukotrienes and their roles in inflammation and asthma [22]. Moreover, at that time, microsomal CYP enzymes were investigated primarily because of their recognized roles in drug and xenobiotic metabolism and their corresponding importance in pharmacology and toxicology. Thus, the discovery of CYP eicosanoids and their potential roles in the pathophysiology of hypertension indicated that microsomal CYP enzymes may be involved in important biological actions beyond drug metabolism and raised the hope of finding novel mechanisms regulating cardiovascular and renal function.
6.2 Reaction and Substrate Specificity of PUFA-Metabolizing CYP Enzymes in Human, Rat and Mouse
6.2.1 CYP Enzymes Involved in 20-HETE Generation
20-HETE is produced by ω-hydroxylation of AA. The capacity of catalyzing this reaction type is widespread among members of the CYP4A and CYP4F subfamilies [17, 23]. In addition to 20-HETE, typically minor amounts of 19-HETE are also generated. The resulting 20-HETE/19-HETE ratio may range from more than 20:1 to 8:1 and is an inherent feature of the individual CYP4A and CYP4F enzymes.
In the human, CYP4A11 and CYP4F2 contribute to renal and hepatic 20-HETE formation [24, 25]. A functional variant of CYP4A11 characterized by phenylalanine-to-serine substitution at amino acid position 434 is associated with essential hypertension (T8590C polymorphism of the CYP4A11 gene, compare Sect. 6.4.1) [26]. CYP4A22, the only other member of the human CYP4A subfamily, lacks hydroxylase activity, presumably due to an amino acid substitution at position 130 that is occupied by glycine in all other CYP4A enzymes but by serine in CYP4A22 [26]. However, recently discovered genetic polymorphisms of CYP4A22 include potential gain-of-function mutations (Gly130Ser) making this gene of particular interest for understanding interindividual differences in 20-HETE production [27, 28]. CYP4F3, originally identified as leukotriene B4 (LTB4) ω-hydroxylase in human blood cells, is a further interesting candidate for the production of 20-HETE in man. Alternative splicing of the CYP4F3 pre-mRNA occurs in the liver, kidney and other tissues resulting in a shift of substrate specificity of the mature enzyme from LTB4 to AA [29, 30]. In vitro, the corresponding CYP4F3B variant displayed significantly higher AA ω-hydroxylase activity than CYP4A11 and CYP4F2 [31].
Recently, CYP2U1, a human CYP enzyme specifically expressed in the thymus and brain, was shown to function as an ω- and (ω−1)-hydroxylase of AA and other PUFAs [32], indicating that also CYP enzymes beyond the CYP4A and CYP4F subfamily members can contribute to 20-HETE production, in particular, in less investigated tissues and physiological conditions. However, exciting novel results can also be expected identifying the endogenous substrates and reaction specificities of CYP4V2 and CYP4F12, the still “orphan” members of the human CYP4 family. Polymorphisms in CYP4V2 and CYP4F12 genes are associated with ocular (Bietti’s crystalline corneoretinal dystrophy) and skin disease (lamellar ichthyosis), respectively [33].
The rat genome encodes four members of the CYP4A subfamily. Among them, CYP4A1 is the most active AA ω-hydroxylase followed by CYP4A2, CYP4A3 and CYP4A8 [34, 35]. Rat CYP4F enzymes shown to generate 20-HETE include CYP4F1 and CYP4F2 [36]. Based on protein expression data and immunoinhibition experiments, it has been suggested that CYP4A1 is the major AA ω-hydroxylase in the rat heart and kidney, whereas CYP4A2 and/or CYP4F1/4 are the major 20-HETE producing enzymes in the rat lung and liver [37]. In the rat kidney, CYP4A1, CYP4A2 and CYP4A3 are expressed both in different segments of the nephron and in preglomerular arterioles [17, 38]. CYP4A8 was specifically localized to the renal and cerebral vasculature, where its enhanced expression is associated with androgen-induced hypertension in the normal rat and the severity of ischemic stroke in SHR, respectively [39, 40].
Compared to human and rat, the mouse genome contains the most extended cluster of CYP4A genes (http://drnelson.uthsc.edu/4ABX.2005.rat.pdf). The individual genes are located within the so-called Cyp4abx cluster on chromosome 4 [41]. Among the functional Cyp4a enzymes identified, Cyp4a12a is the predominant 20-HETE generating enzyme in the kidney of male mice [42]. In comparison, Cyp4a10 that is expressed in both male and female mice displays only a weak AA ω-hydroxylase activity. The female-specific Cyp4a14 lacks the ability of hydroxylating AA but shows significant ω-hydroxylase activity with lauric acid as substrate [42]. Surprisingly, Cyp4a14 gene disruption resulted in increased renal AA ω-hydroxylase activities and caused hypertension in male mice [43]. The mechanism obviously involves increased plasma androgen levels in the Cyp4a14 gene-disrupted mice followed by androgen-induced upregulation of the 20-HETE producing Cyp4a12. Subsequent studies proved that Cyp4a12a overexpression increases 20-HETE levels in preglomerular arterioles and is alone sufficient to elevate blood pressure in mice [44]. Providing a further example of the complex regulation of CYP-eicosanoid formation in mice, deletion of the Cyp4a10 gene caused salt-sensitive hypertension, associated with impaired regulation of the EET-generating Cyp2c44 and of the kidney epithelial sodium channel [45]. The mouse kidney also expresses a series of Cyp4f enzymes. However, their ability to metabolize AA has not yet been demonstrated [46].
6.2.2 CYP Enzymes Involved in EET Generation
Studies with purified or recombinant CYP enzymes demonstrated that, in particular, various members of the CYP2C and CYP2J subfamilies can function as AA epoxygenases [17, 47]. The CYP2C (compare: http://drnelson.uthsc.edu/rat2C.pdf) and CYP2J subfamilies (compare: http://drnelson.uthsc.edu/2Jrat.pdf) evolved differently in human, rat and mouse, making it difficult to identify orthologous genes and to transfer results from animal studies directly to human cardiovascular disease [41].
In the human, the CYP2C subfamily consists of four members (CYP2C8, CYP2C9, CYP2C18 and CYP2C19) and there is only a single CYP2J gene (CYP2J2). All corresponding CYP enzymes are able to produce EETs but differ in their catalytic activities, regio- and stereoselectivities as well as tissue specificities of expression. CYP2C8 and CYP2C9 have been considered as the major source of EETs in the human kidney and liver [48]. CYP2C8 generates 11,12- and 14,15-EET in a ratio of about 1.25:1 and preferentially produces the R,S enantiomers of both metabolites with a selectivity greater than 80 % [49, 50]. In porcine coronary arteries, antisense oligonucleotides downregulating a CYP2C8-related enzyme decreased bradykinin-induced EET formation and vascular relaxation [51]. This experiment provided direct evidence for the involvement of CYP2C enzymes in vascular EET formation and confirmed the concept that EETs function as endothelium-derived hyperpolarizing factors (EDHF) in various vascular beds [52–54]. Endothelial-specific overexpression of CYP2C8 lowers blood pressure and attenuates hypertension-induced renal injury in mice [55]. Surprisingly, the same CYP2C8 transgenic mice are more susceptible to myocardial infarction injury than wild-type (WT) mice [56]. This detrimental effect was explained by CYP2C8-mediated enhanced formation of reactive oxygen species (ROS) and cardiodepressive linoleic acid (LA) metabolites [56]. Compared to CYP2C8, CYP2C9 is less regio- and stereoselective and metabolizes AA to mixtures of 8(S),9(R)-, 11(S),12(R)- and 14(R),15(S)-EETs with optical purities of 66, 69 and 63 %, respectively [49, 50]. In addition to its ability of producing EETs, CYP2C9 was identified as a functionally significant source of ROS in coronary arteries [57]. In line with this finding, inhibition of CYP2C9 with sulfaphenazole improves endothelium-dependent, nitric oxide–mediated vasodilatation in patients with coronary artery disease [58]. The few studies with CYP2C18 and CYP2C19 show that these enzymes produce 8,9-, 11,12- and 14,15-EET [59, 60].
CYP2J2 has been identified as the major AA epoxygenase of the human heart [61] but is also expressed in other tissues including the vasculature, gastrointestinal tract and islets of Langerhans cells in the pancreas [62]. CYP2J2 generates all four regioisomeric EETs. The enzyme shows enantioselectivity in producing 14(R),15(S)-EET with an optical purity of 76 % but forms 8,9- and 11,12-EET as racemic mixtures [61]. Compared to CYP2C subfamily members, recombinant CYP2J2 displays rather weak enzymatic activities [60]. Nonetheless, transgenic mice with tissue-specific overexpression of CYP2J2 were developed as one of the most successful tools for studying the diverse beneficial effects of enhanced endogenous EET formation in cardiovascular disease [63, 64] (compare Sect. 6.4). Unlike CYP2C8 and CYP2C9, CYP2J2 is presumably not a relevant source of ROS [65].
The rat genome harbors 11 functional CYP2C genes. CYP2C23 has been identified as the predominant renal AA epoxygenase [66, 67]. This enzyme produces 8,9-, 11,12- and 14,15-EET in a ratio of 1:2:0.7. The enzyme shows a high degree of stereoselectivity and generates 8(R),9(S)-, 11(R),12(S)- and 14(S),15(R)-EET with optical purities of 95, 85, and 75 % [66]. CYP2C23 protein expression and activity is upregulated in the rat kidney upon excessive dietary salt intake [68]. A deficiency in CYP2C23-mediated renal EET formation is associated with the development of angiotensin II-induced hypertension and renal failure in the rat [69–71]. CYP2C11 was identified as the major AA epoxygenase in the liver of male rats [72]. However, CYP2C11 is also expressed in the heart, kidney and lung [37]. Moreover, CYP2C11 attracted particular interest as an EET-generating CYP enzyme in astrocytes and its potential role in the regulation of cerebral blood flow [73, 74]. Compared with CYP2C23, CYP2C11 is less regio- and stereoselective. CYP2C11 metabolizes AA to 8,9-, 11,12- and 14,15-EETs and also produces significant amounts of mid-chain HETEs [68].
The rat CYP2J gene cluster comprises five functional genes. Among them, CYP2J3 has been identified as a major AA epoxygenase in the heart [75]. Recombinant CYP2J3 metabolized AA to 14,15-, 11,12- and 8, 9-EETs and 19-HETE as the principal reaction products [75]. CYP2J4 is expressed in rat liver, intestine, olfactory mucosa, kidney, heart, and lung and can contribute to EET and HETE formation in these organs [76, 77].
Analysis of the mouse genome indicated the presence of 15 functional Cyp2c genes. Among them, Cyp2c44 is the enzyme most closely related to rat CYP2C23 [78]. Cyp2c44 metabolizes AA primarily to 8,9-, 11,12- and 14,15-EETs in a ratio of about 1:3:1 and shows a high stereoselectivity in producing the R,S-enantiomers of 8,9- and 11,12-EET with optical purities of 95 and 94 %, respectively [78]. Cyp2c44 is expressed in the liver, kidney and adrenals. Recent studies on Cyp2c44 knockout mice revealed an important role of this enzyme in the regulation of renal tubular salt reabsorption via EET-mediated inhibition of the epithelial sodium channel (ENaC) [79]. This function of Cyp2c44-derived EETs is essential for dopamine-induced natriuresis/diuresis and for preventing sodium reabsorption and hypertension in response to high dietary potassium intake [80, 81]. Other members of the mouse CYP2C subfamily shown to metabolize AA primarily to EETs include Cyp2c29, Cyp2c38, Cyp2c39, Cyp2c50 and Cyp2c54 [82, 83]. Cyp2c37 metabolizes AA to 12-HETE [82]. Cyp2c55 produces both EETs and HETEs [83]. Cyp2c40, a major Cyp2c enzyme expressed in the murine gastrointestinal tract, produces 16-HETE > 14,15-EET ≫ 8,9-EET > 11,12-EET in a moderate stereoselective manner with preference for 16(R)-HETE (66 %), 14(R),15(S)-EET (62 %), 11(S),12(R)-EET (70 %) and 8(S),9(R)-EET (86 %) [84]. The biological functions of most of these murine Cyp2c enzymes have not been characterized. Cyp2c29 apparently resembles human CYP2C9 regarding its capacity of producing both EETs and ROS in the vasculature [85]. Moreover, Cyp2c29 is involved in hypoxic pulmonary vasoconstriction [86]. Recently, Cyp2c knockout mice were developed by deleting the whole Cyp2c gene cluster [87]. This model can become important for studying the in vivo functions of Cyp2c genes and for establishing transgenic mice expressing selected human CYP2C enzymes.
Seven functional CYP2J genes (Cyp2j5, Cyp2j6, Cyp2j8, Cyp2j9, Cyp2j11, Cyp2j12 and Cyp2j13) are predicted by the sequence of the mouse genome [41]. Among them, Cyp2j5 has been most extensively characterized. Recombinant Cyp2j5 metabolizes AA to 14,15-, 11,12- and 8,9-EETs and 11- and 15-HETE [88]. Renal expression of Cyp2j5 is upregulated by androgens and downregulated by estrogens [89]. Female Cyp2j5 knockout mice show reduced plasma 17β-estradiol levels and increased blood pressure that can be normalized by estrogen replacement [90]. Recombinant Cyp2j6 was inactive with AA as substrate but metabolized benzphetamine [91]. Cyp2j9 was identified as an AA (ω−l)-hydroxylase predominantly expressed in the mouse brain [92]. Recently, the remaining four members of the murine Cyp2j subfamily (Cyp2j8, Cyp2j11, Cyp2j12 and Cyp2j13) were also cloned and heterologously coexpressed with the human NADPH-CYP oxidoreductase in insect cells [93]. The recombinant enzymes metabolized AA as well as LA (18:2 n-6) to enzyme-specific sets of epoxy and hydroxy metabolites [93].
Based on the studies summarized above, members of the CYP2C and CYP2J subfamilies are clearly the first candidates when searching for the identity of AA epoxygenases involved in the generation of biologically active EETs. However, it is important to note that other CYP enzymes share this catalytic ability [47]. Providing an example, Cyp2b19, a CYP enzyme specifically expressed in mouse skin keratinocytes, metabolizes AA and generates 14,15- and 11,12-EETs, and 11-, 12- and 15-HETEs. Cyp2b19-catalyzed AA metabolism is highly stereoselective for 11(S),12(R)- and 14(S),15(R)-EET, and 11(S)-, 12(R)- and 15(R)-HETE [94]. Cyp2b19 is the major source of endogenous EETs in mouse skin [95] and its enzymatic action can contribute to the regulation of epidermal cornification [96]. CYP2B12 is presumably the rat homolog of murine Cyp2b19 [97]. The human epidermis expresses various genes of the CYP1–4 families including CYP2B6, but the functional counterpart to Cyp2b19 remains to be identified [98]. In contrast to murine Cyp2b19, CYP2B6, the single representative of the CYP2B subfamily in humans, shows only a very weak AA epoxygenase activity [48]. Interestingly, however, CYP2B6 is remarkably active in epoxidizing the 14,15-and 11,12-double bonds of N-arachidonoylethanolamine (anandamide) [99]. Extending the uncertainties in predicting the reaction specificity of individual CYP enzymes solely based on their subfamily membership, human CYP1A2 also functions predominantly as an AA epoxygenase [48, 59]. Moreover, CYP2S1, one of the most recently discovered human CYP enzymes, is expressed in macrophages and metabolizes AA to EETs [100].
6.2.3 CYP Enzymes Involved in Subterminal AA Hydroxylation
The principle metabolites generated by subterminal hydroxylation are 16-, 17-, 18- and 19- HETE. Human CYP enzymes preferentially metabolizing AA to 19-HETE include CYP1A1 and CYP2E1. CYP1A1 generates 19-, 18-, 17- and 16-HETE in a ratio of 5:3:1:1.5, and also minor amounts of 14,15-EET [101]. CYP2E1 metabolizes AA predominantly to 19 and 18-HETE comprising 46 and 32 % of the total products formed [102]. CYP2E1 produces 19(R)-and 19(S)-HETE in a ratio of about 70:30 and 18(R)-HETE with an optical purity of essentially 100 % [102]. 19-HETE counteracts the vasoconstrictory and proinflammatory effects of 20-HETE [103], suggesting that changes in vascular CYP1A1 and CYP2E1 expression may contribute to the regulation of blood pressure. In line with this hypothesis, the SHR model of genetic hypertension shows reduced CYP2E1 expression [104]. As mentioned above, murine Cyp2j9 provides a unique example of an enzyme that almost exclusively metabolizes AA to 19-HETE [92], whereas other CYP2J subfamily members function predominantly as epoxygenases and produce 19-HETE only as a minor product. CYP4F8 and CYP4F12, two human CYP enzymes primarily involved in prostaglandin metabolism, metabolize AA by (ω−2)/(ω−3)-hydroxylation and produce 18-HETE as the main product [105]. Murine Cyp2c40 is currently the only CYP enzyme known to convert AA predominantly to 16-HETE [84].
6.2.4 CYP Enzymes Involved in the Generation of Mid-Chain HETEs
The enzymatic mechanism and biological significance of CYP-catalyzed AA conversion to mid-chain HETEs (5-, 8-, 9-, 11-, 12- and 15-HETE) is only partially understood. This class of CYP-dependent AA metabolites could be directly formed by hydroxylation with double bond migration or by bisallylic oxidation at C7, C10 or C13 followed by rearrangement to the corresponding dienols [106–108]. Mid-chain HETEs were identified as products of NADPH-dependent AA metabolism by liver microsomes as well as various recombinant CYP enzymes including CYP1A2, CYP2C8, CYP2C9 and CYP3A4 [109, 110]. In general, CYPs producing mid-chain HETEs function simultaneously as AA epoxygenases. A CYP enzyme exclusively catalyzing this reaction type has not yet been identified. Among the mid-chain HETEs generated by CYP enzymes, 12(R)-HETE attracted particular attention. 12-HETE can be further metabolized to a keto intermediate followed by a keto-reduction reaction yielding the dehydro-metabolite, 12-hydroxyeicosatrienoic acid (12-HETrE) [111]. 12-HETrE was detected in human tear film and follow-up studies in animal models implicated CYP4B1 and 12-HETrE as important components in corneal inflammation and neovascularization [112–114].
6.2.5 Long-Chain Omega-3 Fatty Acids as Alternative Substrates of AA Metabolizing CYP Enzymes
Traditionally, AA (20:4 n-6) has been considered as the main precursor of CYP eicosanoids. However, AA metabolizing CYP enzymes show rather broad substrate specificities and are able to function as hydroxylases or epoxygenases with virtually all PUFAs of both the n-6 and n-3 families (Fig. 6.2) [115]. Which of the various PUFAs becomes accessible and is actually metabolized largely depends on (1) the relative abundance of the individual PUFAs; (2) the substrate specificity of the phospholipases that release free PUFAs from membrane phospholipids and thus initiate their metabolism by CYP enzymes and other eicosanoid generating oxygenases; and (3) the substrate and reaction specificities of the CYP enzymes expressed in a given tissue.

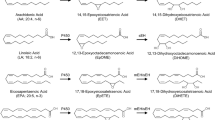
Biosynthesis of long-chain n-6 and n-3 PUFAs. The biosynthetic pathway of long chain n-6 and n-3 polyunsaturated fatty acids provides a series of substrates that can be metabolized by CYP epoxygenases and CYP hydroxylases (For further details, compare Sect. 6.2.5)
To (1)
Mammals unlike plants, marine phytoplankton and nematodes are unable to produce and interconvert n-6 and n-3 PUFAs [116–118]. Accordingly, these two PUFA families are essential components of the mammalian diet and the relative abundance of individual PUFAs in the body is determined by their dietary intake and subsequent tissue-specific mechanisms of metabolism, distribution and uptake [119]. “Western diets” typically contain n-6 and n-3 PUFAs in a ratio of about 15:1, whereas the genetic constitution of our ancestors presumably evolved in a nutritional environment with an n-6/n-3 PUFA ratio of nearly 1:1 [120]. Importantly, the relative deficiency of n-3 PUFAs in the modern human diet has been linked to an increased risk of cardiovascular disease and inflammatory disorders [116, 121–123].
AA is directly available from meat and dairy products or can be synthesized from linoleic acid (LA; 18:2 n-6) that is abundant in vegetable oils (Fig. 6.2). In line with the prevalence of n-6 PUFAs in the “Western diet”, AA is indeed the predominant long-chain PUFA in most organs and tissues, except the brain and retina that are able to largely maintain high levels of docosahexaenoic acid (DHA; 22:n-6) even when the diet provides only small amounts of n-3 PUFAs [124–126]. Long-chain n-3 PUFAs, such as DHA and eicosapentaenoic acid (EPA; 20:5 n-3), can be synthesized from alpha-linolenic acid (ALA; 18:3 n-6) that is contained among others in leafy green vegetables (Fig. 6.2). However, the enzymatic steps converting ALA to EPA and further to DHA have limited efficiencies in human [119]. Fish oil and other seafood are a rich direct source of EPA and DHA due to the marine food chain starting with EPA/DHA producing phytoplankton [117]. Based on the accumulating evidence showing that EPA and DHA have beneficial effects in various cardiac disorders, the use of EPA/DHA supplements is recommended for the management of patients after myocardial infarction and for the treatment of hyperlipidemia [123, 127].
To (2)
Under basal conditions, AA is predominantly esterified into the sn-2 position of membrane phospholipids and thus not accessible to CYP enzymes and other eicosanoid generating oxygenases. However, free AA becomes readily available in response to extracellular stimuli that activate phospholipases A2 (PLA2) that in turn release AA from the membrane stores [128]. In most tissues, extracellular signal-induced activation of the cytosolic calcium-dependent cPLA2 initiates AA release and eicosanoid formation. EPA and DHA are also incorporated into the sn-2 position of membrane phospholipids and thereby partially replace AA. The classical cPLA2 releases AA and EPA with almost equal efficiencies but is largely inactive in liberating DHA [129]. In the brain, AA and DHA are released by different mechanisms using cPLA2 for AA and a calcium-independent phospholipase A2 (most likely iPLA2β) for DHA [130, 131]. The identity of the PLA2 enzymes releasing DHA in other tissues remains to be elucidated. Recently, the endogenous levels of oxidized PUFA metabolites were compared in the livers of iPLA2γ knockout and WT mice. Interestingly, deletion of iPLA2γ was associated with a marked decrease of DHA- but not of LA- or AA-derived CYP epoxygenase metabolites [132].
To (3)
The capacity of CYP enzymes to oxidize EPA and DHA was first shown with rat renal and hepatic microsomes [133, 134]. Recent studies with recombinant CYP enzymes clearly demonstrate that, in fact, all major AA metabolizing CYP enzymes accept these n-3 PUFAs as efficient alternative substrates (Table 6.1) [31, 59, 60, 135, 136].
CYP2C and CYP2J enzymes that epoxidize AA to EETs also metabolize EPA to epoxyeicosatetraenoic acids (EEQs) and DHA to epoxydocosapentaenoic acids (EDPs). The (ω−3) double bond distinguishing EPA and DHA from AA is the preferred site of attack by most of the classical CYP epoxygenases. CYP2C enzymes are in general almost equally efficient when utilizing AA, EPA or DHA as substrates. Surprisingly, however, EPA is the clearly preferred substrate of CYP2J2, which is the predominant AA epoxygenase in the human heart. Moreover, CYP2J2 shows only a moderate regiospecificity when metabolizing AA but predominantly produces 17,18-EEQ from EPA.
CYP4A and CYP4F enzymes, hydroxylating AA to 20-HETE, metabolize EPA to 20-hydroxyeicosapentaenoic acid (20-HEPE) and DHA to 22-hydroxydocosahexaenoic acid (22-HDoHE). Human CYP4A11 is most active with EPA, whereas CYP4F2 prefers DHA over AA and EPA [60]. CYP4A enzymes display remarkably increased (ω−1)-hydroxylase activities when metabolizing EPA or DHA instead of AA. Moreover, some of them even attack the (ω−3) double bond. For example, CYP4A11 metabolizes AA to 20-HETE and 19-HETE in a ratio of 82:18, EPA to 20-HEPE, 19-HEPE and 17,18-EEQ in a ratio of 28:62:10 and DHA to 22-HDoHE, 21-HDoHE and 19,20-EDP in a ratio of 48:44:8 [60]. Similarly, murine recombinant Cyp4a12a hydroxylates AA to 20-HETE and 19-HETE (80:20) but metabolizes EPA to 20-HEPE, 19-HEPE, and 17,18-EEQ in a ratio of 12:32:56 [42]. CYP1A1, CYP2E1 and other enzymes converting AA predominantly to 19-HETE or 18-HETE (CYP4F8 and CYP4F12) show pronounced (ω−3)-epoxygenase activities with EPA and DHA (for references, see Table 6.1).
Taken together, it can be concluded that the capacity of utilizing EPA and DHA as alternative substrates is shared by virtually all of the AA-metabolizing CYP enzymes belonging to the subfamilies 1A, 2C, 2E, 2J, 2U, 4A and 4F. The CYP enzymes generally respond to the altered double-bond structure and chain-length of their fatty acid substrates with remarkable changes in the regioselectivity and, in part, also in the type of the catalyzed oxygenation reaction. Moreover, 17,18-EEQ and 19,20-EDP, the unique epoxy metabolites of EPA and DHA, are formed with pronounced stereoselectivities [137]. CYP1A1, CYP1A2, CYP2E1, CYP2C9, CYP2C11, CYP2C19, CYP2C23 and CYP2J2 as well as murine Cyp4a12a and rat CYP4A1 preferentially generate the corresponding R,S enantiomers, whereas CYP2C8 and CYP2D6 show stereoselectivities in favor of producing 17(S),18(R)-EEQ and 19(S),20(R)-EDP [42, 135–137]. Noteworthy, CYP1A1 metabolizes AA to 19-HETE as the main product and epoxidizes EPA to 17(R),18(S)-EEQ with an optical purity greater than 98 % [101]. These substrate-dependent features of the PUFA metabolizing CYP enzymes may have important physiological implications, considering that the biological activities of CYP eicosanoids are dependent on the regio- and stereoisomeric position of their functional epoxy or hydroxy groups.
6.2.6 Effect of Dietary Omega-3 Fatty Acids on the Endogenous CYP-Eicosanoid Profile
First studies investigating the effects of marine omega-3 fatty acids (EPA and DHA) on eicosanoid formation were focused on potential changes in the production and activity of COX- and LOX-dependent metabolites. These studies were stimulated by the seminal observation in the 1970s of significantly lower myocardial infarction rates in Greenland Inuit’s, who traditionally live on EPA/DHA-rich sea food, compared to Danish controls [138]. Subsequent world-wide epidemiological studies revealed the general existence of striking cardiovascular mortality differences between populations living on n-6 PUFA- versus n-3 PUFA-rich diets [121]. Giving first insight into the mechanisms that might explain the low myocardial infarction rate among Inuit’s, EPA was shown to compete with AA yielding less proaggregatory (thromboxane A3 versus thromboxane A2) and less proinflammatory eicosanoids (leukotriene B5 versus leukotriene B4) via the COX- and LOX-dependent pathways [138, 139]. In contrast, prostacyclin I3, formed from EPA, acts with the same potency as vasodilator and inhibitor of platelet aggregation as its AA-derived counterpart prostacyclin I2. Indeed, a favorable shift of the thromboxane/prostacyclin ratio to a more antiaggregatory and vasodilatory state was shown in Intuits as well as in persons after long-term intake of high amounts of EPA (10–15 g/day) [140, 141]. Many of these studies were performed before the discovery of CYP eicosanoids and of other novel classes of lipid mediators, such as the resolvins, that could bring new twists in the search for EPA- and DHA-derived metabolites mediating the cardiovascular benefits of marine omega-3 fatty acids [142].
First evidence for the in vivo formation of EPA- and DHA-derived CYP epoxygenase metabolites was provided by the detection of EEQs and EDPs in human urine and plasma samples [143, 144]. Marked increases in the plasma levels of EPA- and DHA-derived epoxides and their vicinal diols were observed in healthy volunteers treated for 4 weeks with 4 g of an EPA/DHA-supplement [144] and in asthmatic patients who received for 3 weeks 4 g EPA + 2 g DHA per day [145]. Considering that AA remained the predominant long-chain PUFA despite EPA/DHA supplementation, these studies indicate that EPA and DHA were metabolized in vivo with significantly higher relative efficiencies compared to AA. Even without dietary intervention, the individual differences in the serum concentrations of EPA-derived CYP epoxygenase metabolites correlated well with the EPA content in red blood cells as shown in a recent study comparing the metabolite profiles in hyper- and normolipidemic humans [146].
We analyzed EPA/DHA-supplementation induced tissue-specific changes of the endogenous CYP-eicosanoid profile in the rat [60]. The animals received standard chow supplemented with 5 % sunflower oil (n-6 PUFA-rich diet), or additionally with 2.5 % OMACOR®-oil (a formulation of EPA/DHA-ethylesters containing 480 mg EPA and 360 mg DHA/g). The n-6 PUFA-rich diet resulted in a 10–20-fold excess of AA over EPA and DHA in most organs and tissues except the brain that maintained an almost 1:1 ratio of AA and DHA. In the heart, the AA content was about sevenfold higher than that of EPA + DHA. After EPA/DHA supplementation, the AA levels were generally reduced by 40–50 % and partially replaced by EPA and DHA in a tissue-specific manner. These changes in the relative PUFA levels correlated with marked changes in the endogenous CYP-eicosanoid profile (Table 6.2). For example, the ratio of EETs:EEQs:EDPs was shifted from 93:0:7 to 49:34:17 in the kidney and from 86:0:14 to 26:13:61 in the left ventricle. EPA/DHA-supplementation also modulated the endogenous formation of ω-hydroxylase products and resulted in a tissue-specific replacement of 20-HETE for 20-HEPE and 22-HDoHE (Table 6.2). The corresponding metabolite/precursor fatty acid ratios indicate that the CYP epoxygenases expressed in the different tissues metabolized EPA with a two to fourfold higher efficiency and DHA with almost the same efficiency, compared with AA [60]. Noteworthy, 17,18-EEQ and 19,20-EDP became the predominant CYP epoxygenase metabolites in most tissues. This finding is in line with the intrinsic feature of many individual CYP enzymes to catalyze preferentially the epoxidation of the (ω−3) double bond when having access to EPA and DHA as substrates (compare Sect. 6.2.5). Moreover, also a recent study in growing piglets identified the vicinal diols of 17,18-EEQ and 19,20-EDP as the epoxygenase-derived metabolites most markedly increased upon dietary n-3 PUFA supplementation [147].
Taken together, these studies demonstrate that the formation of endogenous CYP eicosanoids is highly susceptible to changes in the dietary n-6/n-3 PUFA ratio. Thus, the traditional view that AA is the main source of biologically active epoxy and hydroxy metabolites applies primarily to human populations and laboratory animals living on n-6 PUFA-rich (“Western”) diets. However, EPA and DHA may readily become superior sources of CYP-dependent eicosanoids upon n-3 PUFA supplementation or a high dietary intake of fish oil and other marine foodstuffs.
The currently known biological activities of EPA- and DHA-derived CYP metabolites partially resemble those of their AA-derived counterparts, appear in part unique or can even produce opposite effects [148]. The epoxy metabolites of all three PUFAs share vasodilatory properties. However, the potencies of EEQs and EDPs may largely exceed those of EETs in some vascular beds [135, 149]. Interestingly, Cyp1a1 knockout mice display increased blood pressure presumably due to a reduced capacity of producing vasodilatory metabolites from n-3 PUFAs [150]. Potential candidates generated by Cyp1a1 are 17,18-EEQ and 19,20-EDP that efficiently relax murine aortic segments when added at picomolar concentrations [150]. Antiinflammatory effects were first revealed for 11,12- and 14,15-EET but are also exerted by EPA epoxides as exemplified by 17,18-EEQ [151, 152]. 17,18-EEQ and 19,20-EDP inhibit the Ca2+- and isoproterenol-induced increased contractility of neonatal cardiomyocytes, indicating that these metabolites may act as endogenous antiarrhythmic agents [60]. Whereas certain EET regioisomers promote tumor angiogenesis and metastasis, 19,20-EDP and other regioisomeric DHA epoxides inhibit these crucial events in cancerogenesis [153, 154]. Moreover, CYP-dependent EPA- and DHA-derived epoxy metabolites were identified as potent antihyperalgesic agents in an animal model of pain [155]. These findings suggest that EPA- and DHA-derived CYP eicosanoids may serve as mediators in a variety of beneficial effects attributed to fish oil n-3 PUFAs, such as protection against cardiovascular disease, sudden cardiac death and tumor development [123, 156].
6.3 De Novo Biosynthesis and Metabolic Fate of CYP Eicosanoids
6.3.1 CYP Eicosanoids as Second Messengers
As already discussed in Sect. 6.2.5, AA, EPA or DHA become only accessible as substrates to the CYP enzymes after being released from membrane phospholipids. In this way, in vivo generation of CYP eicosanoids is normally strictly coupled to extracellular signals that trigger the activation of phospholipases, which in turn release the potential substrates from intracellular membrane stores. Accordingly, CYP eicosanoids are typically formed as second messengers of diverse hormones, cytokines and growth factors [17]. Examples include bradykinin-induced EET formation in endothelial cells as part of the vasodilatory response [53], VEGF-induced EET formation in angiogenesis [157], and angiotensin II-induced 20-HETE formation in vasoconstriction of renal arterioles [158]. After de novo synthesis, CYP eicosanoids elicit cell type specific signaling pathways but are also subject to rapid further metabolism that may lead to (1) the generation of membrane pools of preformed CYP eicosanoids; (2) the formation of secondary metabolites with new biological activities; or (3) inactivation and degradation (Fig. 6.3).

Biosynthesis and metabolic fate of 17,18-EEQ. The biosynthesis of 17,18-EEQ is initiated by extracellular signals that activate phospholipases A2 (PLA2) in the given tissue. The PLA2 enzymes liberate eicosapentaenoic acid (EPA) from phospholipid (PL) stores and make free EPA accessible as substrate to the CYP enzymes. After its de novo synthesis, 17,18-EEQ triggers intracellular signaling pathways and can be further metabolized via various routes leading to its storage in membrane phospholipids, the formation of secondary metabolites with novel biological activities or to inactivation and degradation (For further details, see Sect. 6.3)
6.3.2 Storage and Release
Unlike COX-dependent prostanoids, CYP-dependent hydroxy and epoxy metabolites are partially re-esterified into the sn-2 position of glycerophospholipids, generating a membrane pool of preformed CYP eicosanoids that is also accessible to PLA2 enzymes [159–161]. This unique feature of CYP eicosanoids is particularly important for their release and action in ischemia/reperfusion injury (compare Sect. 6.4).
6.3.3 Formation of Secondary Metabolites with New Biological Activities Through Actions of COX, LOX and CYP Enzymes
Several CYP eicosanoids such as 20-HETE, 5,6-EET or 17,18-EEQ still contain the double bond structure required for cyclooxygenation and can indeed serve as substrates of COX enzymes [16, 162, 163]. Depending on the COX enzymes and isomerases expressed in a given tissue, this route can result in the formation of 20-hydroxy, 5,6-epoxy or 17,18-epoxy analogs of diverse prostanoid subfamilies including prostaglandins, prostacyclins and thromboxanes. COX-dependent 20-HETE transformation has been proposed as an important mechanism in the regulation of renal microvascular tone [164]. A recent study showed that the proadipogenic effect of 20-HETE depends on its COX-2 mediated transformation to 20-OH-PGE2 [165]. The vasoactivity of 5,6-EET is dependent on the vascular bed and may consist of a vasodilator component of the primary metabolite and a vasoconstrictor component due to COX-dependent secondary metabolite formation [166, 167].
17,18-EEQ provides a thus far unique example of a CYP epoxygenase metabolite that can be further metabolized by LOX enzymes. Recently, 8-OH, 12-OH and 15-OH-17,18-EEQ were identified as endogenous metabolites in the peritoneal fluid of mice after feeding the animals an EPA-rich diet [168]. In vitro, the individual regioisomers can be enzymatically synthesized incubating 17,18-EEQ with purified 8-LOX, 12-LOX and 15-LOX, respectively. Among them, 12(S)-OH-17,18-EEQ (compare Fig. 6.3) displays highly potent antiinflammatory action by limiting neutrophil infiltration in experimental murine peritonitis. In vitro, 12(S)-OH-17,18-EEQ inhibits neutrophil chemotaxis with an EC50 of 0.6 nM [168]. These remarkable findings suggest that the combined actions of CYP epoxygenases and 12-LOX are an important component of the metabolic cascade mediating the antiinflammatory effects of dietary EPA intake. Another EPA-initiated antiinflammatory pathway uses 18-HEPE as a precursor and leads to formation of the E-series of resolvins that are highly potent mediators in the resolution of inflammation [169]. The enzymatic origin of 18-HEPE may include COX (after binding aspirin) or CYP enzymes, whereby the identity of the latter remains to be clarified in humans and mammals [170].
As shown in Fig. 6.4, CYP epoxygenases and CYP hydroxylases also can cooperate in producing secondary metabolites with unique biological activities. EETs are metabolized by CYP4A and CYP4F enzymes to hydroxy EETs (HEETs) and the same class of metabolites is also efficiently produced by CYP2C-catalyzed epoxidation of 20-HETE [71, 171, 172]. Among the CYP enzymes expressed in the rat kidney, CYP4A1 preferentially hydroxylates 11,12-EET [171], whereas CYP2C23 predominantly epoxidizes the 8,9 double bond of 20-HETE [71]. Importantly, the HEETs formed via both pathways act as high-affinity ligands of the peroxisome proliferator-activated receptor alpha (PPARα) that is involved in the regulation of lipid metabolism as well as the control of inflammation [71, 171].

Renal arachidonic acid (AA) metabolism by CYP4A1 and CYP2C23. CYP4A and CYP2C enzymes produce 20-HETE and EETs as primary products. However, they can also cooperate to generate secondary hydroxy-epoxy metabolites (HEETs) that function as high-affinity ligands of the transcription factor PPARα (For further details, compare Sect. 6.3)
6.3.4 Inactivation and Degradation
EETs and related epoxy metabolites derived from other PUFAs are rapidly degraded to the corresponding vicinal diols by the soluble epoxide hydrolase (sEH) [173, 174]. This mechanism leads to a loss of most of the biological activities attributed to EETs, although the vicinal diols can show, in part, overlapping net effects. EETs incorporated into membrane phospholipids or bound in the cytosol to fatty acid binding proteins are largely protected from enzymatic hydrolysis [175, 176]. The sEH enzyme is highly expressed in all major organs and throughout the cardiovascular system [174]. Its expression is further induced by angiotensin II and thus contributes to decreased EET levels in hypertension and cardiac disease [177, 178]. Over the last decade, pharmacological inhibition of sEH-mediated EET hydrolysis became a highly active field of research with great promise for the prevention and treatment of cardiovascular disease [179] (compare Sect. 6.4).
Resembling the metabolic fate of fatty acids, CYP eicosanoids can also become subject to peroxisomal and mitochondrial β-oxidation but also to chain elongation. Thereby, partial β-oxidation as well as chain elongation may produce metabolites with novel biological activities [180].
6.4 CYP Eicosanoids in Cardiovascular Function and Disease
As described in the Introduction, studies in rat models of genetic hypertension led to the concept that imbalances in CYP eicosanoid formation contribute to the pathogenesis of hypertension and target organ damage. Subsequent studies proved this hypothesis in various other animal models of hypertension (see Table 6.3) and provided mechanistic insight into the partially opposing roles of EETs and 20-HETE in the regulation of vascular, renal and cardiac function [16, 17, 54, 180]. The basic concept was successfully extended and specified to a series of other disease conditions such as ischemia-induced injury of the heart, kidney and brain (Table 6.4), cardiac hypertrophy and arrhythmia (Table 6.5), inflammatory disorders, and atherosclerosis [179, 181–186].
6.4.1 Hypertension and Target Organ Damage
Trying to understand the mechanisms linking CYP-eicosanoid formation to blood pressure regulation, it is helpful to distinguish three major types of alterations in CYP-dependent AA metabolism that are associated with the development of hypertension (compare Table 6.3): (1) increased vascular CYP hydroxylase expression and 20-HETE formation resulting in vasoconstriction and vascular inflammation; (2) decreased renal tubular CYP4A expression and 20-HETE formation resulting in impaired renal function; and (3) decreased CYP epoxygenase and/or increased sEH expression leading to decreased EET levels and resulting in impaired vasodilation and renal salt excretion.
Classical genetic models of hypertension or complex models of secondary hypertension such as angiotensin II-infusion hypertension show combinations of these three basic imbalances in CYP-eicosanoid formation. Moreover, these models frequently do not allow deriving the actual cause-and-effect relationships between the disease state and the associated changes in CYP-eicosanoid formation. These problems have been partially overcome due to the recent progress in developing suitable pharmacological tools that specifically target the formation and action of 20-HETE and EETs. These tools include selective inhibitors of CYP hydroxylases [23], CYP epoxygenases [187], and the sEH [179] as well as synthetic agonists and antagonists of 20-HETE [188, 189] and EETs [190–192]. Moreover, genetic engineering has been increasingly used to dissect the tissue-specific actions of CYP eicosanoids and to prove their significance in the development of cardiovascular disease (compare Table 6.3).
To (1)
Androgen-induced hypertension provides a good example of how pharmacological and genetic interventions can be successfully combined for elucidating the prohypertensive and proinflammatory role of 20-HETE [193]. First, androgen treatment was shown to elevate blood pressure in rats. Indicating an important role of 20-HETE, androgen-induced hypertension was associated with increased vascular CYP4A expression and could be ameliorated by treating the animals with an inhibitor of CYP4A-mediated 20-HETE synthesis [194]. Moreover, adenovirus-mediated vascular overexpression of a 20-HETE generating CYP4A enzyme was alone sufficient to cause hypertension and renal injury in rats [195]. Partially explaining these in vivo observations, 20-HETE has been identified (1) as a potent vasoconstrictor by inhibiting calcium-activated potassium (BK) channels in vascular smooth muscle cells [196] and (2) to promote endothelial dysfunction by uncoupling endothelial nitric oxide synthase (eNOS) and activating the proinflammatory transcription factor NF-κB [197]. Beyond these mechanisms, 20-HETE is able to induce angiotensin-converting enzyme expression resulting in enhanced local and circulating angiotensin II-levels that contribute to the systemic prohypertensive effects of vascular 20-HETE overproduction [198, 199].
Other animal models, where hypertension may rely on similar 20-HETE-mediated mechanisms, include cyclosporine-induced hypertension in rats, androgen-induced hypertension in mice, and blood pressure elevation in Cyp4a14 knockout mice that is associated with an upregulation of androgen-inducible Cyp4a12 (for references, see Table 6.3). Increased urinary 20-HETE levels are associated with endothelial dysfunction in humans indicating that 20-HETE may also play an important role in human vascular pathophysiology [200]. Interestingly, the same study found significantly higher 20-HETE levels in men compared to women.
To (2)
There are two major sites of 20-HETE generation in the kidney: (1) preglomerular microvessels where 20-HETE mediates vasoconstriction by inhibiting BK channels and (2) the renal tubule where 20-HETE promotes salt excretion by inhibiting Na+-K+-ATPase in proximal tubules and the Na+-K+-2Cl− cotransporter in the thick ascending loop of Henle [16, 17, 201]. Accordingly, renal tubular 20-HETE deficiency may contribute to the development of hypertension as first suggested based on studies with salt-sensitive Dahl rats [15]. Proving this hypothesis, treatment with a selective inhibitor of 20-HETE formation was sufficient to promote salt-sensitive hypertension in normal Sprague-Dawley rats [202]. Tubular 20-HETE deficiency is obviously also involved in the development of DOCA-salt induced hypertension in mice [203, 204] and some other animal models.
In humans, the T8590C polymorphism leads to the expression of a functional variant of CYP4A11 with reduced AA ω-hydroxylase activity [26]. Carriers of the C-allele show an increased risk of developing essential and salt-sensitive hypertension [26, 205]. Moreover, the CYP4A11 T8590C genotype was suggested to predict responses to medications that affect sodium homeostasis in hypertensive patients [206]. Functional polymorphisms exist also in the human CYP4F2 gene and were recently shown to associate with hypertension and other components of the metabolic syndrome [207].
To (3)
Under physiological conditions, EETs are involved in the regulation of renal blood flow and salt excretion. EETs mediate vasodilator responses and represent the major EDHF in renal arterioles [53, 201, 208]. In distal tubules, EETs inhibit sodium reabsorption by reducing ENaC activity [79, 209]. Adenosine acting via the adenosine A2A receptor (A2AR) promotes renal EET formation in response to high dietary salt increase. The inability to upregulate this pathway is associated with the development of salt-sensitive hypertension in Dahl salt-sensitive rats [210]. Proving the importance of the adenosine-A2AR-EET axis, salt-resistant rats are rendered hypertensive inhibiting the key components of this pathway [211].
EET deficiency caused by downregulation of CYP epoxygenases and/or upregulation of sEH is also an important feature and mediator of angiotensin II-induced hypertension (Table 6.3). For example, in double transgenic rats overexpressing the human angiotensinogen and renin genes, fenofibrate restored CYP2C23-mediated renal EET formation and prevented the development of hypertension and renal injury [71]. Pharmacological inhibition of the sEH enzyme prevented and reversed angiotensin II-infusion hypertension in mice [212]. Direct evidence for the protective role of CYP epoxygenases in angiotensin II-induced hypertension comes from recent studies using transgenic mice with endothelial specific overexpression of the human enzymes CYP2C8 and CYP2J2 [55, 64].
In humans, circulating levels of 20-HETE are increased and those of EETs are decreased in renovascular disease, whereas the urinary excretion of 20-HETE is reduced [213]. Moreover, as reviewed by other authors, genetic association studies indicate that certain functional polymorphisms in the human CYP2J2 and sEH (EPHX2) genes may be linked to an increased risk of developing hypertension, coronary artery disease and stroke [214–216].
6.4.2 Ischemia-Reperfusion Injury
Ischemia-reperfusion (I/R) induced organ damage is a common feature of myocardial infarction, acute kidney injury and stroke. Early events initiating the pathophysiological cascade include ATP depletion and Ca2+-overload followed by a rapid activation of PLA2 enzymes. Ischemia-induced PLA2 activation plays a critical role in I/R injury of the heart and brain [217–219] and has also been demonstrated in the kidney [220]. PLA2 activation results in the generation of potentially toxic lyso-phospholipids as well as accumulation of free AA that in turn may trigger disturbances in eicosanoid formation in the reperfusion phase. Moreover, the activated PLA2 is able to release preformed CYP eicosanoids from their membrane stores as shown for 20-HETE in the kidney [183]. I/R-induced excessive 20-HETE formation was also shown in the heart [221]. Accumulating evidence from various animal models suggests that 20-HETE plays a major detrimental role in I/R-injury, whereas measures increasing EET formation and action exert strong protective effects (Table 6.4).
Heart
First studies leading to the recognition of the detrimental role of 20-HETE in myocardial infarction were performed in dogs. In canine hearts subjected to coronary artery ligation, inhibition of endogenous 20-HETE formation reduced infarct size, whereas exogenous 20-HETE administration exacerbated the injury [222]. Moreover, inhibition of 20-HETE formation enhances the beneficial effects of ischemic preconditioning (IPC) on the severity of myocardial infarction [223].
Initiating research on the protective role of EETs in the heart, exogenous EET administration to isolated perfused hearts was found to improve postischemic functional recovery and also to prevent electrocardiogram abnormalities in the reperfusion phase [75, 224]. EET pretreatments also efficiently reduced myocardial infarction size after transient coronary artery occlusion [182, 225, 226]. Further studies revealed an essential role of EETs in mediating the beneficial effects of pre- and postconditioning [227–229]. Mimicking the effects of exogenous EET administration, cardiomyocyte-specific overexpression of human CYP2J2 in transgenic mice improved recovery of pump function, and ventricular repolarization after ischemia [63]. In line with the cardioprotective effects of EETs, pharmacological inhibition of the sEH enzyme as well as sEH gene deletion ameliorated myocardial I/R-injury in mice [230]. Moreover, a synthetic EET analog was successfully used for protecting isolated murine hearts against global ischemia-induced loss of pump function and myocardial injury [231].
The potential mechanisms underlying the opposing roles of 20-HETE and EETs have been discussed in recent reviews [182, 232, 233]. Accordingly, the detrimental role of 20-HETE in myocardial I/R-injury is certainly multifactorial and involves vasoconstrictor and proinflammatory actions similar to those discussed above in the context of hypertension and vascular injury (compare Sect. 6.4.1). Moreover, 20-HETE induces inherent mechanisms of apoptosis in cardiomyocytes probably by inhibiting the mitochondrial ATP-sensitive potassium channel. EETs oppose the vasoconstrictor and proinflammatory action of 20-HETE and are able to induce prosurvival mechanisms in cardiomyocytes.
Brain
Studies in rat models of brain I/R-injury demonstrated that blockade of 20-HETE synthesis ameliorates cerebral vasospasm following subarachnoid hemorrhage, and reduces infarct size in ischemic stroke [234, 235]. Pharmacological inhibition as well as genetic deletion of the sEH enzyme is protective in mouse models of ischemic stroke [236]. Interestingly, sex-specific expression of the sEH (male>female) has been linked to the pronounced sex difference in the extent of brain injury after cerebral artery occlusion in mice [237]. Based on these and further findings, sEH has been proposed as a novel therapeutic target in stroke [238]. A recent review gives further information on the potential mechanisms of cerebral I/R-injury that are beneficially modulated upon inhibiting 20-HETE or increasing EET levels by inhibiting the sEH [185].
Kidney
I/R-induced acute kidney injury (AKI) leads to increased morbidity and mortality, particularly after cardiovascular surgery and kidney transplantation [239–241]. I/R-induced mechanisms in the kidney include persistent vasoconstriction, inflammation, endothelial dysfunction, and tubular injury [242, 243]. As analyzed in a rat model of AKI, 20-HETE released in the ischemic phase plays an important role in setting the stage for the subsequent events leading to renal failure. Inhibiting the formation or action of 20-HETE during ischemia improved the recovery of renal tissue perfusion and oxygenation in the early reperfusion phase and protected against subsequent inflammatory cell infiltration, tubular epithelial cell apoptosis and decline of renal function [183]. 20-HETE overproduction also exacerbates the cytotoxic and proapoptotic effects of chemical hypoxia on cultured primary renal tubular epithelial cells [244]. In contrast, protective effects of 20-HETE were observed in another rat model of AKI. In this model, systemic long-term inhibition of 20-HETE formation aggravated and antagonizing 20-HETE action in the reperfusion phase ameliorated renal I/R injury [184]. These apparently contradictory results probably reflect the unique dual role of 20-HETE in the kidney that unlike other organs requires 20-HETE for its normal function.
6.4.3 Cardiac Hypertrophy and Arrhythmia
Maladaptive cardiac hypertrophy is associated with structural and electrical remodeling eventually leading to heart failure and increased propensity to ventricular tachyarrhythmia and sudden cardiac death [245]. This disease may occur upon chronic pressure overload due to aortic stenosis but also develops frequently in more complex disease and stress conditions such as hypertension and myocardial infarction or adrenergic overdrive (Table 6.5).
Indicating an important protective role of CYP epoxy metabolites in pressure overload-induced cardiac hypertrophy, pharmacological sEH inhibition prevents and reverses left ventricular hypertrophy after transverse aortic constriction (TAC) in mice [246]. Pressure overload as well as angiotensin II-induced maladaptive cardiac hypertrophy is ameliorated in sEH knockout compared to wild-type mice [247]. Genetic analysis in rats identified the sEH gene (EPHX2) as a susceptibility factor for heart failure [247]. Interestingly, some of the rat strains used for experimental studies carry an EPHX2 promoter variant that decreases basal expression of the sEH enzyme and abolishes its angiotensin II-inducibility [247]. In mice, upregulation of cardiac sEH expression was shown to be essential for angiotensin II-induced cardiac hypertrophy [178].
Direct experimental evidence for a cardioprotective role of enhanced endogenous EET biosynthesis was provided comparing the development of TAC-induced cardiac hypertrophy in CYP2J2-transgenic mice and corresponding wild-type littermates [248]. Cardiomyocyte-specific overexpression of the human CYP epoxygenase markedly improved the survival of the animals and prevented the development of ventricular tachyarrhythmia vulnerability. Reduced arrhythmia susceptibility was related to CYP2J2-mediated protection against hypertrophy-induced delocalization of left ventricular connexin-43. CYP2J2 transgenic mice also displayed improved electrical remodeling in β-adrenergic stimulation-induced cardiac hypertrophy. In this model, CYP2J2 overexpression specifically prevented the development of fibrosis and atrial fibrillation susceptibility [248]. Other studies with CYP2J2 transgenic mice demonstrate that enhanced cardiac EET biosynthesis also protects against doxorubicin-induced cardiotoxicity [249] and the development of heart failure upon long-term infusion of angiotensin II or isoproterenol [250]. In vitro, exogenous administration of 14,15-EET inhibited the hypertrophic response of cultured cardiomyocytes to isoproterenol, whereas 20-HETE was alone sufficient to induce cellular hypertrophy [251].
Taken together, these studies revealed an important role of CYP eicosanoids in the pathogenesis of cardiac hypertrophy, heart failure and arrhythmia. The mechanisms are only partially understood but obviously include opposing roles of 20-HETE and EETs in mediating or suppressing prohypertrophic, proinflammatory and proapoptotic signaling pathways in cardiomyocytes [252]. Moreover, 20-HETE and EETs modulate ion channel activities in cardiomyocytes and thus influence cardiac electrophysiology and Ca2+-handling [253]. Recently, combined inhibition of 20-HETE formation and of EET degradation was shown to attenuate hypertension and cardiac hypertrophy in Ren-2 transgenic rats [254]. This study provides an example for the role of 20-HETE and EETs in conditions of severe hypertension and end-organ damage and also of the promising therapeutic potential of approaches targeting the CYP-eicosanoid pathway in such complex disease states.
6.5 Conclusions
During the last three decades, the work of many laboratories improved our understanding of the physiological and pathophysiological relevance of the CYP-eicosanoid pathway. Extending the initial discoveries showing important roles in hypertension and renal failure, novel and previously unexpected implications have been revealed in myocardial infarction, maladaptive cardiac hypertrophy, acute kidney injury and stroke. Recent progress in CYP-eicosanoid profiling as well as genetic association studies suggest that much of what has been learned from animal experiments is also relevant to human cardiovascular disease. The list is steadily growing and we have to apologize for not explicitly covering in this review many other exciting findings regarding for example the role of CYP-eicosanoids in the gastrointestinal tract [255], lung [256, 257] and liver [258]. There is also significant progress in understanding the contribution of CYP-eicosanoids to the regulation of neurohormone release and pain sensation [259, 260]. Another important avenue of research led from recognizing EETs as stimulators of insulin secretion in isolated rat pancreatic islets [261] via the identification of CYP2J2 as a major epoxygenase in this cell type [262] to the hypothesis that EETs may be the link between endothelial dysfunction and insulin resistance [263]. The most recent paper in this chain demonstrates beneficial effects of sEH inhibition on glucose homeostasis and islet damage in a streptozotocin-induced diabetic mouse model [264].
Beyond their nowadays well-established roles in cardiovascular health and disease, CYP eicosanoids have been recently recognized as mediators of physiological and pathophysiological forms of angiogenesis [153, 265–269]. On the one hand, these novel findings improve our understanding of repair mechanisms and may open new opportunities for promoting wound healing. On the other hand, these findings indicate that alterations in the CYP-eicosanoid pathway may contribute to tumor proliferation and metastasis, age-related macular degeneration and other disease states associated with pathological angiogenesis. They also suggest that interventions into the CYP-eicosanoid pathway aimed at protecting vascular, cardiac and renal function may have detrimental side effects in promoting cancer progression. This concern was specifically raised against therapeutic strategies that increase the endogenous EET levels because, in particular, these AA-derived metabolites could function as “double-edged swords” [270].
The balance of n-6 and n-3 PUFAs in the diet has been recognized as one of the most important modifiable risk factors for the development of cardiovascular disease but also to influence cancerogenesis and pathologic neovascularization in ocular disease. Studies on the substrate and reaction specificity as well as on diet-induced changes in the endogenous CYP-eicosanoid profile clearly demonstrate that CYP enzymes do not metabolize only AA but also a wide range of other n-6 and n-3 PUFAs. In particular, the CYP-dependent metabolism of EPA and DHA generates sets of epoxy metabolites with superior vasodilatory, antiinflammatory and cardioprotective properties compared to the AA-derived counterparts. First studies also suggest that these n-3 PUFA-derived metabolites have unique biological activities in exerting antiarrhythmic effects [60] and suppressing tumor angiogenesis [154]. It is tempting to speculate but remains to be directly shown that the CYP-eicosanoid pathway mediates a variety of the beneficial effects attributed to diets rich in EPA and DHA.
However, there are also important gaps of knowledge. In particular, our understanding of the CYP-eicosanoid induced signaling pathways is incomplete and hampered by the fact that the primary cellular targets of the diverse epoxy and hydroxyl metabolites have not yet been identified. Eicosanoids generated via the COX- and LOX-dependent pathways exert their biological functions by activating G-protein coupled receptors [18]. Accumulating evidence suggests that there are also receptor-like membrane proteins specifically interacting with individual CYP eicosanoids [271–273] and it will be exciting to learn their molecular identities in the near future. Unexpected help in unraveling the components of CYP-eicosanoid mediated signaling pathways in mammals may also come from studies on PUFA-derived signaling in small animal models such as Caenorhabditis elegans and Drosophila melanogaster [118]. Indicating the existence of evolutionary conserved mechanisms, CYP-33E2, a CYP enzyme resembling the human cardiac epoxygenase CYP2J2, is expressed in Caenorhabditis elegans and contributes there to the regulation of pharynx activity, an organ continuously pumping in nematodes [274]. Moreover, CYP eicosanoids are obviously essential for mediating the behavioral response of Caenorhabditis elegans to hypoxia-reoxygenation perhaps via signaling pathways partially resembling those expressed in mammals and mediating the effects of CYP eicosanoids in ischemia-reperfusion injury of the heart, brain and kidney [275].
References
Capdevila J, Parkhill L, Chacos N, Okita R, Masters BS, Estabrook RW (1981) The oxidative metabolism of arachidonic acid by purified cytochromes P-450. Biochem Biophys Res Commun 101:1357–1363
Capdevila J, Chacos N, Werringloer J, Prough RA, Estabrook RW (1981) Liver microsomal cytochrome P-450 and the oxidative metabolism of arachidonic acid. Proc Natl Acad Sci U S A 78:5362–5366
Morrison AR, Pascoe N (1981) Metabolism of arachidonate through NADPH-dependent oxygenase of renal cortex. Proc Natl Acad Sci U S A 78:7375–7378
Oliw EH, Lawson JA, Brash AR, Oates JA (1981) Arachidonic acid metabolism in rabbit renal cortex. Formation of two novel dihydroxyeicosatrienoic acids. J Biol Chem 256:9924–9931
Capdevila JH, Falck JR, Dishman E, Karara A (1990) Cytochrome P-450 arachidonate oxygenase. Methods Enzymol 187:385–394
Capdevila JH, Falck JR, Estabrook RW (1992) Cytochrome P450 and the arachidonate cascade. FASEB J 6:731–736
Capdevila J, Pramanik B, Napoli JL, Manna S, Falck JR (1984) Arachidonic acid epoxidation: epoxyeicosatrienoic acids are endogenous constituents of rat liver. Arch Biochem Biophys 231:511–517
Falck JR, Schueler VJ, Jacobson HR, Siddhanta AK, Pramanik B, Capdevila J (1987) Arachidonate epoxygenase: identification of epoxyeicosatrienoic acids in rabbit kidney. J Lipid Res 28:840–846
Toto R, Siddhanta A, Manna S, Pramanik B, Falck JR, Capdevila J (1987) Arachidonic acid epoxygenase: detection of epoxyeicosatrienoic acids in human urine. Biochim Biophys Acta 919:132–139
Catella F, Lawson JA, Fitzgerald DJ, FitzGerald GA (1990) Endogenous biosynthesis of arachidonic acid epoxides in humans: increased formation in pregnancy-induced hypertension. Proc Natl Acad Sci U S A 87:5893–5897
Karara A, Dishman E, Blair I, Falck JR, Capdevila JH (1989) Endogenous epoxyeicosatrienoic acids. Cytochrome P-450 controlled stereoselectivity of the hepatic arachidonic acid epoxygenase. J Biol Chem 264:19822–19827
McGiff JC (1991) Cytochrome P-450 metabolism of arachidonic acid. Annu Rev Pharmacol Toxicol 31:339–369
Sacerdoti D, Escalante B, Abraham NG, McGiff JC, Levere RD, Schwartzman ML (1989) Treatment with tin prevents the development of hypertension in spontaneously hypertensive rats. Science 243:388–390
Makita K, Takahashi K, Karara A, Jacobson HR, Falck JR, Capdevila JH (1994) Experimental and/or genetically controlled alterations of the renal microsomal cytochrome P450 epoxygenase induce hypertension in rats fed a high salt diet. J Clin Invest 94:2414–2420
Roman RJ, Alonso-Galicia M, Wilson TW (1997) Renal P450 metabolites of arachidonic acid and the development of hypertension in Dahl salt-sensitive rats. Am J Hypertens 10:63S–67S
McGiff JC, Quilley J (1999) 20-HETE and the kidney: resolution of old problems and new beginnings. Am J Physiol 277:R607–R623
Roman RJ (2002) P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev 82:131–185
Funk CD (2001) Prostaglandins and leukotrienes: advances in eicosanoid biology. Science 294:1871–1875
Buczynski MW, Dumlao DS, Dennis EA (2009) Thematic review series: proteomics. An integrated omics analysis of eicosanoid biology. J Lipid Res 50:1015–1038
Bergstroem S, Ryhage R, Samuelsson B, Sjoevall J (1963) Prostaglandins and related factors. 15. The structures of prostaglandin E1, F1α, and F1β. J Biol Chem 238:3555–3564
Vane JR (1971) Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat New Biol 231:232–235
Samuelsson B (1983) Leukotrienes: mediators of immediate hypersensitivity reactions and inflammation. Science 220:568–575
Kroetz DL, Xu F (2005) Regulation and inhibition of arachidonic acid ω-hydroxylases and 20-HETE formation. Annu Rev Pharmacol Toxicol 45:413–438
Powell PK, Wolf I, Jin R, Lasker JM (1998) Metabolism of arachidonic acid to 20-hydroxy-5,8,11, 14-eicosatetraenoic acid by P450 enzymes in human liver: involvement of CYP4F2 and CYP4A11. J Pharmacol Exp Ther 285:1327–1336
Lasker JM, Chen WB, Wolf I, Bloswick BP, Wilson PD, Powell PK (2000) Formation of 20-hydroxyeicosatetraenoic acid, a vasoactive and natriuretic eicosanoid, in human kidney. Role of Cyp4F2 and Cyp4A11. J Biol Chem 275:4118–4126
Gainer JV, Bellamine A, Dawson EP, Womble KE, Grant SW, Wang Y, Cupples LA, Guo CY, Demissie S, O’Donnell CJ, Brown NJ, Waterman MR, Capdevila JH (2005) Functional variant of CYP4A11 20-hydroxyeicosatetraenoic acid synthase is associated with essential hypertension. Circulation 111:63–69
Hiratsuka M, Nozawa H, Katsumoto Y, Moteki T, Sasaki T, Konno Y, Mizugaki M (2006) Genetic polymorphisms and haplotype structures of the CYP4A22 gene in a Japanese population. Mutat Res 599:98–104
Lino Cardenas CL, Renault N, Farce A, Cauffiez C, Allorge D, Lo-Guidice JM, Lhermitte M, Chavatte P, Broly F, Chevalier D (2011) Genetic polymorphism of CYP4A11 and CYP4A22 genes and in silico insights from comparative 3D modelling in a French population. Gene 487:10–20
Christmas P, Jones JP, Patten CJ, Rock DA, Zheng Y, Cheng SM, Weber BM, Carlesso N, Scadden DT, Rettie AE, Soberman RJ (2001) Alternative splicing determines the function of CYP4F3 by switching substrate specificity. J Biol Chem 276:38166–38172
Corcos L, Lucas D, Le Jossic-Corcos C, Dreano Y, Simon B, Plee-Gautier E, Amet Y, Salaun JP (2012) Human cytochrome P450 4F3: structure, functions, and prospects. Drug Metabol Drug Interact 27:63–71
Fer M, Corcos L, Dreano Y, Plee-Gautier E, Salaun JP, Berthou F, Amet Y (2008) Cytochromes P450 from family 4 are the main omega hydroxylating enzymes in humans: CYP4F3B is the prominent player in PUFA metabolism. J Lipid Res 49:2379–2389
Chuang SS, Helvig C, Taimi M, Ramshaw HA, Collop AH, Amad M, White JA, Petkovich M, Jones G, Korczak B (2004) CYP2U1, a novel human thymus- and brain-specific cytochrome P450, catalyzes ω- and (ω-1)-hydroxylation of fatty acids. J Biol Chem 279:6305–6314
Kelly EJ, Nakano M, Rohatgi P, Yarov-Yarovoy V, Rettie AE (2011) Finding homes for orphan cytochrome P450s: CYP4V2 and CYP4F22 in disease states. Mol Interv 11:124–132
Nguyen X, Wang MH, Reddy KM, Falck JR, Schwartzman ML (1999) Kinetic profile of the rat CYP4A isoforms: arachidonic acid metabolism and isoform-specific inhibitors. Am J Physiol 276:R1691–R1700
Yamaguchi Y, Kirita S, Hasegawa H, Aoyama J, Imaoka S, Minamiyama S, Funae Y, Baba T, Matsubara T (2002) Contribution of CYP4A8 to the formation of 20-hydroxyeicosatetraenoic acid from arachidonic acid in rat kidney. Drug Metab Pharmacokinet 17:109–116
Xu F, Falck JR, Ortiz de Montellano PR, Kroetz DL (2004) Catalytic activity and isoform-specific inhibition of rat cytochrome P450 4F enzymes. J Pharmacol Exp Ther 308:887–895
El-Sherbeni AA, Aboutabl ME, Zordoky BN, Anwar-Mohamed A, El-Kadi AO (2013) Determination of the dominant arachidonic acid cytochrome P450 monooxygenases in rat heart, lung, kidney, and liver: protein expression and metabolite kinetics. AAPS J 15:112–122
Marji JS, Wang MH, Laniado-Schwartzman M (2002) Cytochrome P-450 4A isoform expression and 20-HETE synthesis in renal preglomerular arteries. Am J Physiol Renal Physiol 283:F60–F67
Singh H, Schwartzman ML (2008) Renal vascular cytochrome P450-derived eicosanoids in androgen-induced hypertension. Pharmacol Rep 60:29–37
Dunn KM, Renic M, Flasch AK, Harder DR, Falck J, Roman RJ (2008) Elevated production of 20-HETE in the cerebral vasculature contributes to severity of ischemic stroke and oxidative stress in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 295:H2455–H2465
Nelson DR, Zeldin DC, Hoffman SM, Maltais LJ, Wain HM, Nebert DW (2004) Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenetics 14:1–18
Muller DN, Schmidt C, Barbosa-Sicard E, Wellner M, Gross V, Hercule H, Markovic M, Honeck H, Luft FC, Schunck WH (2007) Mouse Cyp4a isoforms: enzymatic properties, gender- and strain-specific expression, and role in renal 20-hydroxyeicosatetraenoic acid formation. Biochem J 403:109–118
Holla VR, Adas F, Imig JD, Zhao X, Price E Jr, Olsen N, Kovacs WJ, Magnuson MA, Keeney DS, Breyer MD, Falck JR, Waterman MR, Capdevila JH (2001) Alterations in the regulation of androgen-sensitive Cyp 4a monooxygenases cause hypertension. Proc Natl Acad Sci U S A 98:5211–5216
Wu CC, Mei S, Cheng J, Ding Y, Weidenhammer A, Garcia V, Zhang F, Gotlinger K, Manthati VL, Falck JR, Capdevila JH, Schwartzman ML (2013) Androgen-sensitive hypertension associates with upregulated vascular CYP4A12-20-HETE synthase. J Am Soc Nephrol 24:1288–1296
Nakagawa K, Holla VR, Wei Y, Wang WH, Gatica A, Wei S, Mei S, Miller CM, Cha DR, Price E Jr, Zent R, Pozzi A, Breyer MD, Guan Y, Falck JR, Waterman MR, Capdevila JH (2006) Salt-sensitive hypertension is associated with dysfunctional Cyp4a10 gene and kidney epithelial sodium channel. J Clin Invest 116:1696–1702
Stec DE, Flasch A, Roman RJ, White JA (2003) Distribution of cytochrome P-450 4A and 4F isoforms along the nephron in mice. Am J Physiol Renal Physiol 284:F95–F102
Zeldin DC (2001) Epoxygenase pathways of arachidonic acid metabolism. J Biol Chem 276:36059–36062
Rifkind AB, Lee C, Chang TK, Waxman DJ (1995) Arachidonic acid metabolism by human cytochrome P450s 2C8, 2C9, 2E1, and 1A2: regioselective oxygenation and evidence for a role for CYP2C enzymes in arachidonic acid epoxygenation in human liver microsomes. Arch Biochem Biophys 320:380–389
Daikh BE, Lasker JM, Raucy JL, Koop DR (1994) Regio- and stereoselective epoxidation of arachidonic acid by human cytochromes P450 2C8 and 2C9. J Pharmacol Exp Ther 271:1427–1433
Zeldin DC, DuBois RN, Falck JR, Capdevila JH (1995) Molecular cloning, expression and characterization of an endogenous human cytochrome P450 arachidonic acid epoxygenase isoform. Arch Biochem Biophys 322:76–86
Fisslthaler B, Popp R, Kiss L, Potente M, Harder DR, Fleming I, Busse R (1999) Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature 401:493–497
Campbell WB, Gebremedhin D, Pratt PF, Harder DR (1996) Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ Res 78:415–423
Campbell WB, Falck JR (2007) Arachidonic acid metabolites as endothelium-derived hyperpolarizing factors. Hypertension 49:590–596
Campbell WB, Fleming I (2010) Epoxyeicosatrienoic acids and endothelium-dependent responses. Pflugers Arch 459:881–895
Lee CR, Imig JD, Edin ML, Foley J, DeGraff LM, Bradbury JA, Graves JP, Lih FB, Clark J, Myers P, Perrow AL, Lepp AN, Kannon MA, Ronnekleiv OK, Alkayed NJ, Falck JR, Tomer KB, Zeldin DC (2010) Endothelial expression of human cytochrome P450 epoxygenases lowers blood pressure and attenuates hypertension-induced renal injury in mice. FASEB J 24:3770–3781
Edin ML, Wang Z, Bradbury JA, Graves JP, Lih FB, DeGraff LM, Foley JF, Torphy R, Ronnekleiv OK, Tomer KB, Lee CR, Zeldin DC (2011) Endothelial expression of human cytochrome P450 epoxygenase CYP2C8 increases susceptibility to ischemia-reperfusion injury in isolated mouse heart. FASEB J 25:3436–3447
Fleming I, Michaelis UR, Bredenkotter D, Fisslthaler B, Dehghani F, Brandes RP, Busse R (2001) Endothelium-derived hyperpolarizing factor synthase (Cytochrome P450 2C9) is a functionally significant source of reactive oxygen species in coronary arteries. Circ Res 88:44–51
Fichtlscherer S, Dimmeler S, Breuer S, Busse R, Zeiher AM, Fleming I (2004) Inhibition of cytochrome P450 2C9 improves endothelium-dependent, nitric oxide-mediated vasodilatation in patients with coronary artery disease. Circulation 109:178–183
Fer M, Dreano Y, Lucas D, Corcos L, Salaun JP, Berthou F, Amet Y (2008) Metabolism of eicosapentaenoic and docosahexaenoic acids by recombinant human cytochromes P450. Arch Biochem Biophys 471:116–125
Arnold C, Markovic M, Blossey K, Wallukat G, Fischer R, Dechend R, Konkel A, von Schacky C, Luft FC, Muller DN, Rothe M, Schunck WH (2010) Arachidonic acid-metabolizing cytochrome P450 enzymes are targets of ω-3 fatty acids. J Biol Chem 285:32720–31733
Wu S, Moomaw CR, Tomer KB, Falck JR, Zeldin DC (1996) Molecular cloning and expression of CYP2J2, a human cytochrome P450 arachidonic acid epoxygenase highly expressed in heart. J Biol Chem 271:3460–3468
Scarborough PE, Ma J, Qu W, Zeldin DC (1999) P450 subfamily CYP2J and their role in the bioactivation of arachidonic acid in extrahepatic tissues. Drug Metab Rev 31:205–234
Seubert J, Yang B, Bradbury JA, Graves J, Degraff LM, Gabel S, Gooch R, Foley J, Newman J, Mao L, Rockman HA, Hammock BD, Murphy E, Zeldin DC (2004) Enhanced postischemic functional recovery in CYP2J2 transgenic hearts involves mitochondrial ATP-sensitive K+ channels and p42/p44 MAPK pathway. Circ Res 95:506–514
Askari A, Thomson SJ, Edin ML, Zeldin DC, Bishop-Bailey D (2013) Roles of the epoxygenase CYP2J2 in the endothelium. Prostaglandins Other Lipid Mediat 107:56–63
Spiecker M, Liao JK (2005) Vascular protective effects of cytochrome P450 epoxygenase-derived eicosanoids. Arch Biochem Biophys 433:413–420
Karara A, Makita K, Jacobson HR, Falck JR, Guengerich FP, DuBois RN, Capdevila JH (1993) Molecular cloning, expression, and enzymatic characterization of the rat kidney cytochrome P-450 arachidonic acid epoxygenase. J Biol Chem 268:13565–13570
Imaoka S, Wedlund PJ, Ogawa H, Kimura S, Gonzalez FJ, Kim HY (1993) Identification of CYP2C23 expressed in rat kidney as an arachidonic acid epoxygenase. J Pharmacol Exp Ther 267:1012–1016
Holla VR, Makita K, Zaphiropoulos PG, Capdevila JH (1999) The kidney cytochrome P-450 2C23 arachidonic acid epoxygenase is upregulated during dietary salt loading. J Clin Invest 104:751–760
Kaergel E, Muller DN, Honeck H, Theuer J, Shagdarsuren E, Mullally A, Luft FC, Schunck WH (2002) P450-dependent arachidonic acid metabolism and angiotensin II-induced renal damage. Hypertension 40:273–279
Zhao X, Pollock DM, Zeldin DC, Imig JD (2003) Salt-sensitive hypertension after exposure to angiotensin is associated with inability to upregulate renal epoxygenases. Hypertension 42:775–780
Muller DN, Theuer J, Shagdarsuren E, Kaergel E, Honeck H, Park JK, Markovic M, Barbosa-Sicard E, Dechend R, Wellner M, Kirsch T, Fiebeler A, Rothe M, Haller H, Luft FC, Schunck WH (2004) A peroxisome proliferator-activated receptor-α activator induces renal CYP2C23 activity and protects from angiotensin II-induced renal injury. Am J Pathol 164:521–532
Capdevila JH, Karara A, Waxman DJ, Martin MV, Falck JR, Guenguerich FP (1990) Cytochrome P-450 enzyme-specific control of the regio- and enantiofacial selectivity of the microsomal arachidonic acid epoxygenase. J Biol Chem 265:10865–10871
Alkayed NJ, Narayanan J, Gebremedhin D, Medhora M, Roman RJ, Harder DR (1996) Molecular characterization of an arachidonic acid epoxygenase in rat brain astrocytes. Stroke 27:971–979
Medhora M, Narayanan J, Harder D (2001) Dual regulation of the cerebral microvasculature by epoxyeicosatrienoic acids. Trends Cardiovasc Med 11:38–42
Wu S, Chen W, Murphy E, Gabel S, Tomer KB, Foley J, Steenbergen C, Falck JR, Moomaw CR, Zeldin DC (1997) Molecular cloning, expression, and functional significance of a cytochrome P450 highly expressed in rat heart myocytes. J Biol Chem 272:12551–12559
Zhang QY, Ding X, Kaminsky LS (1997) cDNA cloning, heterologous expression, and characterization of rat intestinal CYP2J4. Arch Biochem Biophys 340:270–278
Yaghi A, Bradbury JA, Zeldin DC, Mehta S, Bend JR, McCormack DG (2003) Pulmonary cytochrome P-450 2J4 is reduced in a rat model of acute Pseudomonas pneumonia. Am J Physiol Lung Cell Mol Physiol 285:L1099–L1105
DeLozier TC, Tsao CC, Coulter SJ, Foley J, Bradbury JA, Zeldin DC, Goldstein JA (2004) CYP2C44, a new murine CYP2C that metabolizes arachidonic acid to unique stereospecific products. J Pharmacol Exp Ther 310:845–854
Pidkovka N, Rao R, Mei S, Gong Y, Harris RC, Wang WH, Capdevila JH (2013) Epoxyeicosatrienoic acids (EETs) regulate epithelial sodium channel activity by extracellular signal-regulated kinase 1/2 (ERK1/2)-mediated phosphorylation. J Biol Chem 288:5223–5231
Zhang MZ, Wang Y, Yao B, Gewin L, Wei S, Capdevila JH, Harris RC (2013) Role of epoxyeicosatrienoic acids (EETs) in mediation of dopamine’s effects in the kidney. Am J Physiol Renal Physiol 305:F1680–F1686
Sun P, Antoun J, Lin DH, Yue P, Gotlinger KH, Capdevila J, Wang WH (2012) Cyp2c44 epoxygenase is essential for preventing the renal sodium absorption during increasing dietary potassium intake. Hypertension 59:339–347
Luo G, Zeldin DC, Blaisdell JA, Hodgson E, Goldstein JA (1998) Cloning and expression of murine CYP2Cs and their ability to metabolize arachidonic acid. Arch Biochem Biophys 357:45–57
Wang H, Zhao Y, Bradbury JA, Graves JP, Foley J, Blaisdell JA, Goldstein JA, Zeldin DC (2004) Cloning, expression, and characterization of three new mouse cytochrome P450 enzymes and partial characterization of their fatty acid oxidation activities. Mol Pharmacol 65:1148–1158
Tsao CC, Foley J, Coulter SJ, Maronpot R, Zeldin DC, Goldstein JA (2000) CYP2C40, a unique arachidonic acid 16-hydroxylase, is the major CYP2C in murine intestinal tract. Mol Pharmacol 58:279–287
Sun D, Yang YM, Jiang H, Wu H, Ojaimi C, Kaley G, Huang A (2012) Roles of CYP2C29 and RXRγ in vascular EET synthesis of female mice. Am J Physiol Regul Integr Comp Physiol 298:R862–R869
Pokreisz P, Fleming I, Kiss L, Barbosa-Sicard E, Fisslthaler B, Falck JR, Hammock BD, Kim IH, Szelid Z, Vermeersch P, Gillijns H, Pellens M, Grimminger F, van Zonneveld AJ, Collen D, Busse R, Janssens S (2006) Cytochrome P450 epoxygenase gene function in hypoxic pulmonary vasoconstriction and pulmonary vascular remodeling. Hypertension 47:762–770
Scheer N, Kapelyukh Y, Chatham L, Rode A, Buechel S, Wolf CR (2012) Generation and characterization of novel cytochrome P450 Cyp2c gene cluster knockout and CYP2C9 humanized mouse lines. Mol Pharmacol 82:1022–1029
Ma J, Qu W, Scarborough PE, Tomer KB, Moomaw CR, Maronpot R, Davis LS, Breyer MD, Zeldin DC (1999) Molecular cloning, enzymatic characterization, developmental expression, and cellular localization of a mouse cytochrome P450 highly expressed in kidney. J Biol Chem 274:17777–17788
Ma J, Graves J, Bradbury JA, Zhao Y, Swope DL, King L, Qu W, Clark J, Myers P, Walker V, Lindzey J, Korach KS, Zeldin DC (2004) Regulation of mouse renal CYP2J5 expression by sex hormones. Mol Pharmacol 65:730–743
Athirakul K, Bradbury JA, Graves JP, DeGraff LM, Ma J, Zhao Y, Couse JF, Quigley R, Harder DR, Zhao X, Imig JD, Pedersen TL, Newman JW, Hammock BD, Conley AJ, Korach KS, Coffman TM, Zeldin DC (2008) Increased blood pressure in mice lacking cytochrome P450 2J5. FASEB J 22:4096–4108
Ma J, Bradbury JA, King L, Maronpot R, Davis LS, Breyer MD, Zeldin DC (2002) Molecular cloning and characterization of mouse CYP2J6, an unstable cytochrome P450 isoform. Biochem Pharmacol 64:1447–1460
Qu W, Bradbury JA, Tsao CC, Maronpot R, Harry GJ, Parker CE, Davis LS, Breyer MD, Waalkes MP, Falck JR, Chen J, Rosenberg RL, Zeldin DC (2001) Cytochrome P450 CYP2J9, a new mouse arachidonic acid ω-1 hydroxylase predominantly expressed in brain. J Biol Chem 276:25467–25479
Graves JP, Edin ML, Bradbury JA, Gruzdev A, Cheng J, Lih FB, Masinde TA, Qu W, Clayton NP, Morrison JP, Tomer KB, Zeldin DC (2013) Characterization of four new mouse cytochrome P450 enzymes of the CYP2J subfamily. Drug Metab Dispos 41:763–773
Keeney DS, Skinner C, Travers JB, Capdevila JH, Nanney LB, King LE Jr, Waterman MR (1998) Differentiating keratinocytes express a novel cytochrome P450 enzyme, CYP2B19, having arachidonate monooxygenase activity. J Biol Chem 273:32071–32079
Du L, Yermalitsky V, Ladd PA, Capdevila JH, Mernaugh R, Keeney DS (2005) Evidence that cytochrome P450 CYP2B19 is the major source of epoxyeicosatrienoic acids in mouse skin. Arch Biochem Biophys 435:125–133
Ladd PA, Du L, Capdevila JH, Mernaugh R, Keeney DS (2003) Epoxyeicosatrienoic acids activate transglutaminases in situ and induce cornification of epidermal keratinocytes. J Biol Chem 278:35184–35192
Keeney DS, Skinner C, Wei S, Friedberg T, Waterman MR (1998) A keratinocyte-specific epoxygenase, CYP2B12, metabolizes arachidonic acid with unusual selectivity, producing a single major epoxyeicosatrienoic acid. J Biol Chem 273:9279–9284
Du L, Neis MM, Ladd PA, Lanza DL, Yost GS, Keeney DS (2006) Effects of the differentiated keratinocyte phenotype on expression levels of CYP1-4 family genes in human skin cells. Toxicol Appl Pharmacol 213:135–144
Sridar C, Snider NT, Hollenberg PF (2011) Anandamide oxidation by wild-type and polymorphically expressed CYP2B6 and CYP2D6. Drug Metab Dispos 39:782–788
Fromel T, Kohlstedt K, Popp R, Yin X, Awwad K, Barbosa-Sicard E, Thomas AC, Lieberz R, Mayr M, Fleming I (2013) Cytochrome P4502S1: a novel monocyte/macrophage fatty acid epoxygenase in human atherosclerotic plaques. Basic Res Cardiol 108:319–330
Schwarz D, Kisselev P, Ericksen SS, Szklarz GD, Chernogolov A, Honeck H, Schunck WH, Roots I (2004) Arachidonic and eicosapentaenoic acid metabolism by human CYP1A1: highly stereoselective formation of 17(R),18(S)-epoxyeicosatetraenoic acid. Biochem Pharmacol 67:1445–1457
Laethem RM, Balazy M, Falck JR, Laethem CL, Koop DR (1993) Formation of 19(S)-, 19(R)-, and 18(R)-hydroxyeicosatetraenoic acids by alcohol-inducible cytochrome P450 2E1. J Biol Chem 268:12912–12918
Cheng J, Ou JS, Singh H, Falck JR, Narsimhaswamy D, Pritchard KA Jr, Schwartzman ML (2008) 20-Hydroxyeicosatetraenoic acid causes endothelial dysfunction via eNOS uncoupling. Am J Physiol Heart Circ Physiol 294:H1018–H1026
Zhang F, Deng H, Kemp R, Singh H, Gopal VR, Falck JR, Laniado-Schwartzman M, Nasjletti A (2005) Decreased levels of cytochrome P450 2E1-derived eicosanoids sensitize renal arteries to constrictor agonists in spontaneously hypertensive rats. Hypertension 45:103–108
Stark K, Wongsud B, Burman R, Oliw EH (2005) Oxygenation of polyunsaturated long chain fatty acids by recombinant CYP4F8 and CYP4F12 and catalytic importance of Tyr-125 and Gly-328 of CYP4F8. Arch Biochem Biophys 441:174–181
Capdevila JH, Falck JR, Harris RC (2000) Cytochrome P450 and arachidonic acid bioactivation. Molecular and functional properties of the arachidonate monooxygenase. J Lipid Res 41:163–181
Oliw EH, Bylund J, Herman C (1996) Bisallylic hydroxylation and epoxidation of polyunsaturated fatty acids by cytochrome P450. Lipids 31:1003–1021
Brash AR, Boeglin WE, Capdevila JH, Yeola S, Blair IA (1995) 7-HETE, 10-HETE, and 13-HETE are major products of NADPH-dependent arachidonic acid metabolism in rat liver microsomes: analysis of their stereochemistry, and the stereochemistry of their acid-catalyzed rearrangement. Arch Biochem Biophys 321:485–492
Hornsten L, Bylund J, Oliw EH (1996) Dexamethasone induces bisallylic hydroxylation of polyunsaturated fatty acids by rat liver microsomes. Arch Biochem Biophys 332:261–268
Bylund J, Kunz T, Valmsen K, Oliw EH (1998) Cytochromes P450 with bisallylic hydroxylation activity on arachidonic and linoleic acids studied with human recombinant enzymes and with human and rat liver microsomes. J Pharmacol Exp Ther 284:51–60
Yamamoto S, Nishimura M, Conners MS, Stoltz RA, Falck JR, Chauhan K, Laniado-Schwartzman M (1994) Oxidation and keto reduction of 12-hydroxy-5,8,10,14-eicosatetraenoic acids in bovine corneal epithelial microsomes. Biochim Biophys Acta 1210:217–225
Mieyal PA, Dunn MW, Schwartzman ML (2001) Detection of endogenous 12-hydroxyeicosatrienoic acid in human tear film. Invest Ophthalmol Vis Sci 42:328–332
Mezentsev A, Mastyugin V, Seta F, Ashkar S, Kemp R, Reddy DS, Falck JR, Dunn MW, Laniado-Schwartzman M (2005) Transfection of cytochrome P4504B1 into the cornea increases angiogenic activity of the limbal vessels. J Pharmacol Exp Ther 315:42–50
Seta F, Patil K, Bellner L, Mezentsev A, Kemp R, Dunn MW, Schwartzman ML (2007) Inhibition of VEGF expression and corneal neovascularization by siRNA targeting cytochrome P450 4B1. Prostaglandins Other Lipid Mediat 84:116–127
Konkel A, Schunck WH (2011) Role of cytochrome P450 enzymes in the bioactivation of polyunsaturated fatty acids. Biochim Biophys Acta 1814:210–222
De Caterina R (2011) n-3 Fatty acids in cardiovascular disease. N Engl J Med 364:2439–2450
Martins DA, Custodio L, Barreira L, Pereira H, Ben-Hamadou R, Varela J, Abu-Salah KM (2013) Alternative sources of n-3 long-chain polyunsaturated fatty acids in marine microalgae. Mar Drugs 11:2259–2281
Vrablik TL, Watts JL (2013) Polyunsaturated fatty acid derived signaling in reproduction and development: insights from Caenorhabditis elegans and Drosophila melanogaster. Mol Reprod Dev 80:244–259
Arterburn LM, Hall EB, Oken H (2006) Distribution, interconversion, and dose response of n-3 fatty acids in humans. Am J Clin Nutr 83:1467S–1476S
Simopoulos AP (2008) The importance of the ω-6/ω-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Exp Biol Med (Maywood) 233:674–688
Lands WE (2005) Dietary fat and health: the evidence and the politics of prevention: careful use of dietary fats can improve life and prevent disease. Ann N Y Acad Sci 1055:179–192
Calder PC (2006) n-3 Polyunsaturated fatty acids, inflammation, and inflammatory diseases. Am J Clin Nutr 83:1505S–1519S
Kris-Etherton PM, Harris WS, Appel LJ (2002) Fish consumption, fish oil, ω-3 fatty acids, and cardiovascular disease. Circulation 106:2747–2757
SanGiovanni JP, Chew EY (2005) The role of ω-3 long-chain polyunsaturated fatty acids in health and disease of the retina. Prog Retin Eye Res 24:87–138
Uauy R, Hoffman DR, Peirano P, Birch DG, Birch EE (2001) Essential fatty acids in visual and brain development. Lipids 36:885–895
Rapoport SI, Igarashi M (2009) Can the rat liver maintain normal brain DHA metabolism in the absence of dietary DHA? Prostaglandins Leukot Essent Fatty Acids 81:119–123
Saravanan P, Davidson NC, Schmidt EB, Calder PC (2010) Cardiovascular effects of marine ω-3 fatty acids. Lancet 376:540–550
Leslie CC (2004) Regulation of arachidonic acid availability for eicosanoid production. Biochem Cell Biol 82:1–17
Wada M, DeLong CJ, Hong YH, Rieke CJ, Song I, Sidhu RS, Yuan C, Warnock M, Schmaier AH, Yokoyama C, Smyth EM, Wilson SJ, FitzGerald GA, Garavito RM, de Sui X, Regan JW, Smith WL (2007) Enzymes and receptors of prostaglandin pathways with arachidonic acid-derived versus eicosapentaenoic acid-derived substrates and products. J Biol Chem 282:22254–22266
Rosa AO, Rapoport SI (2009) Intracellular- and extracellular-derived Ca2+ influence phospholipase A2-mediated fatty acid release from brain phospholipids. Biochim Biophys Acta 1791:697–705
Cheon Y, Kim HW, Igarashi M, Modi HR, Chang L, Ma K, Greenstein D, Wohltmann M, Turk J, Rapoport SI, Taha AY (2012) Disturbed brain phospholipid and docosahexaenoic acid metabolism in calcium-independent phospholipase A2-VIA (iPLA2 β-knockout mice. Biochim Biophys Acta 1821:1278–1286
Liu X, Moon SH, Mancuso DJ, Jenkins CM, Guan S, Sims HF, Gross RW (2013) Oxidized fatty acid analysis by charge-switch derivatization, selected reaction monitoring, and accurate mass quantitation. Anal Biochem 442:40–50
Van Rollins M, Frade PD, Carretero OA (1988) Oxidation of 5,8,11,14,17-eicosapentaenoic acid by hepatic and renal microsomes. Biochim Biophys Acta 966:133–149
VanRollins M (1995) Epoxygenase metabolites of docosahexaenoic and eicosapentaenoic acids inhibit platelet aggregation at concentrations below those affecting thromboxane synthesis. J Pharmacol Exp Ther 274:798–804
Lauterbach B, Barbosa-Sicard E, Wang MH, Honeck H, Kargel E, Theuer J, Schwartzman ML, Haller H, Luft FC, Gollasch M, Schunck WH (2002) Cytochrome P450-dependent eicosapentaenoic acid metabolites are novel BK channel activators. Hypertension 39:609–613
Barbosa-Sicard E, Markovic M, Honeck H, Christ B, Muller DN, Schunck WH (2005) Eicosapentaenoic acid metabolism by cytochrome P450 enzymes of the CYP2C subfamily. Biochem Biophys Res Commun 329:1275–1281
Lucas D, Goulitquer S, Marienhagen J, Fer M, Dreano Y, Schwaneberg U, Amet Y, Corcos L (2010) Stereoselective epoxidation of the last double bond of polyunsaturated fatty acids by human cytochromes P450. J Lipid Res 51:1125–1133
Dyerberg J, Bang HO, Stoffersen E, Moncada S, Vane JR (1978) Eicosapentaenoic acid and prevention of thrombosis and atherosclerosis? Lancet 2:117–119
Terano T, Salmon JA, Moncada S (1984) Biosynthesis and biological activity of leukotriene B5. Prostaglandins 27:217–232
Fischer S, Weber PC, Dyerberg J (1986) The prostacyclin/thromboxane balance is favourably shifted in Greenland Eskimos. Prostaglandins 32:235–241
von Schacky C, Fischer S, Weber PC (1985) Long-term effects of dietary marine ω-3 fatty acids upon plasma and cellular lipids, platelet function, and eicosanoid formation in humans. J Clin Invest 76:1626–1631
Calder PC (2009) Polyunsaturated fatty acids and inflammatory processes: new twists in an old tale. Biochimie 91:791–795
Knapp HR, Miller AJ, Lawson JA (1991) Urinary excretion of diols derived from eicosapentaenoic acid during n-3 fatty acid ingestion by man. Prostaglandins 42:47–54
Shearer GC, Harris WS, Pedersen TL, Newman JW (2010) Detection of ω-3 oxylipins in human plasma and response to treatment with ω-3 acid ethyl esters. J Lipid Res 51:2074–2081
Lundstrom SL, Yang J, Brannan JD, Haeggstrom JZ, Hammock BD, Nair P, O’Byrne P, Dahlen SE, Wheelock CE (2013) Lipid mediator serum profiles in asthmatics significantly shift following dietary supplementation with ω-3 fatty acids. Mol Nutr Food Res 57:1378–1389
Schuchardt JP, Schmidt S, Kressel G, Dong H, Willenberg I, Hammock BD, Hahn A, Schebb NH (2013) Comparison of free serum oxylipin concentrations in hyper- vs. normolipidemic men. Prostaglandins Leukot Essent Fatty Acids 89:19–29
Bruins MJ, Dane AD, Strassburg K, Vreeken RJ, Newman JW, Salem N Jr, Tyburczy C, Brenna JT (2013) Plasma oxylipin profiling identifies polyunsaturated vicinal diols as responsive to arachidonic acid and docosahexaenoic acid intake in growing piglets. J Lipid Res 54:1598–1607
Westphal C, Konkel A, Schunck WH (2011) CYP-eicosanoids−a new link between ω-3 fatty acids and cardiac disease? Prostaglandins Other Lipid Mediat 96:99–108
Ye D, Zhang D, Oltman C, Dellsperger K, Lee HC, Van Rollins M (2002) Cytochrome P-450 epoxygenase metabolites of docosahexaenoate potently dilate coronary arterioles by activating large-conductance calcium-activated potassium channels. J Pharmacol Exp Ther 303:768–776
Agbor LN, Walsh MT, Boberg JR, Walker MK (2012) Elevated blood pressure in cytochrome P4501A1 knockout mice is associated with reduced vasodilation to ω-3 polyunsaturated fatty acids. Toxicol Appl Pharmacol 264:351–360
Node K, Huo Y, Ruan X, Yang B, Spiecker M, Ley K, Zeldin DC, Liao JK (1999) Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 285:1276–1279
Morin C, Sirois M, Echave V, Albadine R, Rousseau E (2010) 17,18-Epoxyeicosatetraenoic acid targets PPARγ and p38 mitogen-activated protein kinase to mediate its anti-inflammatory effects in the lung: role of soluble epoxide hydrolase. Am J Respir Cell Mol Biol 43:564–575
Panigrahy D, Edin ML, Lee CR, Huang S, Bielenberg DR, Butterfield CE, Barnes CM, Mammoto A, Mammoto T, Luria A, Benny O, Chaponis DM, Dudley AC, Greene ER, Vergilio JA, Pietramaggiori G, Scherer-Pietramaggiori SS, Short SM, Seth M, Lih FB, Tomer KB, Yang J, Schwendener RA, Hammock BD, Falck JR, Manthati VL, Ingber DE, Kaipainen A, D’Amore PA, Kieran MW, Zeldin DC (2012) Epoxyeicosanoids stimulate multiorgan metastasis and tumor dormancy escape in mice. J Clin Invest 122:178–191
Zhang G, Panigrahy D, Mahakian LM, Yang J, Liu JY, Stephen Lee KS, Wettersten HI, Ulu A, Hu X, Tam S, Hwang SH, Ingham ES, Kieran MW, Weiss RH, Ferrara KW, Hammock BD (2013) Epoxy metabolites of docosahexaenoic acid (DHA) inhibit angiogenesis, tumor growth, and metastasis. Proc Natl Acad Sci U S A 110:6530–6535
Morisseau C, Inceoglu B, Schmelzer K, Tsai HJ, Jinks SL, Hegedus CM, Hammock BD (2010) Naturally occurring monoepoxides of eicosapentaenoic acid and docosahexaenoic acid are bioactive antihyperalgesic lipids. J Lipid Res 51:3481–3490
Lavie CJ, Milani RV, Mehra MR, Ventura HO (2009) ω-3 Polyunsaturated fatty acids and cardiovascular diseases. J Am Coll Cardiol 54:585–594
Webler AC, Michaelis UR, Popp R, Barbosa-Sicard E, Murugan A, Falck JR, Fisslthaler B, Fleming I (2008) Epoxyeicosatrienoic acids are part of the VEGF-activated signaling cascade leading to angiogenesis. Am J Physiol Cell Physiol 295:C1292–C1301
Alonso-Galicia M, Maier KG, Greene AS, Cowley AW Jr, Roman RJ (2002) Role of 20-hydroxyeicosatetraenoic acid in the renal and vasoconstrictor actions of angiotensin II. Am J Physiol Regul Integr Comp Physiol 283:R60–R68
Karara A, Dishman E, Falck JR, Capdevila JH (1991) Endogenous epoxyeicosatrienoyl-phospholipids. A novel class of cellular glycerolipids containing epoxidized arachidonate moieties. J Biol Chem 266:7561–7569
Carroll MA, Balazy M, Huang DD, Rybalova S, Falck JR, McGiff JC (1997) Cytochrome P450-derived renal HETEs: storage and release. Kidney Int 51:1696–1702
Kaduce TL, Fang X, Harmon SD, Oltman CL, Dellsperger KC, Teesch LM, Gopal VR, Falck JR, Campbell WB, Weintraub NL, Spector AA (2004) 20-Hydroxyeicosatetraenoic acid (20-HETE) metabolism in coronary endothelial cells. J Biol Chem 279:2648–2656
Oliw EH (1991) 17R(18S)epoxyeicosatetraenoic acid, a cytochrome P-450 metabolite of 20:5n-3 in monkey seminal vesicles, is metabolized to novel prostaglandins. Biochem Biophys Res Commun 178:1444–1450
Oliw EH, Okamoto S, Hornsten L, Sato F (1992) Biosynthesis of prostaglandins from 17(18)epoxy-eicosatetraenoic acid, a cytochrome P-450 metabolite of eicosapentaenoic acid. Biochim Biophys Acta 1126:261–268
Cheng MK, McGiff JC, Carroll MA (2003) Renal arterial 20-hydroxyeicosatetraenoic acid levels: regulation by cyclooxygenase. Am J Physiol Renal Physiol 284:F474–F479
Kim DH, Puri N, Sodhi K, Falck JR, Abraham NG, Shapiro J, Schwartzman ML (2013) Cyclooxygenase-2 dependent metabolism of 20-HETE increases adiposity and adipocyte enlargement in mesenchymal stem cell-derived adipocytes. J Lipid Res 54:786–793
Yang W, Gauthier KM, Reddy LM, Sangras B, Sharma KK, Nithipatikom K, Falck JR, Campbell WB (2005) Stable 5,6-epoxyeicosatrienoic acid analog relaxes coronary arteries through potassium channel activation. Hypertension 45:681–686
Moreland KT, Procknow JD, Sprague RS, Iverson JL, Lonigro AJ, Stephenson AH (2007) Cyclooxygenase (COX)-1 and COX-2 participate in 5,6-epoxyeicosatrienoic acid-induced contraction of rabbit intralobar pulmonary arteries. J Pharmacol Exp Ther 321:446–454
Kubota T, Arita M, Isobe Y, Iwamoto R, Goto T, Yoshioka T, Urabe D, Inoue M, Arai H (2014) Eicosapentaenoic acid is converted via ω-3 epoxygenation to the anti-inflammatory metabolite 12-hydroxy-17,18-epoxyeicosatetraenoic acid. FASEB J 28:586–593
Serhan CN, Chiang N, Van Dyke TE (2008) Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol 8:349–361
Arita M, Clish CB, Serhan CN (2005) The contributions of aspirin and microbial oxygenase to the biosynthesis of anti-inflammatory resolvins: novel oxygenase products from ω-3 polyunsaturated fatty acids. Biochem Biophys Res Commun 338:149–157
Cowart LA, Wei S, Hsu MH, Johnson EF, Krishna MU, Falck JR, Capdevila JH (2002) The CYP4A isoforms hydroxylate epoxyeicosatrienoic acids to form high affinity peroxisome proliferator-activated receptor ligands. J Biol Chem 277:35105–35112
Le Quere V, Plee-Gautier E, Potin P, Madec S, Salaun JP (2004) Human CYP4F3s are the main catalysts in the oxidation of fatty acid epoxides. J Lipid Res 45:1446–1458
Morisseau C, Hammock BD (2013) Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu Rev Pharmacol Toxicol 53:37–58
Harris TR, Hammock BD (2013) Soluble epoxide hydrolase: gene structure, expression and deletion. Gene 526:61–74
Widstrom RL, Norris AW, Spector AA (2001) Binding of cytochrome P450 monooxygenase and lipoxygenase pathway products by heart fatty acid-binding protein. Biochemistry 40:1070–1076
Widstrom RL, Norris AW, Van Der Veer J, Spector AA (2003) Fatty acid-binding proteins inhibit hydration of epoxyeicosatrienoic acids by soluble epoxide hydrolase. Biochemistry 42:11762–11767
Ai D, Fu Y, Guo D, Tanaka H, Wang N, Tang C, Hammock BD, Shyy JY, Zhu Y (2007) Angiotensin II up-regulates soluble epoxide hydrolase in vascular endothelium in vitro and in vivo. Proc Natl Acad Sci U S A 104:9018–9023
Ai D, Pang W, Li N, Xu M, Jones PD, Yang J, Zhang Y, Chiamvimonvat N, Shyy JY, Hammock BD, Zhu Y (2009) Soluble epoxide hydrolase plays an essential role in angiotensin II-induced cardiac hypertrophy. Proc Natl Acad Sci U S A 106:564–569
Imig JD, Hammock BD (2009) Soluble epoxide hydrolase as a therapeutic target for cardiovascular diseases. Nat Rev Drug Discov 8:794–805
Spector AA, Norris AW (2007) Action of epoxyeicosatrienoic acids on cellular function. Am J Physiol Cell Physiol 292:C996–C1012
Wu CC, Gupta T, Garcia V, Ding Y, Schwartzman ML (2014) 20-HETE and blood pressure regulation: clinical implications. Cardiol Rev 22:1–12
Seubert JM, Zeldin DC, Nithipatikom K, Gross GJ (2007) Role of epoxyeicosatrienoic acids in protecting the myocardium following ischemia/reperfusion injury. Prostaglandins Other Lipid Mediat 82:50–59
Hoff U, Lukitsch I, Chaykovska L, Ladwig M, Arnold C, Manthati VL, Fuller TF, Schneider W, Gollasch M, Muller DN, Flemming B, Seeliger E, Luft FC, Falck JR, Dragun D, Schunck WH (2011) Inhibition of 20-HETE synthesis and action protects the kidney from ischemia/reperfusion injury. Kidney Int 79:57–65
Regner KR, Zuk A, Van Why SK, Shames BD, Ryan RP, Falck JR, Manthati VL, McMullen ME, Ledbetter SR, Roman RJ (2009) Protective effect of 20-HETE analogues in experimental renal ischemia reperfusion injury. Kidney Int 75:511–517
Imig JD, Simpkins AN, Renic M, Harder DR (2011) Cytochrome P450 eicosanoids and cerebral vascular function. Expert Rev Mol Med 13:e7. doi:10.1017/51462399411001773
Deng Y, Theken KN, Lee CR (2010) Cytochrome P450 epoxygenases, soluble epoxide hydrolase, and the regulation of cardiovascular inflammation. J Mol Cell Cardiol 48:331–341
Brand-Schieber E, Falck JF, Schwartzman M (2000) Selective inhibition of arachidonic acid epoxidation in vivo. J Physiol Pharmacol 51:655–672
Yu M, Cambj-Sapunar L, Kehl F, Maier KG, Takeuchi K, Miyata N, Ishimoto T, Reddy LM, Falck JR, Gebremedhin D, Harder DR, Roman RJ (2004) Effects of a 20-HETE antagonist and agonists on cerebral vascular tone. Eur J Pharmacol 486:297–306
Williams JM, Murphy S, Burke M, Roman RJ (2010) 20-hydroxyeicosatetraeonic acid: a new target for the treatment of hypertension. J Cardiovasc Pharmacol 56:336–344
Gauthier KM, Falck JR, Reddy LM, Campbell WB (2004) 14,15-EET analogs: characterization of structural requirements for agonist and antagonist activity in bovine coronary arteries. Pharmacol Res 49:515–524
Gauthier KM, Deeter C, Krishna UM, Reddy YK, Bondlela M, Falck JR, Campbell WB (2002) 14,15-Epoxyeicosa-5(Z)-enoic acid: a selective epoxyeicosatrienoic acid antagonist that inhibits endothelium-dependent hyperpolarization and relaxation in coronary arteries. Circ Res 90:1028–1036
Imig JD, Elmarakby A, Nithipatikom K, Wei S, Capdevila JH, Tuniki VR, Sangras B, Anjaiah S, Manthati VL, Reddy DS, Falck JR (2010) Development of epoxyeicosatrienoic acid analogs with in vivo anti-hypertensive actions. Front Physiol 1(article 157):1–8
Wu CC, Schwartzman ML (2011) The role of 20-HETE in androgen-mediated hypertension. Prostaglandins Other Lipid Mediat 96:45–53
Singh H, Cheng J, Deng H, Kemp R, Ishizuka T, Nasjletti A, Schwartzman ML (2007) Vascular cytochrome P450 4A expression and 20-hydroxyeicosatetraenoic acid synthesis contribute to endothelial dysfunction in androgen-induced hypertension. Hypertension 50:123–129
Inoue K, Sodhi K, Puri N, Gotlinger KH, Cao J, Rezzani R, Falck JR, Abraham NG, Laniado-Schwartzman M (2009) Endothelial-specific CYP4A2 overexpression leads to renal injury and hypertension via increased production of 20-HETE. Am J Physiol Renal Physiol 297:F875–F884
Imig JD, Zou AP, Stec DE, Harder DR, Falck JR, Roman RJ (1996) Formation and actions of 20-hydroxyeicosatetraenoic acid in rat renal arterioles. Am J Physiol 270:R217–R227
Cheng J, Wu CC, Gotlinger KH, Zhang F, Falck JR, Narsimhaswamy D, Schwartzman ML (2010) 20-Hydroxy-5,8,11,14-eicosatetraenoic acid mediates endothelial dysfunction via IκB kinase-dependent endothelial nitric-oxide synthase uncoupling. J Pharmacol Exp Ther 332:57–65
Sodhi K, Wu CC, Cheng J, Gotlinger K, Inoue K, Goli M, Falck JR, Abraham NG, Schwartzman ML (2010) CYP4A2-induced hypertension is 20-hydroxyeicosatetraenoic acid- and angiotensin II-dependent. Hypertension 56:871–878
Cheng J, Garcia V, Ding Y, Wu CC, Thakar K, Falck JR, Ramu E, Schwartzman ML (2012) Induction of angiotensin-converting enzyme and activation of the renin-angiotensin system contribute to 20-hydroxyeicosatetraenoic acid-mediated endothelial dysfunction. Arterioscler Thromb Vasc Biol 32:1917–1924
Ward NC, Rivera J, Hodgson J, Puddey IB, Beilin LJ, Falck JR, Croft KD (2004) Urinary 20-hydroxyeicosatetraenoic acid is associated with endothelial dysfunction in humans. Circulation 110:438–443
Imig JD (2013) Epoxyeicosatrienoic acids, 20-hydroxyeicosatetraenoic acid, and renal microvascular function. Prostaglandins Other Lipid Mediat 104–105:2–7
Hoagland KM, Flasch AK, Roman RJ (2003) Inhibitors of 20-HETE formation promote salt-sensitive hypertension in rats. Hypertension 42:669–673
Honeck H, Gross V, Erdmann B, Kargel E, Neunaber R, Milia AF, Schneider W, Luft FC, Schunck WH (2000) Cytochrome P450-dependent renal arachidonic acid metabolism in desoxycorticosterone acetate-salt hypertensive mice. Hypertension 36:610–616
Zhou Y, Luo P, Chang HH, Huang H, Yang T, Dong Z, Wang CY, Wang MH (2008) Clofibrate attenuates blood pressure and sodium retention in DOCA-salt hypertension. Kidney Int 74:1040–1048
Laffer CL, Gainer JV, Waterman MR, Capdevila JH, Laniado-Schwartzman M, Nasjletti A, Brown NJ, Elijovich F (2008) The T8590C polymorphism of CYP4A11 and 20-hydroxyeicosatetraenoic acid in essential hypertension. Hypertension 51:767–772
Williams JS, Hopkins PN, Jeunemaitre X, Brown NJ (2011) CYP4A11 T8590C polymorphism, salt-sensitive hypertension, and renal blood flow. J Hypertens 29:1913–1918
Fava C, Montagnana M, Danese E, Sjogren M, Almgren P, Guidi GC, Hedblad B, Engstrom G, Minuz P, Melander O (2012) The functional variant V433M of the CYP4F2 and the metabolic syndrome in Swedes. Prostaglandins Other Lipid Mediat 98:31–36
Imig JD, Navar LG, Roman RJ, Reddy KK, Falck JR (1996) Actions of epoxygenase metabolites on the preglomerular vasculature. J Am Soc Nephrol 7:2364–2370
Wei Y, Sun P, Wang Z, Yang B, Carroll MA, Wang WH (2006) Adenosine inhibits ENaC via cytochrome P-450 epoxygenase-dependent metabolites of arachidonic acid. Am J Physiol Renal Physiol 290:F1163–F1168
Carroll MA (2012) Role of the adenosine2A receptor-epoxyeicosatrienoic acid pathway in the development of salt-sensitive hypertension. Prostaglandins Other Lipid Mediat 98:39–47
Liclican EL, McGiff JC, Falck JR, Carroll MA (2008) Failure to upregulate the adenosine2A receptor-epoxyeicosatrienoic acid pathway contributes to the development of hypertension in Dahl salt-sensitive rats. Am J Physiol Renal Physiol 295:F1696–F1704
Jung O, Brandes RP, Kim IH, Schweda F, Schmidt R, Hammock BD, Busse R, Fleming I (2005) Soluble epoxide hydrolase is a main effector of angiotensin II-induced hypertension. Hypertension 45:759–765
Minuz P, Jiang H, Fava C, Turolo L, Tacconelli S, Ricci M, Patrignani P, Morganti A, Lechi A, McGiff JC (2008) Altered release of cytochrome P450 metabolites of arachidonic acid in renovascular disease. Hypertension 51:1379–1385
Theken KN, Lee CR (2007) Genetic variation in the cytochrome P450 epoxygenase pathway and cardiovascular disease risk. Pharmacogenomics 8:1369–1383
Spiecker M, Liao J (2006) Cytochrome P450 epoxygenase CYP2J2 and the risk of coronary artery disease. Trends Cardiovasc Med 16:204–208
Zordoky BN, El-Kadi AO (2010) Effect of cytochrome P450 polymorphism on arachidonic acid metabolism and their impact on cardiovascular diseases. Pharmacol Ther 125:446–463
Bonventre JV, Huang Z, Taheri MR, O’Leary E, Li E, Moskowitz MA, Sapirstein A (1997) Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature 390:622–625
Tabuchi S, Uozumi N, Ishii S, Shimizu Y, Watanabe T, Shimizu T (2003) Mice deficient in cytosolic phospholipase A2 are less susceptible to cerebral ischemia/reperfusion injury. Acta Neurochir Suppl 86:169–172
Saito Y, Watanabe K, Fujioka D, Nakamura T, Obata JE, Kawabata K, Watanabe Y, Mishina H, Tamaru S, Kita Y, Shimizu T, Kugiyama K (2012) Disruption of group IVA cytosolic phospholipase A2 attenuates myocardial ischemia-reperfusion injury partly through inhibition of TNF-α-mediated pathway. Am J Physiol Heart Circ Physiol 302:H2018–H2030
Nakamura H, Nemenoff RA, Gronich JH, Bonventre JV (1991) Subcellular characteristics of phospholipase A2 activity in the rat kidney. Enhanced cytosolic, mitochondrial, and microsomal phospholipase A2 enzymatic activity after renal ischemia and reperfusion. J Clin Invest 87:1810–1818
Nithipatikom K, DiCamelli RF, Kohler S, Gumina RJ, Falck JR, Campbell WB, Gross GJ (2001) Determination of cytochrome P450 metabolites of arachidonic acid in coronary venous plasma during ischemia and reperfusion in dogs. Anal Biochem 292:115–124
Nithipatikom K, Gross ER, Endsley MP, Moore JM, Isbell MA, Falck JR, Campbell WB, Gross GJ (2004) Inhibition of cytochrome P450 ω-hydroxylase: a novel endogenous cardioprotective pathway. Circ Res 95:e65–e71
Nithipatikom K, Endsley MP, Moore JM, Isbell MA, Falck JR, Campbell WB, Gross GJ (2006) Effects of selective inhibition of cytochrome P-450 ω-hydroxylases and ischemic preconditioning in myocardial protection. Am J Physiol Heart Circ Physiol 290:H500–H505
Batchu SN, Law E, Brocks DR, Falck JR, Seubert JM (2009) Epoxyeicosatrienoic acid prevents postischemic electrocardiogram abnormalities in an isolated heart model. J Mol Cell Cardiol 46:67–74
Gross GJ, Hsu A, Falck JR, Nithipatikom K (2007) Mechanisms by which epoxyeicosatrienoic acids (EETs) elicit cardioprotection in rat hearts. J Mol Cell Cardiol 42:687–691
Gross GJ, Gauthier KM, Moore J, Falck JR, Hammock BD, Campbell WB, Nithipatikom K (2008) Effects of the selective EET antagonist, 14,15-EEZE, on cardioprotection produced by exogenous or endogenous EETs in the canine heart. Am J Physiol Heart Circ Physiol 294:H2838–H2844
Gross GJ, Gauthier KM, Moore J, Campbell WB, Falck JR, Nithipatikom K (2009) Evidence for role of epoxyeicosatrienoic acids in mediating ischemic preconditioning and postconditioning in dog. Am J Physiol Heart Circ Physiol 297:H47–H52
Yu GG, Zeng XJ, Wang HX, Lu LQ, Zheng SP, Ma LQ, Chang J, Wang J, Zhang DM, Du FH, Zhang LK (2011) Cytochrome P450 2J3/epoxyeicosatrienoic acids mediate the cardioprotection induced by ischaemic post-conditioning, but not preconditioning, in the rat. Clin Exp Pharmacol Physiol 38:63–70
Gross GJ, Baker JE, Moore J, Falck JR, Nithipatikom K (2011) Abdominal surgical incision induces remote preconditioning of trauma (RPCT) via activation of bradykinin receptors (BK2R) and the cytochrome P450 epoxygenase pathway in canine hearts. Cardiovasc Drugs Ther 25:517–522
Motoki A, Merkel MJ, Packwood WH, Cao Z, Liu L, Iliff J, Alkayed NJ, Van Winkle DM (2008) Soluble epoxide hydrolase inhibition and gene deletion are protective against myocardial ischemia-reperfusion injury in vivo. Am J Physiol Heart Circ Physiol 295:H2128–H2134
Batchu SN, Lee SB, Qadhi RS, Chaudhary KR, El-Sikhry H, Kodela R, Falck JR, Seubert JM (2011) Cardioprotective effect of a dual acting epoxyeicosatrienoic acid analogue towards ischaemia reperfusion injury. Br J Pharmacol 162:897–907
Gross GJ, Falck JR, Gross ER, Isbell M, Moore J, Nithipatikom K (2005) Cytochrome P450 and arachidonic acid metabolites: role in myocardial ischemia/reperfusion injury revisited. Cardiovasc Res 68:18–25
Nithipatikom K, Gross GJ (2010) Review article: epoxyeicosatrienoic acids: novel mediators of cardioprotection. J Cardiovasc Pharmacol Ther 15:112–119
Miyata N, Seki T, Tanaka Y, Omura T, Taniguchi K, Doi M, Bandou K, Kametani S, Sato M, Okuyama S, Cambj-Sapunar L, Harder DR, Roman RJ (2005) Beneficial effects of a new 20-hydroxyeicosatetraenoic acid synthesis inhibitor, TS-011 [N-(3-chloro-4-morpholin-4-yl) phenyl-N’-hydroxyimido formamide], on hemorrhagic and ischemic stroke. J Pharmacol Exp Ther 314:77–85
Renic M, Klaus JA, Omura T, Kawashima N, Onishi M, Miyata N, Koehler RC, Harder DR, Roman RJ (2009) Effect of 20-HETE inhibition on infarct volume and cerebral blood flow after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab 29:629–639
Zhang W, Otsuka T, Sugo N, Ardeshiri A, Alhadid YK, Iliff JJ, DeBarber AE, Koop DR, Alkayed NJ (2008) Soluble epoxide hydrolase gene deletion is protective against experimental cerebral ischemia. Stroke 39:2073–2078
Zhang W, Iliff JJ, Campbell CJ, Wang RK, Hurn PD, Alkayed NJ (2009) Role of soluble epoxide hydrolase in the sex-specific vascular response to cerebral ischemia. J Cereb Blood Flow Metab 29:1475–1481
Zhang W, Koerner IP, Noppens R, Grafe M, Tsai HJ, Morisseau C, Luria A, Hammock BD, Falck JR, Alkayed NJ (2007) Soluble epoxide hydrolase: a novel therapeutic target in stroke. J Cereb Blood Flow Metab 27:1931–1940
Lameire N, Van Biesen W, Vanholder R (2005) Acute renal failure. Lancet 365:417–430
Aydin Z, van Zonneveld AJ, de Fijter JW, Rabelink TJ (2007) New horizons in prevention and treatment of ischaemic injury to kidney transplants. Nephrol Dial Transplant 22:342–346
Karkouti K, Wijeysundera DN, Yau TM, Callum JL, Cheng DC, Crowther M, Dupuis JY, Fremes SE, Kent B, Laflamme C, Lamy A, Legare JF, Mazer CD, McCluskey SA, Rubens FD, Sawchuk C, Beattie WS (2009) Acute kidney injury after cardiac surgery: focus on modifiable risk factors. Circulation 119:495–502
Sutton TA, Fisher CJ, Molitoris BA (2002) Microvascular endothelial injury and dysfunction during ischemic acute renal failure. Kidney Int 62:1539–1549
Bonventre JV, Zuk A (2004) Ischemic acute renal failure: an inflammatory disease? Kidney Int 66:480–485
Nilakantan V, Maenpaa C, Jia G, Roman RJ, Park F (2008) 20-HETE-mediated cytotoxicity and apoptosis in ischemic kidney epithelial cells. Am J Physiol Renal Physiol 294:F562–F570
Wang Y, Hill JA (2010) Electrophysiological remodeling in heart failure. J Mol Cell Cardiol 48:619–632
Xu D, Li N, He Y, Timofeyev V, Lu L, Tsai HJ, Kim IH, Tuteja D, Mateo RK, Singapuri A, Davis BB, Low R, Hammock BD, Chiamvimonvat N (2006) Prevention and reversal of cardiac hypertrophy by soluble epoxide hydrolase inhibitors. Proc Natl Acad Sci U S A 103:18733–18738
Monti J, Fischer J, Paskas S, Heinig M, Schulz H, Gosele C, Heuser A, Fischer R, Schmidt C, Schirdewan A, Gross V, Hummel O, Maatz H, Patone G, Saar K, Vingron M, Weldon SM, Lindpaintner K, Hammock BD, Rohde K, Dietz R, Cook SA, Schunck WH, Luft FC, Hubner N (2008) Soluble epoxide hydrolase is a susceptibility factor for heart failure in a rat model of human disease. Nat Genet 40:529–537
Westphal C, Spallek B, Konkel A, Marko L, Qadri F, Degraff LM, Schubert C, Bradbury JA, Regitz-Zagrosek V, Falck JR, Zeldin DC, Muller DN, Schunck WH, Fischer R (2013) CYP2J2 overexpression protects against arrhythmia susceptibility in cardiac hypertrophy. PLoS One 8:e73490. doi:10.1371/journal.pone.0073490
Zhang Y, El-Sikhry H, Chaudhary KR, Batchu SN, Shayeganpour A, Jukar TO, Bradbury JA, Graves JP, DeGraff LM, Myers P, Rouse DC, Foley J, Nyska A, Zeldin DC, Seubert JM (2009) Overexpression of CYP2J2 provides protection against doxorubicin-induced cardiotoxicity. Am J Physiol Heart Circ Physiol 297:H37–H46
Wang X, Ni L, Yang L, Duan Q, Chen C, Edin ML, Zeldin DC, Wang DW (2014) CYP2J2-derived epoxyeicosatrienoic acids suppress endoplasmic reticulum stress in heart failure. Mol Pharmacol 85:105–115
Tse MM, Aboutabl ME, Althurwi HN, Elshenawy OH, Abdelhamid G, El-Kadi AO (2013) Cytochrome P450 epoxygenase metabolite, 14,15-EET, protects against isoproterenol-induced cellular hypertrophy in H9c2 rat cell line. Vascul Pharmacol 58:363–373
Alsaad AM, Zordoky BN, Tse MM, El-Kadi AO (2013) Role of cytochrome P450-mediated arachidonic acid metabolites in the pathogenesis of cardiac hypertrophy. Drug Metab Rev 45:173–195
Xiao YF (2007) Cyclic AMP-dependent modulation of cardiac L-type Ca2+ and transient outward K+ channel activities by epoxyeicosatrienoic acids. Prostaglandins Other Lipid Mediat 82:11–18
Certikova Chabova V, Walkowska A, Kompanowska-Jezierska E, Sadowski J, Kujal P, Vernerova Z, Vanourkova Z, Kopkan L, Kramer HJ, Falck JR, Imig JD, Hammock BD, Vaneckova I, Cervenka L (2010) Combined inhibition of 20-hydroxyeicosatetraenoic acid formation and of epoxyeicosatrienoic acids degradation attenuates hypertension and hypertension-induced end-organ damage in Ren-2 transgenic rats. Clin Sci (Lond) 118:617–632
Zeldin DC, Foley J, Goldsworthy SM, Cook ME, Boyle JE, Ma J, Moomaw CR, Tomer KB, Steenbergen C, Wu S (1997) CYP2J subfamily cytochrome P450s in the gastrointestinal tract: expression, localization, and potential functional significance. Mol Pharmacol 51:931–943
Jacobs ER, Zeldin DC (2001) The lung HETEs (and EETs) up. Am J Physiol Heart Circ Physiol 280:H1–H10
Loot AE, Fleming I (2011) Cytochrome P450-derived epoxyeicosatrienoic acids and pulmonary hypertension: central role of transient receptor potential C6 channels. J Cardiovasc Pharmacol 57:140–147
Sacerdoti D, Gatta A, McGiff JC (2003) Role of cytochrome P450-dependent arachidonic acid metabolites in liver physiology and pathophysiology. Prostaglandins Other Lipid Mediat 72:51–71
Terashvili M, Tseng LF, Wu HE, Narayanan J, Hart LM, Falck JR, Pratt PF, Harder DR (2008) Antinociception produced by 14,15-epoxyeicosatrienoic acid is mediated by the activation of β-endorphin and met-enkephalin in the rat ventrolateral periaqueductal gray. J Pharmacol Exp Ther 326:614–622
Wagner K, Inceoglu B, Hammock BD (2011) Soluble epoxide hydrolase inhibition, epoxygenated fatty acids and nociception. Prostaglandins Other Lipid Mediat 96:76–83
Falck JR, Manna S, Moltz J, Chacos N, Capdevila J (1983) Epoxyeicosatrienoic acids stimulate glucagon and insulin release from isolated rat pancreatic islets. Biochem Biophys Res Commun 114:743–749
Zeldin DC, Foley J, Boyle JE, Moomaw CR, Tomer KB, Parker C, Steenbergen C, Wu S (1997) Predominant expression of an arachidonate epoxygenase in islets of Langerhans cells in human and rat pancreas. Endocrinology 138:1338–1346
Mustafa S, Sharma V, McNeill JH (2009) Insulin resistance and endothelial dysfunction: are epoxyeicosatrienoic acids the link? Exp Clin Cardiol 14:e41–e50
Chen L, Fan C, Zhang Y, Bakri M, Dong H, Morisseau C, Maddipati KR, Luo P, Wang CY, Hammock BD, Wang MH (2013) Beneficial effects of inhibition of soluble epoxide hydrolase on glucose homeostasis and islet damage in a streptozotocin-induced diabetic mouse model. Prostaglandins Other Lipid Mediat 104–105:42–48
Cheranov SY, Karpurapu M, Wang D, Zhang B, Venema RC, Rao GN (2008) An essential role for SRC-activated STAT-3 in 14,15-EET-induced VEGF expression and angiogenesis. Blood 111:5581–5591
Fleming I (2011) The cytochrome P450 pathway in angiogenesis and endothelial cell biology. Cancer Metastasis Rev 30:541–555
Panigrahy D, Greene ER, Pozzi A, Wang DW, Zeldin DC (2011) EET signaling in cancer. Cancer Metastasis Rev 30:525–540
Pozzi A, Popescu V, Yang S, Mei S, Shi M, Puolitaival SM, Caprioli RM, Capdevila JH (2010) The anti-tumorigenic properties of peroxisomal proliferator-activated receptor α are arachidonic acid epoxygenase-mediated. J Biol Chem 285:12840–12850
Yang S, Wei S, Pozzi A, Capdevila JH (2009) The arachidonic acid epoxygenase is a component of the signaling mechanisms responsible for VEGF-stimulated angiogenesis. Arch Biochem Biophys 489:82–91
Wang D, Dubois RN (2012) Epoxyeicosatrienoic acids: a double-edged sword in cardiovascular diseases and cancer. J Clin Invest 122:19–22
Spector AA (2009) Arachidonic acid cytochrome P450 epoxygenase pathway. J Lipid Res 50(Suppl):S52–S56
Yang W, Tuniki VR, Anjaiah S, Falck JR, Hillard CJ, Campbell WB (2008) Characterization of epoxyeicosatrienoic acid binding site in U937 membranes using a novel radiolabeled agonist, 20-125i-14,15-epoxyeicosa-8(Z)-enoic acid. J Pharmacol Exp Ther 324:1019–1027
Chen Y, Falck JR, Manthati VL, Jat JL, Campbell WB (2011) 20-Iodo-14,15-epoxyeicosa-8(Z)-enoyl-3-azidophenylsulfonamide: photoaffinity labeling of a 14,15-epoxyeicosatrienoic acid receptor. Biochemistry 50:3840–3848
Kosel M, Wild W, Bell A, Rothe M, Lindschau C, Steinberg CE, Schunck WH, Menzel R (2011) Eicosanoid formation by a cytochrome P450 isoform expressed in the pharynx of Caenorhabditis elegans. Biochem J 435:689–700
Ma DK, Rothe M, Zheng S, Bhatla N, Pender CL, Menzel R, Horvitz HR (2013) Cytochrome P450 drives a HIF-regulated behavioral response to reoxygenation by C. elegans. Science 341:554–558
Harmon SD, Fang X, Kaduce TL, Hu S, Raj Gopal V, Falck JR, Spector AA (2006) Oxygenation of ω-3 fatty acids by human cytochrome P450 4F3B: effect on 20-hydroxyeicosatetraenoic acid production. Prostaglandins Leukot Essent Fatty Acids 75:169–177
Kulas J, Schmidt C, Rothe M, Schunck WH, Menzel R (2008) Cytochrome P450-dependent metabolism of eicosapentaenoic acid in the nematode Caenorhabditis elegans. Arch Biochem Biophys 472:65–75
Xiao B, Li X, Yan J, Yu X, Yang G, Xiao X, Voltz JW, Zeldin DC, Wang DW (2010) Overexpression of cytochrome P450 epoxygenases prevents development of hypertension in spontaneously hypertensive rats by enhancing atrial natriuretic peptide. J Pharmacol Exp Ther 334:784–794
Koeners MP, Wesseling S, Ulu A, Sepulveda RL, Morisseau C, Braam B, Hammock BD, Joles JA (2011) Soluble epoxide hydrolase in the generation and maintenance of high blood pressure in spontaneously hypertensive rats. Am J Physiol Endocrinol Metab 300:E691–E698
Zhao X, Pollock DM, Inscho EW, Zeldin DC, Imig JD (2003) Decreased renal cytochrome P450 2C enzymes and impaired vasodilation are associated with angiotensin salt-sensitive hypertension. Hypertension 41:709–714
Wang MH, Smith A, Zhou Y, Chang HH, Lin S, Zhao X, Imig JD, Dorrance AM (2003) Downregulation of renal CYP-derived eicosanoid synthesis in rats with diet-induced hypertension. Hypertension 42:594–599
Sodhi K, Inoue K, Gotlinger KH, Canestraro M, Vanella L, Kim DH, Manthati VL, Koduru SR, Falck JR, Schwartzman ML, Abraham NG (2009) Epoxyeicosatrienoic acid agonist rescues the metabolic syndrome phenotype of HO-2-null mice. J Pharmacol Exp Ther 331:906–916
Herse F, Lamarca B, Hubel CA, Kaartokallio T, Lokki AI, Ekholm E, Laivuori H, Gauster M, Huppertz B, Sugulle M, Ryan MJ, Novotny S, Brewer J, Park JK, Kacik M, Hoyer J, Verlohren S, Wallukat G, Rothe M, Luft FC, Muller DN, Schunck WH, Staff AC, Dechend R (2012) Cytochrome P450 subfamily 2J polypeptide 2 expression and circulating epoxyeicosatrienoic metabolites in preeclampsia. Circulation 126:2990–2999
Huang H, Chang HH, Xu Y, Reddy DS, Du J, Zhou Y, Dong Z, Falck JR, Wang MH (2006) Epoxyeicosatrienoic acid inhibition alters renal hemodynamics during pregnancy. Exp Biol Med (Maywood) 231:1744–1752
Blanton A, Nsaif R, Hercule H, Oyekan A (2006) Nitric oxide/cytochrome P450 interactions in cyclosporin A-induced effects in the rat. J Hypertens 24:1865–1872
Fava C, Montagnana M, Melander O (2009) Overexpression of cytochrome P450 4F2 in mice increases 20-hydroxyeicosatetraenoic acid production and arterial blood pressure. Kidney Int 76:913, author reply 913–914
Liu X, Zhao Y, Wang L, Yang X, Zheng Z, Zhang Y, Chen F, Liu H (2009) Overexpression of cytochrome P450 4F2 in mice increases 20-hydroxyeicosatetraenoic acid production and arterial blood pressure. Kidney Int 75:1288–1296
Ding Y, Wu CC, Garcia V, Dimitrova I, Weidenhammer A, Joseph G, Zhang F, Manthati VL, Falck JR, Capdevila JH, Schwartzman ML (2013) 20-HETE induces remodeling of renal resistance arteries independent of blood pressure elevation in hypertension. Am J Physiol Renal Physiol 305:F753–F763
Gross GJ, Hsu A, Gross ER, Falck JR, Nithipatikom K (2013) Factors mediating remote preconditioning of trauma in the rat heart: central role of the cytochrome P450 epoxygenase pathway in mediating infarct size reduction. J Cardiovasc Pharmacol Ther 18:38–45
Koerner IP, Zhang W, Cheng J, Parker S, Hurn PD, Alkayed NJ (2008) Soluble epoxide hydrolase: regulation by estrogen and role in the inflammatory response to cerebral ischemia. Front Biosci 13:2833–2841
Acknowledgments
This review is dedicated to the late John Charles (Jack) McGiff, MD and Professor of Pharmacology. Professor McGiff made seminal discoveries on the role of CYP eicosanoids in blood pressure regulation and he inspired many students and colleagues to follow his way of successfully combining biochemistry and pathophysiology for understanding the mechanisms of complex cardiovascular diseases. This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG): Schu822/5; FOR 1054 and Schu822/7-1; FOR 1368.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Westphal, C., Konkel, A., Schunck, WH. (2015). Cytochrome P450 Enzymes in the Bioactivation of Polyunsaturated Fatty Acids and Their Role in Cardiovascular Disease. In: Hrycay, E., Bandiera, S. (eds) Monooxygenase, Peroxidase and Peroxygenase Properties and Mechanisms of Cytochrome P450. Advances in Experimental Medicine and Biology, vol 851. Springer, Cham. https://doi.org/10.1007/978-3-319-16009-2_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-16009-2_6
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-16008-5
Online ISBN: 978-3-319-16009-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)