
Abstract
Summary
Odanacatib is a cathepsin K inhibitor investigated for the treatment of postmenopausal osteoporosis. Phase 2 data indicate that 50 mg once weekly inhibits bone resorption and increases bone mineral density, with only a transient decrease in bone formation. We describe the background, design and participant characteristics for the phase 3 registration trial.
Introduction
Odanacatib (ODN) is a selective cathepsin K inhibitor being evaluated for the treatment of osteoporosis. In a phase 2 trial, ODN 50 mg once weekly reduced bone resorption while preserving bone formation and progressively increased BMD over 5 years. We describe the phase III Long-Term ODN Fracture Trial (LOFT), an event-driven, randomized, blinded placebo-controlled trial, with preplanned interim analyses to permit early termination if significant fracture risk reduction was demonstrated. An extension was planned, with participants remaining on their randomized treatment for up to 5 years, then transitioning to open-label ODN.
Methods
The three primary outcomes were radiologically determined vertebral, hip, and clinical non-vertebral fractures. Secondary end points included clinical vertebral fractures, BMD, bone turnover markers, and safety and tolerability, including bone histology. Participants were women, 65 years or older, with a BMD T-score ≤−2.5 at the total hip (TH) or femoral neck (FN) or with a prior radiographic vertebral fracture and a T-score ≤−1.5 at the TH or FN. They were randomized to ODN or placebo tablets. All received weekly vitamin D3 (5600 international units (IU)) and daily calcium supplements as needed to ensure a daily intake of approximately 1200 mg.
Results
Altogether, 16,713 participants were randomized at 387 centers. After a planned interim analysis, an independent data monitoring committee recommended that the study be stopped early due to robust efficacy and a favorable benefit/risk profile. Following the base study closeout, 8256 participants entered the study extension.
Conclusions
This report details the background and study design of this fracture end point trial and describes the baseline characteristics of its participants.
Similar content being viewed by others

Introduction
Osteoporosis is a metabolic bone disease characterized by a chronic excess of bone resorption relative to bone formation, leading to progressive bone loss, deterioration of bone microarchitecture, and increased risk of fracture [1].
Bone formation and resorption are tightly coordinated during the remodeling cycle. Osteocytes exhibit a mechanosensory capacity and are major regulators of bone remodeling [2]. Osteoclast-mediated bone resorption releases growth factors from the bone matrix [3, 4] that increase osteoblast progenitor number, recruitment, differentiation, and activity. In addition, osteoclasts regulate osteoblasts directly via cell-cell contact [3, 5] and by secreting additional factors [6] that increase osteoblast number and/or activity. Osteoclast number and activity are regulated by receptor activator of nuclear factor kappa-B ligand (RANKL), a membrane-bound protein produced by neighboring osteocytes and osteoblasts [7]. Thus, the products of osteoclast activity play a key role in the complex interactive regulation of bone remodeling.
With the exception of parathyroid hormone (PTH) and its analogs, all agents currently used in the treatment of osteoporosis, such as bisphosphonates, selective estrogen receptor modulators, and the anti-RANKL antibody, act primarily by decreasing osteoclast-mediated bone resorption [8], allowing osteoblasts to increase bone mass by filling in the remodeling space [9, 10]. Furthermore, slower bone remodeling allows increased bone mineralization, at least for some agents, and leads to a reduction in cortical porosity [9–12]. However, inhibition of osteoclast activity also leads to a secondary reduction in bone formation [8, 13], limiting the ultimate increase in bone mass.
The mechanism of osteoclast-mediated bone resorption is well characterized [7]. Osteoclasts attach to the bone surface and secrete hydrogen ions and proteolytic enzymes into the remodeling space. The acidic environment dissolves bone mineral and exposes bone matrix proteins. Cathepsin K (CatK) is the primary osteoclast-produced protease involved in the degradation of type I collagen and other bone matrix proteins [14]; its pH optimum is in the range of the acidic remodeling space.
Congenital absence of CatK in humans results in pycnodysostosis, a rare autosomal recessive osteochondrodysplasia. This disease is characterized not only by high bone mass but also increased risk of fractures and bone deformities [15]. Importantly, the increased fragility and bone deformities are not present in heterozygotes [15]. Targeted disruption of the CatK gene in mice produced a high bone mass phenotype [16] while overexpression of CatK increased bone turnover and decreased trabecular bone volume [17].
The effect of CatK inhibition has been studied in estrogen-deficient non-human primates and rabbits. In these studies, CatK inhibitors significantly reduced bone resorption while relatively preserving bone formation, resulting in both increased BMD and bone strength, in contrast to congenital absence of CatK [16]. CatK inhibition decreased the removal of bone matrix protein but did not reduce osteoclast number or affect other osteoclast activities, such as the production of osteoblast-stimulating factors [16].
Odanacatib (ODN, MK-0822) is a selective, orally administered CatK inhibitor. Unlike basic CatK inhibitors, ODN is neutral, so it does not accumulate in the acidic environment of lysosomes, which could lead to off-target inhibition of other cathepsins [18, 19]. ODN is metabolized by CYP3A4, has an apparent terminal half-life of approximately 40 to 80 h [20, 21], and its absorption is not impaired by food intake [20, 21].
A phase IIb dose-ranging trial and its extensions have demonstrated progressive dose-dependent increases in BMD in postmenopausal women with low BMD [22–24]. ODN substantially reduced biochemical markers of bone resorption. However, markers of bone formation decreased to a lesser degree than was previously observed with other anti-resorptive treatments and returned towards baseline within 2 years, on continued treatment [22–24]. Histomorphometry of bone biopsies from participants in the phase IIb study indicated that osteoclast number was maintained [22], confirming preclinical results [16]. Lack of reduction of serum TRAP5b levels with ODN treatment in the phase IIb trial confirmed osteoclast viability. Discontinuation of ODN treatment resulted in increased bone turnover and reduction in BMD, which returned to baseline within 1 to 2 years [23]. Based on BMD data after 12 months from the phase IIb trial, ODN 50 mg once weekly was selected as the dosing regimen for the phase III trial [22].
A prospective study was designed to assess the anti-fracture efficacy and safety of ODN in postmenopausal women with osteoporosis. This report describes the design of this study and presents the key baseline characteristics of its participants.
Study design
Overview of study design
The study, odanacatib Protocol 18, was designed by the sponsor, Merck & Co. Inc., Whitehouse Station, NJ, USA, in collaboration with a Scientific Advisory Committee (SAC) that provided input on the clinical development program.
The study was planned as a multinational, randomized, double-blind, event-driven placebo-controlled trial of approximately 5 years duration and was approved by governing ethical review committees (ERCs) and institutional review boards (IRBs). Provision was made for early termination of the study if criteria for reduction of fracture risk versus placebo were met in prespecified interim analyses (see “Statistical methods”).
Study participants were women at least 65 years of age and postmenopausal for at least 5 years, who were osteoporosis treatment-naïve or had been exposed to minimal prior osteoporosis treatment. Two categories of participants were eligible (Table 1):
-
Women with a prior radiographic vertebral fracture were required to have a BMD T-score ≤−1.5 at either the femoral neck (FN) or total hip (TH).
-
Those without a prior vertebral fracture were required to have a BMD T-score ≤−2.5 at the FN or TH, using the NHANES III 1998 database (reference values are those for Caucasian young adult women).
In either category, women with prior hip fractures at any time or clinical fragility fractures within the prior 2 years or whose BMD T-score was <−4.0 at either the FN or TH were excluded to minimize individual participant risk, unless they were unable or unwilling to use available osteoporosis treatments.
This trial was designed to test the hypotheses that treatment with ODN 50 mg once weekly reduces the risk of radiographically detected vertebral, clinical hip, and clinical non-vertebral fractures (all primary end points) in postmenopausal women with osteoporosis. Secondary end points included the incidence of clinical vertebral fractures; change in height; BMD at the TH, FN, trochanter, lumbar spine (LS), 1/3 radius; biochemical markers of bone formation and resorption; and safety and tolerability, including bone histology.
Participants were randomized in a 1:1 ratio to take either ODN 50 mg or matching placebo once weekly, without regard to food or physical position. They were given 5600 international units (IU) of vitamin D3 weekly and calcium supplements as needed to ensure a total daily calcium intake of approximately 1200 mg. A dietary calcium questionnaire was administered at intake and at yearly intervals during the trial. Following screening and randomization visits, study visits in the clinic were scheduled every 3 months in the first year of the study and every 6 months thereafter. Telephone contact alternated with clinic visits every 3 months after the first year.
Rationale for study design
This trial was designed with primary fracture end points, consistent with regulatory requirements for new drugs for the treatment of postmenopausal osteoporosis. It employed a placebo control group, consistent with previous and current registration trials of drugs for that indication. This approach has been the subject of extensive discussion [25], including a US FDA Advisory Committee Meeting [26] which provided a frame of reference for recent trial design. Placebo-controlled pivotal trials in osteoporosis were considered acceptable, provided that appropriate limits are placed on the severity of disease in those individuals who could otherwise be treated with available medication. Background treatment with adequate calcium and vitamin D3 intake for all participants was also regarded as an important consideration. Limits regarding the range of acceptable BMD and fractures prior to study entry as well as monitoring for excessive on-study bone density decreases have been generally consistent in more recently completed [27] and ongoing [28, 29] phase III trials.
Accordingly, the present study excluded participants at relatively high risk for fracture, using specific criteria based on BMD and prior fracture history, and employed safeguards to protect participant safety. The trial was overseen by an external data monitoring committee (DMC) that periodically reviewed unblinded safety data. BMD was measured yearly and identified participants with excessive bone loss (EBL) (predefined as >7 % from baseline at any time at the LS or TH). If the decrease was confirmed, the participant was then discontinued from study medication and treated conventionally. Adverse events of interest were referred for adjudication by panels of experts with relevant scientific expertise (see section on “Adverse event reporting and adjudication”).
The study was powered to detect a significant effect of ODN to reduce the risk of hip fractures. Because of the safety criteria at enrollment, the baseline individual hip fracture risk in the study population was limited. Hence, the sample size estimate was about twice that of the largest osteoporosis trial. The event-driven design of the study allowed for some uncertainty in the estimate of the rate at which participants would experience fracture events.
Recruitment, follow-up, and assessments
Recruitment
Informed consent was obtained from all study participants before any study procedure was performed. It was required that a qualified physician discuss alternative treatment options with each participant. At the request of a regulatory agency, participant enrollment was conducted in two phases to allow expansion of the safety experience for ODN before the entire study was enrolled.
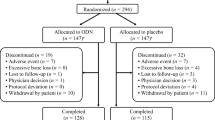
The first phase (the lead cohort) randomized 1499 participants between November 2007 and March 2008. After 9 months of treatment, the lead cohort safety data were evaluated by the DMC (see below for details), which recommended that the enrollment of the remainder of the participants (the main cohort) could commence. The enrollment of 15,214 participants in the main cohort occurred from January to November 2009 (Fig. 1). Thus, a total of 16,713 women were recruited at 387 investigational sites in 40 countries.

CONSORT Diagram. The asterisk indicates 642 randomized participants were excluded from all analyses due to the following reasons: allocation of duplicate allocation numbers (n = 3), took no study medication (n = 156), or because their study site was closed and excluded from all efficacy and safety data analyses (n = 483). The primary all-patients-as-treated analysis will be based on 16,071 participants, of whom 1373 were in the lead cohort. The dagger indicates 245 of the 8256 patients were found to be ineligible because they had experienced excessive bone loss detected at the transitional visit
At one investigative site, sponsor inspections identified issues that materially affected data integrity. The sponsor determined that these findings warranted exclusion of that site’s data from the primary efficacy and safety analyses and informed the US FDA. (All data, including those excluded from the primary analyses, are to be provided in regulatory submissions). Data from 483 participants were affected, including 111 in the lead cohort. An additional 159 participants were excluded from the analysis because they never took study medication (N = 156) or because they had been assigned duplicate allocation numbers (N = 3). Thus, the number of evaluable participants was 16,071, including 1373 in the lead cohort.
Randomization and stratification
Randomization was performed by a computer-generated allocation schedule using an interactive voice recognition system (IVRS), which randomly assigned each participant to ODN or placebo. Participants were assigned to one of two strata, according to vertebral fracture status. Participants without prior vertebral fracture were assigned to the no-prior-vertebral-fracture stratum. At least two thirds of the participants in the no-prior-fracture stratum were required to be at least 70 years of age. Participants with a prior vertebral fracture were assigned to the prior-vertebral fracture stratum. IVRS was used to keep track of enrollment and the number of participants randomized into each stratum and to close a stratum as needed. IVRS was also available for emergency unblinding if this were required for an individual participant’s safety.
Blinding
Study participants, investigators and their staff, and sponsor’s personnel were blinded to treatment allocation. Only the DMC and support statistical personnel (see below) had access to unblinded data. Selected adverse events (AEs) and changes in selected laboratory results were provided to the FDA in an unblinded fashion, while maintaining the blind for study participants, investigators, and other sponsor personnel.
Assessments
Prior to randomization, a medical history, including assessment of concomitant medications, physical examination, BMD measurements by dual-energy X-ray absorptiometry (DXA), lateral spine radiographs for vertebral fracture assessment, laboratory assessments, and review of inclusion and exclusion criteria were performed. Follow-up assessments were conducted as follows: limited physical examination including vital signs and laboratory assessment every 3 months during the first year, and then every 6 months. Drug accounting and AE assessments were performed at every visit. Lateral spine radiographs were performed at baseline, month 6, month 12, and every 12 months thereafter. BMD was measured at baseline and yearly thereafter. Height was assessed using a calibrated stadiometer at baseline and yearly.
Radiographic vertebral fracture assessment
Spine radiographs were evaluated at a central site (Synarc, Newark, CA, USA). Screening spine radiographs were evaluated for the presence or absence of a vertebral fracture using the Genant semi-quantitative scale [30]. Only vertebral fractures in the region T4 to L4 were counted. Participants with Genant grade 1–3 deformities (mild, moderate, or severe) were included in the stratum with prior vertebral fracture.
Follow-up radiographs were evaluated to detect incident (new or worsening) vertebral fractures, first using the Genant semi-quantitative method and upon detection of a fracture, morphometric analysis for confirmation.
BMD measurements
BMD was measured using Lunar, Hologic, or Norland densitometers, at femoral sites (total hip, femoral neck, and trochanter) and the lumbar spine in all participants and at the 1/3 radius and total body (excluding head) in a randomly selected 10 % subset of participants from sites with appropriate equipment. The central DXA assessment site (Synarc, Newark, CA, USA) performed QC assessment of all DXA scanners and analysis of all DXA scans.
Laboratory assessments
Serum chemistry, including serum calcium (total and corrected for serum albumin), phosphate, magnesium, liver function tests, hematology, and urinalysis were performed at baseline and follow-up. Serum 25-hydroxyvitamin D, PTH, and biochemical markers of bone turnover were measured in the same subset of participants who underwent 1/3 radius and total body DXA measurements as described above. The following bone turnover markers were measured by Synarc Labs (Lyon, France) on specimens obtained in the fasting state: urinary N-telopeptides of type I collagen (NTx, VITROS ECL/ECIQ, Ortho Clinical Diagnostics, Rochester NY, USA), serum C-telopeptides of type I collagen (CTx, Elecsys-CrossLaps/serum ECL, Roche Diagnostics, Mannheim Germany), serum procollagen type 1 N-terminal propeptide (P1NP, Elecsys/serum ECL kit, Roche Diagnostics, Mannheim Germany), and serum bone-specific alkaline phosphatase (BSAP, Access Ostase, Beckman Coulter, Brea, CA, USA). Fasting urine and serum specimens were archived from each participant at all visits.
Bone biopsy
Transilial bone biopsy was an optional procedure in participants who provided a separate consent. A total of 341 biopsies were obtained from 272 participants. Forty biopsies were obtained at baseline and 301 after 12, 24, or 36 months. Double-fluorochrome labeling with either tetracycline or demeclocycline was used to assess bone formation. The biopsy specimens were analyzed at the Osteoporosis Research Center, Creighton University, Omaha, NE, USA using histomorphometric and micro-CT techniques [31]. Biopsy examiners were blinded to participant identity, treatment allocation and time-point. Histomorphometric variables were measured, calculated, and expressed according to the guidelines of the American Society of Bone and Mineral research (ASBMR) Nomenclature Committee [32].
Clinical fracture end point assessment
Clinical vertebral and non-vertebral fractures were key end points of the study. Therefore, all fractures reported as AEs were adjudicated by a clinical adjudication committee (CAC) of radiologists at the central evaluation site (Synarc, Newark, CA, USA). Based on clinical history and radiographs, the CAC determined whether a fracture was confirmed; its anatomic location and laterality; and whether it was osteoporotic or due to trauma, stress, or other pathology (neoplasm or infection). The adjudication involved a two-round process. A unanimous vote from three CAC members, chosen at random from a panel of four, who reviewed the participant data independently via internet-based software (Virtual Panel System, Parexel International, Waltham, MA, USA), was required to complete the adjudication during the first review. If the vote were not unanimous, then all four CAC members would review the case in a conference (second review) and a three-fourths majority vote was required to confirm the fracture. The CAC could also determine that insufficient data were available to complete the adjudication of a fracture event. Fractures that were possible atypical femoral shaft fracture events were adjudicated according to the ASBMR criteria and were always discussed by four CAC members in a one-round process [33, 34].
Adverse event reporting and adjudication
At each clinic or telephone visit, participants were queried about interval AEs and the responses recorded. Participants were also instructed to report any serious AEs such as hospitalization to the investigational sites immediately. Changes in laboratory parameters were evaluated by the investigators to determine whether they were clinically significant and reportable as AEs.
Several categories of specific AEs were designated for adjudication by external CAC committees (cardiovascular, cerebrovascular, dental, skin thickening and hardening, delayed fracture union, and serious respiratory events) to confirm their occurrence and specific attributes. These AEs, rationale for their adjudication, data collected for the adjudication, and specific attributes to be adjudicated are summarized in Table 2. These AEs were adjudicated in a two-round process similar to that previously described for fractures. Adjudication of all AEs was managed by a central site (Parexel International, Waltham, MA, USA), which was responsible for collection of all clinical data, electronic distribution to the CAC members, coordination of CAC member conferences, collation of adjudication results, and final data transfer to the sponsor.
Discontinuation criteria
Participants were discontinued from blinded study drug if the investigator considered that the risk outweighed the benefit of continued participation on blinded therapy for an individual participant. Additionally, participants were discontinued from blinded study drug if they experienced excessive bone loss, as described above; initiated treatment with a strong CYP3A4 inducer (e.g., carbamazepine, phenytoin, rifampin, St. John’s wort) for longer than 6 months; commenced treatment with other drugs for osteoporosis; or had persistent elevations in serum transaminases or bilirubin. The sponsor provided alendronate 70 mg OW to patients who were discontinued from study drug, although other treatment options were not prohibited. Participants were followed for outcome and safety monitoring through the end of the base trial even if blinded study therapy was discontinued.
Data monitoring committee
The ODN clinical development program, including this fracture outcome study, was monitored by a DMC, which consisted of seven experts not involved in the conduct of the study, including six physicians, five of whom were experts in osteoporosis with subspecialty expertise in endocrinology, rheumatology, geriatrics, and epidemiology; a dermatologist; and a non-physician statistician. The DMC was supported by a statistician and statistical programmer from the sponsor. Only the DMC and their support statistical personnel had access to unblinded data from the trial, with the exception that safety data were provided to the FDA in an unblinded fashion as described above. Since September 2007, the DMC has met at intervals of approximately 4 months to review safety data from the ODN program, including data from this trial, and to make recommendations on the conduct of the trials. The DMC was also responsible for the review of the two planned interim efficacy analyses and had the option of recommending to the sponsor to terminate the study early due to futility or due to robust efficacy and a favorable benefit-risk profile of the drug.
Ancillary studies
Population pharmacokinetics study
Three specimens were collected from each participant in the lead cohort for the measurement of plasma ODN levels to confirm findings from phase I. Information was collected with regard to timing of the collection and last dose of ODN and characteristics of the meal prior to last dose.
Special imaging sub-study
A sub-study of 164 participants investigated the effect of ODN on volumetric BMD of the hip and spine and, specifically, on the trabecular and cortical components at each site, using quantitative computed tomography (QCT, instruments from GE Medical Systems, Phillips, Siemens, and Toshiba). High-resolution peripheral QCT (Scanco Xtreme CT) was employed to investigate the effect of ODN on bone microarchitecture at the distal radius and distal tibia in some of these participants. Imaging procedures were performed yearly, and all participants in this study had biochemical markers of bone turnover measured at these time-points. Only participants in this sub-study were asked to consent to optional bone biopsies at baseline and 12 months.
Sarcopenia end point sub-study
This was an exploratory sub-study of 556 participants designed to identify molecular signatures of sarcopenia using DNA and RNA biomarkers in the blood and to examine changes in appendicular lean body mass, measured by total body DXA, and physical function, measured by the Short Physical Performance Battery (SPPB).
Participation in the special imaging and sarcopenia sub-studies was optional and involved separate consent forms from those used for the main study.
Statistical methods
Data analysis
The primary end points of the study were the first morphometrically confirmed vertebral fracture per participant, first osteoporotic clinical hip fracture confirmed by adjudication, and first osteoporotic clinical non-vertebral fracture confirmed by adjudication. There was a hierarchical order from morphometric vertebral fractures (the first primary end point) to the other two end points; there was no order set between hip and non-vertebral fractures.
The all-patients-as-treated population (n = 16,071) was used for safety analyses and included all participants who took at least one dose of study medication and had at least one follow-up visit with the exclusions noted above. Treatment groups were compared using the Miettinen and Nurminen method [35], adjusted for differential follow-up period, for prespecified sets of AEs of clinical relevance.
The full-analysis population was used for efficacy analyses. With the exclusions noted above in “Recruitment,” it included all randomized participants who took at least one dose of study medication regardless of protocol violation, adherence to study medication, or early discontinuation from the study and had at least one follow-up efficacy assessment. The follow-up period was from randomization to study termination. The number of patients included in the efficacy analyses varied according to the number and timing of observations for each end point.
Morphometrically confirmed vertebral fracture data were evaluated with a generalized linear model for binary data with the complementary log-log transformation of the probability of an event up to the time-point. Clinical hip and non-vertebral fracture data were analyzed by time-to-event methods using Kaplan-Meier estimates and a Cox proportional hazards model.
A step-down procedure [36] controlled for increased false positive error risk due to multiple primary end points, in the order of (1) morphometric vertebral fractures and (2) clinical hip and clinical non-vertebral fractures. Significance of the morphometric vertebral fracture primary end point was required before testing the other two primary end points. The Hochberg procedure was used to control for the multiplicity of fracture end points [37].
For BMD end points, treatments were compared using a longitudinal model with terms for treatment, stratum, geographic region, and treatment-time interaction on the percent change from baseline. Log-transformed fractional change from baseline in biochemical markers was analyzed using the same model as BMD. Yearly rate of height loss was analyzed using a longitudinal model. Testing for secondary end points was performed only if significance for the first primary end point (morphometric vertebral fractures) was obtained. For the purpose of addressing the issue of multiplicity adjustment, three groups were considered: (1) BMD measures, (2) biochemical markers of bone resorption, and (3) clinical vertebral fractures and height. Hochberg multiplicity adjustment procedure was used within each of three groups. No adjustment for multiplicity between the three groups was applied.
Sample size considerations
To provide at least 90 % statistical power to demonstrate a risk reduction of vertebral fracture by 50 %, hip fracture by 35 %, or non-vertebral fracture by 20 %, the estimated required numbers of participants with these fractures were 114, 237, and 824, respectively. Sample size estimates were based on the fracture incidence in the alendronate Fracture Intervention Trial (FIT) and assumed that approximately 25 % of participants would have a prior radiographic vertebral fracture [38]. The estimated enrollment requirement was based on hip fracture projections, since these events are least frequent. On this basis, randomization of approximately 16,000 participants was planned.
Interim analyses
Two interim analyses were planned to be conducted, when approximately 70 and 85 % of the targeted 237 participants had experienced an osteoporotic hip fracture event. To adjust for multiplicity, an alpha spending function was used for the interim and final analyses, such that p values of approximately 0.007, 0.018, and 0.046 were required for the first and second interim and final analyses, respectively.
Planned subgroup analyses
Prespecified subgroup analyses for the primary efficacy end points included age (<70 and ≥70 years), race, prior radiographic vertebral fracture, baseline BMD T-score tertiles, baseline biochemical bone turnover marker tertiles, geographic region, baseline renal function, bisphosphonate intolerance, vitamin D status, BMI tertiles, and participation in the lead or main cohort. Subgroup analyses including age, race, bisphosphonate intolerance, and ability or willingness to be treated with available osteoporosis therapies were conducted for the following safety end points: combined clinical as well as combined laboratory AEs and skin disorders and dental disorders reported by investigators as AEs.
Participant baseline characteristics
The numbers of participants screened and enrolled are shown in Fig. 1 and their geographical distribution in Table 3. Baseline characteristics are shown in Table 4. The average age of the study population was 72.8 years, 56.5 % of the participants were Caucasian, and the average number of years since menopause was 25.3. Baseline vertebral fractures were identified in 46.5 % of subjects.
Extension study
In recent years, questions have arisen about the benefits and risks of long-term treatment with osteoporosis medications [39]. At the initiation of the current trial, it was anticipated that up to 5 years of blinded therapy might be required to evaluate its end points, and this duration was specified in the protocol and participant consent forms that were approved by ERCs and IRBs. Provision was made for early termination of the study if significant reductions in fracture risk were demonstrated in either preplanned interim analysis. In order to ensure collection of long-term data, an extension study was planned in which study participants would continue in a blinded fashion on their randomized treatment until they reached the originally anticipated total of 5 years, after which participants would transition to open-label ODN.
Current status
On July 11, 2012, the sponsor announced the decision to terminate the base study early based on the DMC recommendation following its review of the first interim analysis. That analysis was performed after approximately 70 % of the target hip fracture events (158 participants with an incident osteoporotic hip fracture) had occurred. The DMC based its recommendation on the observation of robust efficacy and a favorable benefit/risk profile of ODN relative to placebo. The DMC also recommended that additional safety data be obtained in the previously planned blinded extension study. Following this recommendation, study participants attended a final base study visit and, if eligible, were asked to consent to enter the extension study. The base study was closed out between August and November, 2012, and will form the basis for the primary comparative efficacy and safety analyses.
There were important differences between the base and extension studies in study design and conduct. In the base study, all participants who discontinued study medication were allowed to remain in the trial and be followed unless they withdrew consent. However, participants who were no longer taking study medication at the time of their final base study visit were not eligible to enter the extension. Of 16,713 women initially randomized in the base study, 8256 entered the extension study. Of the approximately 8000 women who did not enter the extension, most were ineligible because they had not completed the base study on therapy or because the investigational site did not participate in this phase of the trial. A total of 1281 women experienced excessive bone loss during the base study. Most of them discontinued prior to entering the extension. However, 245 women were excluded shortly after entering the extension because of excessive bone loss detected at the transitional visit.
The loss of nearly half the original participants created a substantial likelihood that the extension population would differ materially from the original trial population. Participants who experienced excessive bone loss during the base study were discontinued from study drug and were therefore ineligible to enter the study extension. It would be expected that a greater number of participants from the placebo arm were discontinued due to excessive bone loss. Thus, although it was blinded, the extension cannot be presumed to have maintained the randomization.
As osteoporosis requires long-term treatment, extension studies of active treatment, with or without a withdrawal arm, have become customary. Prior trials have not generally extended the randomized placebo phase beyond the primary end point. In the report of the 10-year extended phase III alendronate studies, the authors used the expected effect of aging on fracture rates in the original placebo group in order to provide context for interpretation of the rates observed on active treatment in the latter years [40]. A more sophisticated approach, taking more variables into account, was subsequently developed and validated using data from the alendronate FIT trial [41]. This “virtual twin” model has been used in the analyses of data from the long-term extension of the denosumab phase III trial for which a placebo comparison was no longer available [42]. A similar model will be developed from the placebo group in the ODN base study. This may provide useful information about expected fracture rates and other efficacy and safety results for a virtual long-term placebo group with characteristics matching the active treatment population.
Summary
Inhibition of cathepsin K is an attractive pharmacological approach with therapeutic potential in osteoporosis. Preclinical and initial clinical data provided a strong rationale for the use of ODN in the treatment of osteoporosis, and the phase IIb trial identified 50 mg once weekly as a dose regimen that is effective on BMD end points. The ODN phase III fracture trial was designed to rigorously investigate the efficacy and safety of this selective CatK inhibitor in the treatment of osteoporosis in postmenopausal women.
This placebo-controlled study excluded participants who were at excessively high individual risk of fracture and included safeguards to ensure participant safety. In addition to fractures, the study evaluated multiple end points that not only provide information about ODN but may also help expand our knowledge of osteoporosis. An interim analysis demonstrated sufficiently robust anti-fracture efficacy and favorable benefit-risk profile to warrant early termination of the base study. The blinded extension study enrolled more than 8000 continuing participants and will provide additional data concerning the longer-term safety and efficacy of ODN.
References
Osteoporosis prevention, diagnosis, and therapy. JAMA 2001; 285; 1: 785-95
Bonewald LF (2011) The amazing osteocyte. J Bone Miner Res 26:229–238
Martin TJ, Sims NA (2005) Osteoclast-derived activity in the coupling of bone formation to resorption. Trends Mol Med 11:76–81
Tang Y, Wu X, Lei W, Pang L, Wan C, Shi Z, Zhao L, Nagy TR, Peng X, Hu J, Feng X, Van Hul W, Wan M, Cao X (2009) TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat Med 15:757–765
Zhao C, Irie N, Takada Y, Shimoda K, Miyamoto T, Nishiwaki T, Suda T, Matsuo K (2006) Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metab 4:111–121
Lotinun S, Kiviranta R, Matsubara T, Alzate JA, Neff L, Luth A, Koskivirta I, Kleuser B, Vacher J, Vuorio E, Horne WC, Baron R (2013) Osteoclast-specific cathepsin K deletion stimulates S1P-dependent bone formation. J Clin Invest 123(1):666–681
Boyle WJ, Simonet WS, Lacey DL (2003) Osteoclast differentiation and activation. Nature 423(1):337–342
Baron R, Ferrari S, Russell RG (2011) Denosumab and bisphosphonates: different mechanisms of action and effects. Bone 48(5):677–692
Heaney RP, Yates AJ, Santora AC (1997) Bisphosphonate effects and the bone remodeling transient. J Bone Miner Res 12(1):1143–1151
Hernandez CJ, Beaupre GS, Marcus R, Carter DR (2001) A theoretical analysis of the contributions of remodeling space, mineralization, and bone balance to changes in bone mineral density during alendronate treatment. Bone 29(1):511–516
Roschger P, Rinnerthaler S, Yates J, Rodan GA, Fratzl P, Klaushofer K (2001) Alendronate increases degree and uniformity of mineralization in cancellous bone and decreases the porosity in cortical bone of osteoporotic women. Bone 29(2):185–191
Zebaze RM, Libanati C, Austin M, Ghasem-Zadeh A, Hanley DA, Zanchetta JR, Thomas T, Boutroy S, Bogado CE, Bilezikian JP, Seeman E (2014) Differing effects of denosumab and alendronate on cortical and trabecular bone. Bone 59(2):173–179
Lewiecki EM (2011) New targets for intervention in the treatment of postmenopausal osteoporosis. Nat Rev Rheumatol 7(1):631–638
Costa AG, Cusano NE, Silva BC, Cremers S, Bilezikian JP (2011) Cathepsin K: its skeletal actions and role as a therapeutic target in osteoporosis. Nat Rev Rheumatol 7(1):447–456
Gelb BD, Shi GP, Chapman HA, Desnick RJ (1996) Pycnodysostosis, a lysosomal disease caused by cathepsin K deficiency. Science 273:1236–1238
Duong L (2012) Therapeutic inhibition of cathepsin K—reducing bone resorption while maintaining bone formation. BoneKEy Rep 1:1–8
Kiviranta R, Morko J, Uusitalo H, Aro HT, Vuorio E, Rantakokko J (2001) Accelerated turnover of metaphyseal trabecular bone in mice overexpressing cathepsin K. J Bone Miner Res 16:1444–1452
Falgueyret JP, Desmarais S, Oballa R, Black WC, Cromlish W, Khougaz K, Lamontagne S, Masse F, Riendeau D, Toulmond S, Percival MD (2005) Lysosomotropism of basic cathepsin K inhibitors contributes to increased cellular potencies against off-target cathepsins and reduced functional selectivity. J Med Chem 48(2):7535–7543
Gauthier JY, Chauret N, Cromlish W, Desmarais S, Duong LT, Falgueyret JP, Kimmel DB, Lamontagne S, Leger S, LeRiche T, Li CS, Masse F, McKay DJ, Nicoll-Griffith DA, Oballa RM, Palmer JT, Percival MD, Riendeau D, Robichaud J, Rodan GA, Rodan SB, Seto C, Therien M, Truong VL, Venuti MC, Wesolowski G, Young RN, Zamboni R, Black WC (2008) The discovery of odanacatib (MK-0822), a selective inhibitor of cathepsin K. Bioorg Med Chem Lett 18:923–928
Stoch SA, Zajic S, Stone J, Miller DL, Van Dyck K, Gutierrez MJ, De Decker M, Liu L, Liu Q, Scott BB, Panebianco D, Jin B, Duong LT, Gottesdiener K, Wagner JA (2009) Effect of the cathepsin K inhibitor odanacatib on bone resorption biomarkers in healthy postmenopausal women: two double-blind, randomized, placebo-controlled phase I studies. Clin Pharmacol Ther 86:175–182
Stoch SA, Zajic S, Stone JA, Miller DL, van Bortel L, Lasseter KC, Pramanik B, Cilissen C, Liu Q, Liu L, Scott BB, Panebianco D, Ding Y, Gottesdiener K, Wagner JA (2013) Odanacatib, a selective cathepsin K inhibitor to treat osteoporosis: safety, tolerability, pharmacokinetics and pharmacodynamics—results from single oral dose studies in healthy volunteers. Br J Clin Pharmacol 75:1240–1254
Bone HG, McClung MR, Roux C, Recker RR, Eisman JA, Verbruggen N, Hustad CM, DaSilva C, Santora AC, Ince BA (2010) Odanacatib, a cathepsin-K inhibitor for osteoporosis: a two-year study in postmenopausal women with low bone density. J Bone Miner Res 25:937–947
Eisman JA, Bone HG, Hosking DJ, McClung MR, Reid IR, Rizzoli R, Resch H, Verbruggen N, Hustad CM, DaSilva C, Petrovic R, Santora AC, Ince BA, Lombardi A (2011) Odanacatib in the treatment of postmenopausal women with low bone mineral density: three-year continued therapy and resolution of effect. J Bone Miner Res 26:242–251
Langdahl B, Binkley N, Bone H, Gilchrist N, Resch H, Rodriguez PJ, Denker A, Lombardi A, Le Bailly DT, DaSilva C, Rosenberg E, Leung A (2012) Odanacatib in the treatment of postmenopausal women with low bone mineral density: five years of continued therapy in a phase 2 study. J Bone Miner Res 27(1):2251–2258
Kanis JA, Alexandre JM, Bone HG, Abadie E, Brasseur D, Chassany O, Durrleman S, Lekkerkerker JF, Caulin F (2003) Study design in osteoporosis: a European perspective. J Bone Miner Res 18(24):1133–1138
Clinical trials for new osteoporosis treatments 2002. http://www.fda.gov/ohrms/dockets/ac/cder02 htm#EndocrinologicMetabolicDrugs
Cummings SR, San Martin J, McClung MR, Siris ES, Eastell R, Reid IR, Delmas P, Zoog HB, Austin M, Wang A, Kutilek S, Adami S, Zanchetta J, Libanati C, Siddhanti S, Christiansen C (2009) Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 361:756–765
Study to evaluate the safety and efficacy of BA058 for prevention of fracture in postmenopausal women 2013. http://www.clinicaltrials.gov/ct2/show/NCT01343004?term=BA-058&rank=3
Multicenter, international, randomized, double-blind, placebo-controlled, parallel-group study to assess the efficacy and safety of AMG 785 treatment in postmenopausal women with osteoporosis 2012. http://www.clinicaltrials.gov/ct2/show/NCT01575834?term=AMG+785&rank=13A
Genant HK, Wu CY, van Kuijk C, Nevitt MC (1993) Vertebral fracture assessment using a semiquantitative technique. J Bone Miner Res 8(2):1137–1148
Recker RR, Kimmel DB, Dempster D, Weinstein RS, Wronski TJ, Burr DB (2011) Issues in modern bone histomorphometry. Bone 49(1):955–964
Dempster DW, Compston JE, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR, Parfitt AM (2013) Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res 28(2):2–17
Shane E, Burr D, Ebeling PR, Abrahamsen B, Adler RA, Brown TD, Cheung AM, Cosman F, Curtis JR, Dell R, Dempster D, Einhorn TA, Genant HK, Geusens P, Klaushofer K, Koval K, Lane JM, McKiernan F, McKinney R, Ng A, Nieves J, O’Keefe R, Papapoulos S, Sen HT, van der Meulen MC, Weinstein RS, Whyte M (2010) Atypical subtrochanteric and diaphyseal femoral fractures: report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res 25(1):2267–2294
Shane E, Burr D, Abrahamsen B, Adler RA, Brown TD, Cheung AM, Cosman F, Curtis JR, Dell R, Dempster DW, Ebeling PR, Einhorn TA, Genant HK, Geusens P, Klaushofer K, Lane JM, McKiernan F, McKinney R, Ng A, Nieves J, O’Keefe R, Papapoulos S, Howe TS, van der Meulen MC, Weinstein RS, Whyte MP (2014) Atypical subtrochanteric and diaphyseal femoral fractures: second report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res 29(1):1–23
Miettinen O, Nurminen M (1985) Comparative analysis of two rates. Stat Med 4(2):213–226, 13
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol 57:289–300
Hochberg YA (1988) A sharper Bonferroni procedure for multiple tests of significance. Biometrica 75:800–802
Black DM, Cummings SR, Karpf DB, Cauley JA, Thompson DE, Nevitt MC, Bauer DC, Genant HK, Haskell WL, Marcus R, Ott SM, Torner JC, Quandt SA, Reiss TF, Ensrud KE (1996) Randomised trial of effect of alendronate on risk of fracture in women with existing vertebral fractures. Fracture Intervention Trial Research Group. Lancet 348:1535–1541
Black DM, Bauer DC, Schwartz AV, Cummings SR, Rosen CJ (2012) Continuing bisphosphonate treatment for osteoporosis—for whom and for how long? N Engl J Med 366:2051–2053
Bone HG, Hosking D, Devogelaer JP, Tucci JR, Emkey RD, Tonino RP, Rodriguez-Portales JA, Downs RW, Gupta J, Santora AC, Liberman UA (2004) Ten years’ experience with alendronate for osteoporosis in postmenopausal women. N Engl J Med 350(1):1189–1199
Vittinghoff E, McCulloch CE, Woo C, Cummings SR (2010) Estimating long-term effects of treatment from placebo-controlled trials with an extension period, using virtual twins. Stat Med 29(1):1127–1136
Bone HG, Chapurlat R, Brandi ML, Brown JP, Czerwinski E, Krieg MA, Mellstrom D, Radominski SC, Reginster JY, Resch H, Ivorra JA, Roux C, Vittinghoff E, Daizadeh NS, Wang A, Bradley MN, Franchimont N, Geller ML, Wagman RB, Cummings SR, Papapoulos S (2013) The effect of three or six years of denosumab exposure in women with postmenopausal osteoporosis: results from the FREEDOM extension. J Clin Endocrinol Metab 98(1):4483–4492
Acknowledgments
We would like to thank Boyd B Scott Ph.D., Elizabeth Rosenberg Ph.D., and Keith Kaufman M.D. (Merck & Co. Inc.) for their assistance in writing and critical review of this manuscript and Jennifer Pawlowski, MS, (Merck & Co. Inc.) for her logistical support in the submission of this manuscript.
Funding statement
This study was funded by Merck & Co., Inc., Whitehouse Station, NJ.
Conflicts of interest
Henry Bone received consultancy fees or honoraria from Merck & Co. Inc., Amgen, Aclairo, Takeda, GSK, Radius, Azelon, Purdue Pharmaceuticals, Mission Pharmacal, Akros, Noven, Tarsa, and Novartis; grants from Merck & Co. Inc., Amgen, NPS, Nordic Bioscience, Tarsa, and Novartis; speaker fees from Amgen, Pfizer, ISCD, and Merck & Co. Inc.; and travel support from Merck & Co. Inc.
David Dempster received consulting fees or honoraria from Merck & Co. Inc., Amgen Inc., Eli Lilly & Co and speaker fees from Amgen Inc. and Eli Lilly & Co.
John A. Eisman received investigator fees, consulting fees or honorarium, and travel support from Merck & Co. Inc. He receives similar support and at times untied grant support from Amgen, Eli Lilly, Sanofi-Aventis, Servier, and Novartis.
Susan L. Greenspan received advisory board honoraria from Merck & Co. Inc.; travel support from Merck & Co. Inc.; and grants from Eli Lilly, Amgen, Warner-Chilcott, and Tarsa Therapeutics
Michael McClung received investigator fees from Merck & Co. Inc.; consulting fees or honoraria from Merck & Co., Amgen, Eli Lilly, and Novartis; grants from Amgen; and speaker fees from Amgen Inc. and Warner Chilcott.
Toshitaka Nakamura Received consulting fees or honoraria from Amgen, Eli Lilly, Chugai, Daiichi Sankyo, and Asahi Kasei; grants from Eisai, Takeda, Astellas, and Ono; and speaker fees from Eli Lilly, Chugai, Takeda, and Daiichi Sankyo.
Socrates Papapoulos received consulting fees or honoraria from Merck & Co., Amgen, Novartis, Pfizer, Roche, GSK, and Axsome Therapeutics and speaker fees from Amgen Inc., GSK, Merck & Co., Eli Lilly, and Roche.
Weichung J. Shih declares no conflict of interest, apart from honoraria for participation on the odanacatib scientific advisory committee.
Albert T. Leung, Arthur Santora, Nadia Verbruggen, Andrea Rybak-Feiglin, and Antonio Lombardi are employees of Merck & Co. Inc., Whitehouse Station, NJ, and may own stock or stock options in Merck & Co. Inc.
Scientific advisory committee
-
Henry Bone, Michigan Bone & Mineral Clinic, Detroit, MI, USA
-
David Dempster, Columbia University Medical Center, New York, NY, USA
-
John A Eisman, Garvan Institute of Medical Research, University of Notre Dame Australia, St Vincent’s Hospital and UNSW Australia, Sydney, Australia
-
Susan Greenspan, University of Pittsburgh, Pittsburgh, PA, USA
-
Michael McClung, Oregon Osteoporosis Center, Portland, Oregon, USA
-
Toshitaka Nakamura, University of Occupational and Environmental Health, Kitakyushu, Japan
-
Socrates Papapoulos, Leiden University Medical Center, Leiden, Netherlands
-
Weichung J. Shih, Robert Wood Johnson Medical School, Piscataway, NJ, USA
Data monitoring committee
-
Robert Wallace (chairperson), University of Iowa College of Public Health, Iowa City, IA, USA
-
Cyrus Cooper, MRC Epidemiology Research Centre, Southampton General Hospital, Southampton, UK
-
Kristine Ensrud, Minneapolis Veteran Affairs Medical Center and University of Minnesota, Minneapolis, MN, USA
-
David Kendler, ProHealth Clinical Research, Vancouver, BC, Canada
-
Leonard Milstone, Yale University School of Medicine, New Haven, CT, USA
-
Stuart Ralston, Molecular Medicine Centre, Western General Hospital Edinburgh, UK
-
Bruce Turnbull, Cornell University School of Operations Research and Information Engineering, Ithaca, NY, USA
Author information
Authors and Affiliations
Corresponding author
Additional information
This study was registered with clinicaltrials.gov with registration number NCT00529373.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 1811 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Bone, H.G., Dempster, D.W., Eisman, J.A. et al. Odanacatib for the treatment of postmenopausal osteoporosis: development history and design and participant characteristics of LOFT, the Long-Term Odanacatib Fracture Trial. Osteoporos Int 26, 699–712 (2015). https://doi.org/10.1007/s00198-014-2944-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-014-2944-6