
Abstract
Specific small interfering RNA (siRNA) constructs were used to test for the functional relation of subunits α, β, and γ of the epithelial Na+ channel (ENaC) to the hypertonicity-induced cation channel (HICC) in confluent rat hepatocytes. In current-clamp recordings, hypertonic stress (300 → 400 mosM) increased membrane conductance from 75.4 ± 9.4 to 91.1 ± 11.2 pS (p < 0.001). The effect was completely blocked by 100 μM amiloride and reduced to 46, 30, and 45% of the control value by anti-α-, anti-β-, and anti-γ-rENaC siRNA, respectively. Scanning acoustic microscopy revealed an initial shrinkage of cells from 6.98 ± 0.45 to 6.03 ± 0.43 pl within 2 min. This passive response was then followed by a regulatory volume increase (RVI) by 0.42 ± 0.05 pl (p < 0.001). With anti-α-, anti-β-, and anti-γ-rENaC siRNA, the volume response was reduced to 31, 31, and 36% of the reference level, respectively. It is concluded that all three subunits of the ENaC are functionally related to RVI and HICC activation in rat hepatocytes.
Similar content being viewed by others


Introduction
It became increasingly evident in recent years that the mechanisms of cell volume regulation are employed in a variety of physiological processes in addition to those of mere cell homoeostasis. Among these are the coordination of ion transport across epithelia, the control of cell metabolism and gene expression, and, most notably, the triggering of cell proliferation and apoptosis [3, 13, 19].
Whenever studied in a quantitative fashion, hypertonicity-induced cation channels (HICCs) have proven to be the main mechanism of the regulatory volume increase (RVI) of a shrunken cell (see [16, 17] for review). In rat hepatocytes, for instance, the contribution of the HICC to the RVI process is approx. 4:1:1, when compared to Na+/H+ antiport and Na+–K+–2Cl− symport [22, 23]. For human HeLa cervix carcinoma cells, a ratio of 2:1 is found for HICCs and Na+/H+ antiport, whereas Na+–K+–2Cl− symport is clearly not involved [21]. In HepG2 human liver carcinoma cells, RVI appears to be exclusively mediated by HICCs with no significant contribution of Na+/H+ antiport and Na+–K+–2Cl− symport at all [18].
Despite the obvious significance of HICCs for cell volume control and functioning, very little is known so far as for the actual molecular architecture of these channels. Of note, however, the HICC in hepatocytes is clearly inhibited by micromolar concentrations of amiloride [1, 18, 24], which is the classic blocker of the epithelial Na+ channel (ENaC) [8]. Moreover, in rat hepatocytes, α-, β-, and γ-ENaC are detectable on the messenger RNA (mRNA) as well as on the protein level [1], and anti-sense oligo-nucleotides directed against α-ENaC decrease HICC currents by some 60% [2]. As the use of anti-sense oligo-DNA has quite frequently been reported to lead to false-positive results, however, the latter finding needs to be approved by an independent and more reliable technique. Furthermore, no information is available so far concerning a possible contribution of β- and γ-ENaC to HICC currents.
In the present study, we tested for the actual contributions of α-, β-, and γ-ENaC to HICC currents in rat hepatocytes by use of highly specific small interfering RNAs (siRNAs) directed against each of these subunits. The identical constructs were then employed to functionally correlate HICC inhibition to the actual process of RVI. To this end, the novel and noninvasive technique of scanning acoustic microscopy (SAM) is introduced here, which allows the determination of cell volumes over periods of hours without any staining of cells and at high temporal and spatial resolution.
Materials and methods
Cell culture
Harvesting of rat hepatocytes and cell culture techniques have been described in detail previously [20, 22]. All experiments were conducted on confluent monolayers at day 1 after cell preparation. Only in this configuration, rat hepatocytes exhibit, both, a quasi-physiological RVI as well as the activation of the HICC [6, 9, 22].
siRNA and real-time PCR
siRNAs were purchased from Qiagen (Hilden, Germany); to visualize the effective transfection of cells, each construct was 3′-Alexa488-labeled. siRNAs were complexed to 3.3% Dharmacon DharmaFECT1 transfection reagent (PerbioScience Deutschland, Bonn, Germany) in serum-free “Dulbecco’s Modified Eagle Medium” (DMEM; Invitrogen, Karlsruhe, Germany) that, after 20 min, was 1:3 diluted with a complete DMEM fortified with 10% fetal bovine serum, 2 mM glutamine, 1 mM pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin, 1 μM dexametasone, 10 nM triiodothyronine, 10 nM thyroxine, and 5 μg/ml bovine insulin. Transfection was started with the seeding of cells and was complete after some 24 h.
Three different siRNAs were used to reduce the mRNA level of α-rENaC (GenBank® Accession No. NM_031548), which is the central element in channel functioning [8]. These were anti-α-rENaC siRNA1 sense r(CCC UUA AUC CUU ACA GAU A) and antisense r(UAU CUG UAA GGA UUA AGG G), anti-α-rENaC siRNA2 sense r(GGG UGA UGG UGC AUG GUC A) and antisense r(UGA CCA UGC ACC AUC ACC C), and anti-α-rENaC siRNA3 sense r(GAG GGA ACA GGC UGA GUA A) and antisense r(UUA CUC AGC CUG UUC CCU C). For β-rENaC (NM_012648) sense r(CGG GAA CUA GUG AAC UCA A) and antisense r(UUG AGU UCA CUA GUU CCC G) were used; γ-rENaC (NM_017046) was silenced by sense r(GGA UUU CAA UUG UGC UCA A) and antisense r(UUG AGC ACA AUU GAA AUC C).
RNA was extracted from cultured rat hepatocytes at 24 h after transfection by use of the RNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol. Total RNA was quantified by means of UV-photometry, and purity was determined on the basis of the nucleic acid/protein ratio at optical density (OD) 260/OD 280.
Reverse transcription from mRNA to complementary DNA (cDNA) was performed by use of the Super Script II Reverse Transcription Kit (Invitrogen) following the manufacturer’s protocol, and the cDNA was used as the template for real-time polymerase chain reaction (PCR; see below).
Efficiency of siRNAs against target genes was determined, at the mRNA level, by means of real-time PCR (GeneAmp 5700 Sequence Detection System, Perkin Elmer Applied Biosystems™, Foster City, USA). Results were analyzed on the basis of the comparative threshold cycle method [4, 14] and normalized to the housekeeping gene for β-actin as an internal standard.
Transfection rates were determined by counting fluorescent vs. non-fluorescent cells in areas of 320 × 320 μm (i.e., on some 90 to 110 cells, on the average); this was done eight times for each of the siRNAs used.
Electrophysiology
Intracellular microelectrode and current-clamp techniques were similar to previous reports [20, 22]. Briefly, hepatocytes were impaled with two-channel micro-electrodes: One channel was used for the recording of membrane voltage, the second for the injection of constant current pulses. With these “current clamp”-recordings, changes in cell input resistance become readily detectable. As this parameter, to some extent, also depends on electric cell-to-cell coupling, its reciprocal is denoted as the apparent membrane conductance, here [20]. Because of the sizeable degree of cell coupling in confluent monolayers, patch-clamp recordings on rat hepatocytes are not feasible.
With every siRNA tested, successful transfection was occurring in clusters of some 62 to 82 of fluorescent cells, and, in every instance, a cell in the very center of such a cluster was impaled for the respective current-clamp recording.
Scanning acoustic microscopy
Central element of the SAM (developed at the Fraunhofer Institut Biomedizinische Technik, St. Ingbert, Germany) is an acoustic lens consisting of a sapphire cylinder of 7-mm diameter with a 50-μm spherical cavity (as the focussing element) at its front and with a thin piezo-electric film on its top (see Fig. 1a). A high voltage sinus pulse of 1-ns duration is used to excite the piezo-element yielding an ultrasonic wave with a center frequency close to 1 GHz (a technique that was originally introduced for the use in biological systems by Bereiter-Hahn and colleagues [11, 12, 15]). This sound wave is focused to a small spot of approx. 1 μm traveling vertically toward a cell (or a monolayer of cells) where it is reflected twice, first, at the cell surface (giving the “front-echo”) and, second, at the substrate of the preparation (giving the “substrate-echo”; Fig. 1a). Thus, two echoes are generated that evolve with a small time difference that is proportional to the actual height of a given cell. These echoes are detected by the piezo-element and converted into an electric current that is amplified and then digitized by a fast transient recorder (running at eight billion samples per second). To increase the signal-to-noise ratio of received signals, a 400-fold averaging is performed at every single position. By means of an automatic piezo-electric scanner-unit, spot-recordings of this kind are performed in a raster-like pattern over an overall area of 100 × 100 μm. When set at a spatial resolution of 2 μm, one measurement (at a total of 2,500 positions) is complete in less than 20 s. A typical recording in just one dimension (“x-scan”) is exemplified in Fig. 1a, equivalent to a vertical cross-section through a single cell and with the intensities obtained given in a gray scale. Using the software-package “AMIRA” (Konrad-Zuse-Zentrum für Informationstechnik, Berlin, Germany), a 3D cell surface reconstruction can then be obtained from the various x-scans, as it is shown in Fig. 1b.

SAM. a The principle of the technique. A 1-GHz sound wave is generated with a piezo-element and focussed by a sapphire lens onto a cell where it is reflected twice, at the upper cell surface and at the glass plate (substrate) where the cell is cultured on. Sound echoes are detected with the same device and then transformed into an electric signal (yellow boxes symbolize—from left to right—the high-frequency electric generator, the switch from the stimulation to the detection mode of the system, as well as the amplifier for signal detection). From the differences in time when substrate and front-echoes are recorded, the actual cell height at a given point can be computed. In the lower right, a scan of a single cell in a single dimension (x-scan) is exemplified. b Typical 3D-reconstruction. The upper layer (shown in silver) depicts the 100 × 100 μm surface area of a hepatocyte monolayer under isotonic conditions, the second layer (in gold) shows the equivalent surface at 2 min of hypertonic stress, i.e., at the time when cell shrinkage is maximal (c.f. to Fig. 4a). The lower cross-hatched area represents the level of the substrate with the squares marking the positions where the 50 × 50 = 2,500 single spot-recordings have been done. The elevated structures in the image (where cell shrinkage is less pronounced) reflect areas above cell nuclei. The turret in the back (in silver/gold) represents some kind of cell debris (exhibiting no changes in height at all), an artifact that is routinely corrected for, but that is included here for demonstration purposes. Note that, in this presentation, the resolution of the z-axis is three times that of the x/y-axes
In detail, the returning and recorded waveform y(t) can be described by the equation given below (Eq. 1). The first term describes the echo coming from the cell surface at a time t 1 with the amplitude A 1 and the second term the echo coming from the glass slide at a time t 2 with the amplitude A 2. a(t) describes the pulse shape of the echo from a perfect reflector in the focus of the lens and can be modeled as a Gaussian peak. ω is the center frequency of the ultrasound, which, in this case, is 1 GHz.
Using the Hilbert transform properties of Eq. 1, the envelope of the waveform can be calculated. Combining Eqs. 2 and 3
gives an approximation envelop of Eq. 1 for non overlapping signals of
Thus, the envelope of the received signal can be seen as two Gaussian peaks at the locations t 1 and t 2 representing the front and substrate echoes. These peaks in the signal can readily by identified by a peak detector that fits a quadratic polynomial to a sequential number of time instances. Then, for each peak, the quadratic fit is tested against a certain threshold level. The time difference Δt between t 1 (the arrival time of the front echo) and t 2 (the arrival time of the substrate echo) depends on the local height of the cell d and the local sound speed inside the cell c according to
Finally, the total volume of the cell monolayer (in the area of 100 × 100 μm) can be summarized according to
where V denotes the cell volume n x , n y the number raster lines in x and y directions, and Δx and Δy the distance between consecutive points of the scanning raster.
From this value, the actual volume of a single hepatocyte was computed on the basis of an average cell surface of 962 μm2 as it was determined in many years of research by means of confocal laser-scanning microscopy [22, 23], i.e., by simply multiplying the total volume of the monolayer with a factor of 0.0962 = 962/10,000 μm2.
Experimental solutions
The experimental solutions contained (in mM): NaCl, 144.0; KCl, 2.7; MgCl2, 1.0; CaCl2, 1.8; NaH2PO4, 0.4; Na+–HEPES−, 2.5; HEPES−, 2.5; pH 7.4. Osmolarities were adjusted to 300 mosM (isotonic) and 400 mosM (hypertonic) by addition of mannitol under osmometric control (Knauer, Berlin, Germany). In the current-clamp recordings, 0.5 mM quinine was present to reduce K+ conductance [20, 22, 23]. All compounds were of the highest grade available. Experiments were performed at 36.0 ± 0.5°C.
Statistics
Data are presented as means±SEM with n denoting the number of experiments, except in Fig. 2 where means±SD of two independent experiments are shown. Student’s t-tests for paired and unpaired data were applied as appropriate. *, **, and *** stands for p < 0.05, 0.01, and 0.001, with reference to the respective isotonic value; #, ##, and ### indicate the same levels of significance, but with reference to control data (i.e. TR = transfection reagent).

mRNA content of α-, β-, and γ-ENaC in rat hepatocytes under control conditions (TR transfection reagent) and following transfection with various siRNA constructs, determined by real-time PCR in duplicate. The hatched lines indicate the average amount of cell transfection obtained, as was judged from fluorescence microscopy for comparison
Results and discussion
Real-time PCR
Reduction of the respective mRNA contents was determined by means of real-time PCR experiments performed in duplicate. As depicted in Fig. 2, transfection with siRNA1, siRNA2, and siRNA3 directed against α-rENaC reduced the mRNA content of cells to 52.0 ± 2.0%, 60.0 ± 8.0%, and 37.5 ± 2.5% with reference to control (TR = transfection reagent), respectively. Likewise, siRNAs against β-rENaC and γ-rENaC reduced the mRNA content to 53.5 ± 7.5% and 49.0 ± 2.0% of the control value.
Noteworthy, the higher the efficiency of a transfection was actually found to be (as was determined by fluorescence microscopy; see the hatched lines in Fig. 2), the lower was the remaining expression of a certain gene.
Hypertonicity-induced cation channels
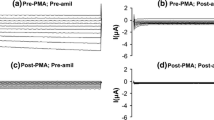
Changing osmolarity from 300 to 400 mosM led to an increase of apparent membrane conductance from 75.4 ± 9.4 to 91.1 ± 11.2 pS, i.e., by 15.7 ± 2.5 pS (n = 12, p < 0.001) under control conditions (TR = transfection reagent; Fig. 3a,b). The effect was complete within 10 to 12 min, but, in every instance, the new steady-state was preceded by a small overshoot very likely reflecting a passive increase of the conductivity of the cytosol upon hypertonic cell shrinkage (Fig. 3a; [20, 22, 23]). In the presence of 100 μM amiloride, which is a specific inhibitor of the HICC in rat hepatocytes [1, 20, 24], membrane conductance was 78.4 ± 17.9 and 75.0 ± 16.1 pS, under isotonic and hypertonic conditions, respectively. In other words, the compound significantly reduced the hypertonic change of conductance to −3.4 ± 6.1 pS (n = 7, p < 0.01; Fig. 3a,b). As the HICC in rat hepatocytes is inhibited by amiloride with an apparent Ki of some 5 μM [1, 24], these results strongly suggest that the observe changes of membrane conductance in response to hypertonic stress, in the control recordings, do in fact reflect the activation of the HICC. Also of note, isotonic membrane conductance was very similar under both experimental conditions indicating that the basal contribution of the HICC to this parameter is rather minor.

Hypertonicity-induced changes of apparent membrane conductance. a Typical current-clamp recordings under control conditions (TR transfection reagent) and in the presence of 100 μM amiloride as a blocker of the HICC (TR + AMI). For the time indicated, osmolarity was increased from 300 (iso) to 400 mosM (hyper). b Summary of hypertonicity-induced changes of membrane conductance (n = 5 to 12, as indicated)
With the above experimental conditions as reference points, we tested for the actual contribution of α-, β-, and γ-ENaC to the HICC in rat hepatocytes. Concerning α-ENaC, three different constructs were tested. As depicted in Fig. 3b, siRNA1 and siRNA2 directed against α-ENaC had no significant effects on the hypertonicity-induced increase of apparent membrane conductance, which equaled 16.6 ± 4.1 pS (n = 5, p = 0.857) and 22.9 ± 6.3 pS (n = 7, p = 0.225), respectively. In cells transfected with siRNA3, however, the membrane response to hypertonic stress was reduced to 7.3 ± 1.6 pS (n = 7, p = 0.028), i.e., to 46.3% of the control recordings on the average. Interestingly, within the group of anti-α-ENaC siRNA constructs used, the relative changes of membrane conductance observed were directly correlating with the actual profile of mRNA contents as was determined by means of real-time PCR (see Fig. 2).
In the next series of measurements, we tested for the possible contribution of β- and γ-ENaC to the HICC. In fact, both the anti-β as well as the anti-γ-ENaC construct significantly reduced HICC conductance to 4.7 ± 1.0 pS (n = 10, p < 0.01) and 7.1 ± 1.9 pS (n = 8; p < 0.05), respectively (Fig. 3b), i.e., to 30.0 and 45.4% of the control value. Taken together, these data strongly suggest that subunits α-, β- as well as γ-ENaC are functionally related to the HICC in rat hepatocytes. Also of note, the results of the experiments with α-ENaC siRNA3, as well as with the siRNAs against β- and γ-ENaC were not significantly different from the experiments with amiloride.
Quantification of cell volume and RVI
To test for the actual effects of a reduction of α-, β-, and γ-ENaC mRNA on rat hepatocyte RVI, hypertonicity-induced changes of cell volume were determined by means of SAM. As depicted in Fig. 4a, increasing osmolarity from 300 to 400 mosM led to an initial shrinkage of cells from 6.98 ± 0.45 pl to 6.03 ± 0.43 pl within 2 min (n = 9, p < 0.001). This passive response was then followed by a partial volume recovery to 6.45 ± 0.44 pl at 10 min of hypertonic stress, equivalent to an RVI value of 0.42 ± 0.05 pl (p < 0.001). Interestingly, this is equivalent to a relative RVI of approx. 44% when referred to the initial amount of cell shrinkage, a value very similar to the one obtained by means of confocal laser-scanning microscopy (i.e., 42.5% [22]). Also of note in this respect, isotonic control volumes determined with the latter technique were found to be 5.9 pl [20, 22], which is only some 15% smaller than the values obtained in the present study by use of SAM.

Hypertonicity-induced cell shrinkage and RVI. a Typical SAM measurements under control conditions (TR) and in the presence of 100 μM Amiloride (TR + AMI). b Summary of cell volume measurements (n = 3 to 12, as indicated). RVI data were computed according to the protocol depicted in (a). See text for details
In the presence of 100 μM amiloride, cell volumes equaled 6.11 ± 0.53 pl and 5.56 ± 0.49 pl under isotonic conditions and at 2 min of hypertonic stress, respectively (n = 12; Fig. 4a). These data are not significantly different from those obtained under control conditions. As expected, however, RVI was reduced to 0.21 ± 0.07 pl, i.e., to 50.1% of the reference value (p < 0.05; Fig. 4b).
Very similar to the results obtained in the current-clamp recordings, siRNA1 and siRNA2 directed against α-ENaC did not exhibit significant effects on the RVI of rat hepatocytes, which was 0.29 ± 0.06 pl (n = 7) and 0.38 ± 0.08 pl (n = 5), respectively (Fig. 4b). In contrast, when cells were transfected with anti-α-ENaC siRNA3, the RVI response was significantly reduced to 0.13 ± 0.06 pl (n = 8, p < 0.01; Fig. 4b), which is 31.3% of the control value. Likewise, the anti-β as well as the anti-γ-ENaC siRNAs both significantly reduced hepatocyte RVI, which was 0.13 ± 0.03 pl (n = 3, p < 0.01) and 0.15 ± 0.03 pl (n = 5, p < 0.01; Fig. 4b), respectively, equivalent to 31.3 and 36.3% of the reference data. Of note, none of the latter three sets of measurements were yielding results that are statistically different to the recordings with amiloride.
Conclusions
In rat hepatocytes, specific siRNA constructs directed against α-, β-, and γ-rENaC significantly reduced amiloride-sensitive HICC currents. Likewise, the process of RVI was inhibited with precisely the same profile of efficiency, altogether rendering a functional correlation between the HICC and the three subunits of the ENaC very likely. It is of note, however, that the HICC in rat hepatocytes is very unlikely to be related to the group of HICCs that are not sensitive to amiloride, as for instance the one that is expressed in HeLa cells [21].
Whether β- and γ-rENaC do indeed contribute to HICC architecture or if they just serve as cofactors for the exocytotic insertion of α-rENaC into the plasma membrane [5, 7, 10] remains to be elucidated. In addition, it is not clear yet what partners of α-rENaC may exist that actually participate in the formation of the HICC.
SAM is introduced here as a novel and potent technique for the quantification of cell volumes being noninvasive and working at a high spatial and temporal resolution. No staining of cells is required, which offers the opportunity of time-lapse experiments over hours and days that may be instrumental in further defining the actual interplay of cell volume regulation and proliferation vs. apoptosis [3, 13, 19].
References
Böhmer C, Wagner CA, Beck S, Moschen I, Melzig J, Werner A, Lin JT, Lang F, Wehner F (2000) The shrinkage-activated Na+ conductance of rat hepatocytes and its possible correlation to rENaC. Cell Physiol Biochem 10:187–194
Böhmer C, Wehner F (2001) The epithelial Na+ channel (ENaC) is related to the hypertonicity-induced Na+ conductance in rat hepatocytes. FEBS Lett 494:125–128
Bortner CD, Cidlowski JA (2004) The role of apoptotic volume decrease and ionic homeostasis in the activation and repression of apoptosis. Pflugers Arch 448:313–318
Bustin SA (2000) Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J Mol Endocrinol 25:169–193
Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, Horisberger JD, Rossier BC (1994) Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature 367:463–467
Corasanti JG, Gleeson D, Boyer JL (1990) Effects of osmotic stresses on isolated rat hepatocytes. I. Ionic mechanisms of cell volume regulation. Am J Physiol 258:G290–G298
Firsov D, Schild L, Gautschi I, Mérillat AM, Schneeberger E, Rossier BC (1996) Cell surface expression of the epithelial Na channel and a mutant causing Liddle syndrome: a quantitative approach. Proc Natl Acad Sci USA 93:15370–15375
Kellenberger S, Schild L (2002) Epithelial sodium channel/degenerin family of ion channels: a variety of functions for a shared structure. Physiol Rev 82:735–767
Kirschner U, van Driessche W, Werner A, Wehner F (2003) Hypertonic activation of phospholemman in solitary rat hepatocytes in primary culture. FEBS Lett 537:151–156
Konstas AA, Korbmacher C (2003) The gamma-subunit of ENaC is more important for channel surface expression than the beta-subunit. Am J Physiol 284:C447–C456
Kundu T, Bereiter-Hahn J, Karl I (2000) Cell property determination from the acoustic microscope generated voltage versus frequency curves. Biophys J 78:2270–2279
Lüers H, Hillmann K, Litniewski J, Bereiter-Hahn J (1991) Acoustic microscopy of cultured cells. Distribution of forces and cytoskeletal elements. Cell Biophys 18:279–293
Okada Y, Maeno E, Shimizu T, Manabe K, Mori S, Nabekura T (2004) Dual roles of plasmalemmal chloride channels in induction of cell death. Pflugers Arch 448:287–295
Ririe KM, Rasmussen RP, Wittwer CT (1997) Product differentiation by analysis of DNA melting curves during the polymerase chain reaction. Anal Biochem 245:154–160
Wagner O, Zinke J, Dancker P, Grill W, Bereiter-Hahn J (1999) Viscoelastic properties of f-actin, microtubules, f-actin/alpha-actinin, and f-actin/hexokinase determined in microliter volumes with a novel nondestructive method. Biophys J 76:2784–2796
Wehner F (2006) Cell volume-regulated cation channels. Contrib Nephrol 152:25–53
Wehner F, Bondarava M, ter Veld F, Endl E, Nürnberger HR, Li T (2006) Hypertonicity-induced cation channels. Acta Physiol 187:21–25
Wehner F, Lawonn P, Tinel H (2002) Ionic mechanisms of regulatory volume increase (RVI) in the human hepatoma cell-line HepG2. Pflugers Arch 443:779–790
Wehner F, Olsen H, Tinel H, Kinne-Saffran E, Kinne RKH (2003) Cell volume regulation: osmolytes, osmolyte transport, and signal transduction. Rev Physiol Biochem Pharmacol 148:1–80
Wehner F, Sauer H, Kinne RKH (1995) Hypertonic stress increases the Na+ conductance of rat hepatocytes in primary culture. J Gen Physiol 105:507–535
Wehner F, Shimizu T, Sabirov R, Okada Y (2003) Hypertonic activation of a non-selective cation conductance in HeLa cells and its contribution to cell volume regulation. FEBS Lett 551:20–24
Wehner F, Tinel H (1998) Role of Na+ conductance, Na+–H+ exchange, and Na+–K+–2Cl− symport in the regulatory volume increase of rat hepatocytes. J Physiol (Lond) 506:127–142
Wehner F, Tinel H (2000) Osmolyte and Na+ transport balances of rat hepatocytes as a function of hypertonic stress. Pflugers Arch 441:12–24
Wehner F, Tinel H, Kinne RKH (1997) Pharmacology of volume activated Na+ conductance in rat hepatocytes. Physiologist 40:A-4
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Plettenberg, S., Weiss, E.C., Lemor, R. et al. Subunits α, β and γ of the epithelial Na+ channel (ENaC) are functionally related to the hypertonicity-induced cation channel (HICC) in rat hepatocytes. Pflugers Arch - Eur J Physiol 455, 1089–1095 (2008). https://doi.org/10.1007/s00424-007-0355-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-007-0355-7