
Summary
Purpose This phase Ib study (NCT01862328) evaluated the maximum tolerated dose (MTD), safety, and efficacy of pevonedistat in combination with standard-of-care chemotherapies in patients with solid tumors. Methods Patients received pevonedistat with docetaxel (arm 1, n = 22), carboplatin plus paclitaxel (arm 2, n = 26), or gemcitabine (arm 3, n = 10) in 21-days (arms 1 and 2) or 28-days (arm 3) cycles. A lead-in cohort (arm 2a, n = 6) determined the arm 2 carboplatin dose. Dose escalation proceeded via continual modified reassessment. Results Pevonedistat MTD was 25 mg/m2 (arm 1) or 20 mg/m2 (arm 2); arm 3 was discontinued due to poor tolerability. Fifteen (23%) patients experienced dose-limiting toxicities during cycle 1 (grade ≥3 liver enzyme elevations, febrile neutropenia, and thrombocytopenia), managed with dose holds or reductions. Drug-related adverse events (AEs) occurred in 95% of patients. Most common AEs included fatigue (56%) and nausea (50%). One drug-related death occurred in arm 3 (febrile neutropenia). Pevonedistat exposure increased when co-administered with carboplatin plus paclitaxel; no obvious changes were observed when co-administered with docetaxel or gemcitabine. Among 54 response-evaluable patients, two had complete responses (arm 2) and 10 had partial responses (three in arm 1, one in arm 2a, six in arm 2); overall response rates were 16% (arm 1) and 35% (arm 2). High ERCC1 expression correlated with clinical benefit in arm 2. Conclusion Pevonedistat with docetaxel or with carboplatin plus paclitaxel was tolerable without cumulative toxicity. Sustained clinical responses were observed in pretreated patients receiving pevonedistat with carboplatin and paclitaxel. ClinicalTrials.gov identifier: NCT01862328.
Similar content being viewed by others

Introduction
The ubiquitin-proteasome system regulates turnover of proteins involved in apoptosis, proliferation, and signal transduction [1,2,3]. Cullin-RING ligases (CRLs), the largest subfamily of E3 ubiquitin ligases, target proteins for proteasomal degradation through the addition of polyubiquitin chains [2]. CRL activation via conjugation to the small ubiquitin-like protein NEDD8 (neural precursor cell expressed, developmentally downregulated 8) is regulated by NEDD8-activating enzyme (NAE). CRLs and associated regulatory proteins are attractive novel targets for the development of antitumor agents [2, 3].
Pevonedistat (TAK-924/MLN4924) is a first-in-class, investigational, small-molecule NAE inhibitor that covalently binds to NEDD8, leading to apoptotic cell death. Pevonedistat has shown single-agent activity in various human solid tumor cell lines and xenograft models, as well as in patients with advanced solid tumors and hematologic malignancies [2, 4,5,6,7,8,9,10,11,12]. Pevonedistat delays completion of platinum-induced DNA repair in a transcription-coupled nucleotide excision repair (NER) and interstrand crosslink repair (ICR) pathway−dependent mechanism [13]. Excision repair cross-complementation group 1 (ERCC1), a component of the NER pathway, is thought to be involved in resistance to platinum-based therapies [14, 15] and predictive of decreased efficacy of platinum therapy [16,17,18,19,20,21]. Pevonedistat enhances the antitumor activity of taxanes and gemcitabine, is synergistic with platinum salts, and is active in platinum-resistant tumors [13, 22,23,24].
The safety and activity of pevonedistat monotherapy were investigated in a phase I study in patients with acute myeloid leukemia (AML) or myelodysplastic syndromes (MDS), which demonstrated that treatment was feasible with clinical activity [11]. Additionally, the efficacy and safety of pevonedistat in combination with the standard-of-care agent azacitidine have been investigated in treatment-naïve patients with AML (ClinicalTrials.gov identifier NCT01814826) [25]. Phase II and III studies of pevonedistat in combination with azacitidine in high-risk MDS, chronic myelomonocytic leukemia, or low-blast count AML are currently ongoing (ClinicalTrials.gov identifiers NCT02610777 and NCT03268954). To further explore the role of pevonedistat in combination with standard-of-care chemotherapies, this phase Ib study aimed to determine the maximum tolerated dose (MTD) and assess the safety, tolerability, and pharmacokinetics of pevonedistat in combination with docetaxel, carboplatin plus paclitaxel, or gemcitabine in patients with advanced solid tumors. The expression of ERCC1 as a candidate biomarker of response to treatment with pevonedistat in combination with carboplatin plus paclitaxel therapy was also investigated.
Methods
Study design and patients
This was an open-label, multicenter, phase Ib, dose-escalation study of pevonedistat in combination with either docetaxel (arm 1), carboplatin plus paclitaxel (arm 2), or gemcitabine (arm 3) in adult patients with confirmed solid tumors who had progressed despite standard therapy or for whom conventional therapy was considered ineffective (ClinicalTrials.gov identifier NCT01862328; Online Resource: Supplementary Fig. 1 a). Patient eligibility criteria are described in the Online Resource. Patients were enrolled between 10 June 2013 and 04 May 2015 at six US centers. This study was approved by the Institutional Review Board and Independent Ethics Committee at each site and conducted in accordance with all applicable regulatory requirements, Good Clinical Practice standards, and the Declaration of Helsinki. All patients provided written informed consent.
Dose escalation in arm 1 consisted of intravenous pevonedistat at a 15 mg/m2 starting dose on days 1, 3, and 5 plus docetaxel 75 mg/m2 on day 1 of 21-day cycles. A lead-in cohort (arm 2a) of six patients received pevonedistat 15 mg/m2 on days 1, 3, and 5 plus carboplatin AUC6 (target area under the concentration-time curve of 6 mg/mL·min) on day 1 to determine the dose of carboplatin in arm 2. If two or more dose-limiting toxicities (DLTs) were reported, carboplatin dosing was to be reduced to AUC5, and paclitaxel be reduced from the planned 200 to 175 mg/m2. Patients in arm 2 received escalating doses of pevonedistat starting at 15 mg/m2 on days 1, 3, and 5, in combination with carboplatin AUC5 plus paclitaxel 175 mg/m2 on day 1 of 21-day cycles. In arm 3, patients received escalating doses of pevonedistat starting at 25 mg/m2 and gemcitabine 1000 mg/m2 on days 1, 8, and 15 of 28-day cycles. Planned pevonedistat doses were higher in arm 3 (versus arm 1 and arm 2) but administration was on a weekly schedule (versus three times a week in arm 1 and arm 2) to match the dosing schedule of gemcitabine, to ensure exposure to both drugs and leverage the mechanism of action of the two drugs. Full dosing details are included in the Online Resource.
Pevonedistat dose escalation (Online Resource: Supplementary Fig. 1 b) proceeded via an adaptive Bayesian continual reassessment method (CRM) based on cycle 1 DLTs (Online Resource). Upon MTD determination in each arm, additional patients were enrolled to confirm the MTD (MTD expansion cohorts). Patients were treated for up to 12 cycles or until disease progression or unacceptable toxicity. Patients with clinical benefit could continue with combination treatment or single-agent pevonedistat beyond 12 cycles.
The primary objective was to establish the MTD of pevonedistat in combination with docetaxel, with carboplatin plus paclitaxel, and with gemcitabine. Secondary objectives included disease response and pharmacokinetics of pevonedistat plus standard-of-care regimens. An exploratory objective was to identify potential biomarkers of response to pevonedistat-containing therapy, including tumor ERCC1 expression.
Assessments
Adverse events (AEs) were evaluated according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.03. DLT determination criteria are described in the Online Resource. Investigator-assessed tumor responses were based on Response Evaluation Criteria In Solid Tumors (RECIST), version 1.1 [26]. Response assessment methods are described in the Online Resource. Blood sampling methods for pharmacokinetic analyses, immunohistochemistry and ERCC1 expression evaluation methods, and statistical methods are reported in the Online Resource.
Results
Patients
At data cutoff (01 April 2016), 64 patients had been enrolled (22 in arm 1, six in arm 2a, 26 in arm 2, and 10 in arm 3) and had received at least one dose of study drug. Patient demographics and baseline characteristics were generally similar between arms (Table 1). The median number of treatment cycles was 4 (range, 1–21), and 26 (41%) patients completed ≥5 cycles (Online Resource: Supplementary Table 1). At data cutoff, 62 (97%) patients had discontinued treatment (mostly due to disease progression [n = 39] and AEs [n = 11]). Two patients remained on study in arm 2, receiving single-agent pevonedistat 20 mg/m2 as maintenance.
DLTs and MTD
Fifty-six (88%) patients were included in the DLT-evaluable population (Table 2). In the dose-escalation phase of arm 1, none of the three patients receiving pevonedistat 15 mg/m2 experienced a DLT, and two of 12 patients receiving pevonedistat 25 mg/m2 experienced ≥1 DLT (grade 3 increased alanine aminotransferase [ALT]/aspartate aminotransferase [AST]). Therefore, the MTD for pevonedistat was established as 25 mg/m2 in combination with docetaxel. Six additional patients (of whom five were DLT evaluable) were enrolled in the expansion portion at this dose to further characterize safety and tolerability (17 total DLT-evaluable patients), with two additional patients experiencing ≥1 DLT (grade 3 ALT/AST elevation).
In arm 2a, two of six patients receiving pevonedistat 15 mg/m2 plus carboplatin AUC6 experienced DLTs: grade 4 thrombocytopenia and grade 3 elevated AST. Due to the toxicity observed in this lead-in cohort, patients enrolled in subsequent arm 2 cohorts received carboplatin AUC5 and paclitaxel 175 mg/m2.
In arm 2, no DLTs were observed in an initial cohort of patients treated with pevonedistat 15 mg/m2. Following the CRM, pevonedistat was escalated to 25 mg/m2, and two of five patients experienced DLTs (grade 3 increased AST and grade 3 increased ALT and AST). This led to dose de-escalation of pevonedistat (15 mg/m2) and, among three additional patients enrolled at this dose level, one DLT (grade 3 febrile neutropenia) was observed. Per protocol, patients were then enrolled to an intermediate dose level of pevonedistat 20 mg/m2, and two of six patients had DLTs (grade 3 increased ALT and AST). Based on all DLTs to date and according to the CRM algorithm, the predicted MTD at this stage was 18.1 mg/m2. As the predicted MTD was between the midlow (17.5 mg/m2) and midhigh (22.5 mg/m2) (see the Online Resource) and because at least six patients had been enrolled at the 20 mg/m2 dose, the CRM algorithm did not suggest further dose escalation or de-escalation and the MTD was declared as 20 mg/m2 (Online Resource: Supplementary Fig. 1 b). Six additional patients were enrolled at this dose (expansion phase) for a total of 12 patients receiving pevonedistat at the MTD (20 mg/m2) in combination with paclitaxel plus carboplatin; one of these six patients reported a DLT (grade 3 increased AST), confirming the MTD in this arm.
In arm 3, eight patients enrolled at the initial dose of pevonedistat 25 mg/m2 were DLT evaluable and three experienced a DLT: one had grade 3 increased ALT and AST, one had grade 4 febrile neutropenia and withdrew from the study, and one had grade 3 febrile neutropenia, withdrew from the study, and died from complications of febrile neutropenia. Due to the number of DLTs observed in this arm, and based on two patients missing >1 treatment dose in cycle 1, as well as the need to delay therapy in cycle 2 to allow recovery from gemcitabine-related myelosuppression toxicity, no additional patients were enrolled in accordance with the CRM algorithm. Pevonedistat dose de-escalation to 15 mg/m2 was not explored, as myelosuppression was considered related to gemcitabine. The MTD of pevonedistat in combination with gemcitabine was not determined, and this drug combination was abandoned.
Of 15 patients with cycle 1 DLTs, 10 had liver function test (LFT) elevations (increased AST/ALT), which were the predominant DLTs reported across all arms regardless of standard-of-care chemotherapy. All LFT elevations were without clinical sequelae and reversible with dose delays or holds; prior therapies for these patients are reported in the Online Resource: Supplementary Table 2.
Safety
All 64 patients experienced ≥1 AE and 53 (83%) had grade ≥3 AEs (Table 3). The most common any-grade AEs were fatigue (56%), nausea (50%), anemia (41%), constipation, diarrhea (34% each), increased AST (31%), and vomiting (30%). The most common grade ≥3 AEs were decreased neutrophil count (22%) and increased AST (20%). Overall, 95% of patients had ≥1 drug-related AE of any grade and 66% had grade ≥3 drug-related AEs (Online Resource: Supplementary Table 3). Across arms, 26 (41%) patients experienced ≥1 serious AE, including febrile neutropenia (9%), dyspnea (6%), abdominal pain, and pneumonia (3% each).
Four patients (arm 2) reported AEs leading to permanent treatment discontinuation. Of these, one patient receiving pevonedistat 15 mg/m2 in combination with carboplatin plus paclitaxel discontinued due to thrombocytopenia, and three patients (two receiving pevonedistat 20 mg/m2 and one 25 mg/m2, both with carboplatin plus paclitaxel) discontinued due to peripheral neuropathy or peripheral sensory neuropathy.
There were five on-study deaths, including one on day 23, cycle 1 that was considered drug-related (arm 3, pevonedistat 25 mg/m2: febrile neutropenia). A further three patients died during cycle 1 and one died during cycle 9, all of which were considered unrelated to treatment (all pevonedistat 25 mg/m2; one each in arm 1 and arm 2, two in arm 3).
Pevonedistat pharmacokinetics
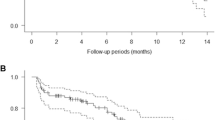
Figure 1 compares the dose-normalized pevonedistat concentrations for patients receiving pevonedistat in combination with docetaxel (n = 16, dose-escalation; n = 6, MTD expansion), carboplatin plus paclitaxel (n = 20 dose-escalation; n = 6, MTD expansion), or gemcitabine (n = 10) with those from patients who received single-agent pevonedistat in previous studies [7,8,9, 11]. Examination of individual patient plasma concentration-time data (Fig. 1a and c) revealed no significant changes in pevonedistat pharmacokinetics when given with docetaxel (arm 1) or gemcitabine (arm 3), as the observed pevonedistat concentrations in the presence of docetaxel or gemcitabine showed considerable overlap with those with pevonedistat alone. By contrast, following the end of infusion and throughout the 48-h sampling period, pevonedistat plasma concentrations were consistently higher in patients receiving carboplatin plus paclitaxel than in patients receiving single-agent pevonedistat (Fig. 1b).

Observed dose-normalized concentration-time profiles of pevonedistat with and without concurrent administration of docetaxel (a), carboplatin plus paclitaxel (b), or gemcitabine (c). The figure shows raw concentration-time data from prior studies of single-agent pevonedistat (circles) [7,8,9, 11] and from the combination therapy in this study (crosses)
Disease response
A summary of best response to treatment is shown in Table 4. In arm 1, among 19 response-evaluable patients, three patients receiving the MTD achieved a partial response (PR), for an overall response rate (ORR) of 16%. Among responders, one had cholangiocarcinoma, two had head and neck cancer (squamous cell carcinoma and salivary gland carcinoma), and all three had lung metastases. One patient in arm 2a (squamous cell carcinoma of the head and neck not otherwise specified) achieved a PR.
In arm 2, among 23 response-evaluable patients, eight patients achieved a PR or complete response (CR; ORR 35%). Two patients with bladder cancer and endometrial cancer receiving pevonedistat 20 mg/m2 (MTD) achieved a CR; at data cutoff, the duration of CR was 8.1 and 8.5 months (9 and 11 cycles), respectively. At the time of reporting, both patients remained on study (cycle 43+ representing the longest treatment duration) receiving single-agent pevonedistat since cycle 9 and 13, respectively. Of the six patients achieving PRs in arm 2, two received pevonedistat 25 mg/m2 and four received pevonedistat 20 mg/m2; all six had progressive disease after PR.
Median duration of response (DOR) for the 12 patients achieving a CR or PR across both arms was 5.9 months (range, 0.03–12.02). Median DOR for the eight patients receiving pevonedistat with carboplatin and paclitaxel (arm 2) was 7.4 months (range, 2.37–12.02). Across 54 response-evaluable patients, 41 achieved stable disease or better (≥SD), of whom 25 received treatment for ≥5 cycles (eight in arm 1, 15 in arm 2, and two in arm 3), with the longest treatment duration being 21 cycles at data cutoff (arm 2, chondrosarcoma). At the pevonedistat MTDs, 10 of 16 patients in arm 1 (25 mg/m2) and all 12 patients in arm 2 (20 mg/m2) achieved ≥SD; of these, six and nine patients, respectively, received study treatment for ≥5 cycles.
Prior therapies in patients achieving a response are presented in the Online Resource: Supplementary Table 4. Of 12 patients achieving a CR or PR, 10 had received prior platinum therapy. None of the responders in arm 1 (pevonedistat plus docetaxel) had received prior docetaxel, and one patient had received prior carboplatin plus paclitaxel. By contrast, all but one patient achieving a CR or PR in arm 2 (pevonedistat in combination with carboplatin and paclitaxel) and one responder in arm 2a (pevonedistat plus carboplatin) had received ≥1 course of platinum and/or taxane. In arm 2, four patients had received both taxane and platinum as prior therapies.
ERCC1 expression
ERCC1 expression levels were determined by calculating the H-score in patients receiving pevonedistat 20 mg/m2 or 15 mg/m2 in arm 2 and arm 2a. The median H-score for all ERCC1-evaluable patients (n = 21) was 170 (range, 65–290), which served as the cutoff to classify patients into ERCC1 high (H-score > 170, n = 10) and low (H-score ≤170, n = 11) expression level groups. High ERCC1 expression appeared to correlate with clinical benefit (CR/PR/SD duration ≥5 cycles; Fig. 2a). There was a trend for patients with high ERCC1 levels to remain on study longer than patients with low ERCC1, with a median time on study of 10.5 and 4.7 months, respectively (Fig. 2b).

Correlation between baseline ERCC1 expression levels* and (a) clinical benefit (CR/PR/durable SD ≥5 cycles) or (b) time on study. (a) *ERCC1 staining intensity was determined by immunohistochemistry and the H-score was calculated. The median H-score of 170 for the ERCC1-evaluable patient population (n = 21) was used as the cutoff to dichotomize patients into ERCC1 high (H-score > 170, n = 10) and low (H-score ≤170, n = 11) expression level groups. (b) The black horizontal lines inside the boxes represent the median time on study for the ERCC1 high and low expression level groups. Time on study (months) was defined as follows: (last study visit date – first dose date +1)/30.4375. This was 10.5 and 4.7 months for patients with ERCC1 high and low expression levels, respectively. Box plots represent interquartile range. Outliers are indicated by the highlighted circles. CR, complete response; ERCC1, Excision repair cross-complementation group 1; PD, progressive disease; PR, partial response; SD, stable disease
Discussion
This phase Ib study was the first to investigate the safety and tolerability of pevonedistat plus standard-of-care chemotherapies in patients with advanced solid tumors. The MTD for pevonedistat was determined to be 25 mg/m2 with docetaxel and 20 mg/m2 with carboplatin plus paclitaxel. The combination of pevonedistat plus gemcitabine was deemed intolerable due to two patient withdrawals and one on-study death (febrile neutropenia), and based on patients missing ≥1 treatment dose (cycle 1) or experiencing gemcitabine-related myelosuppression which led to dose delays in cycle 2. Therefore, enrollment into this cohort ceased. The most frequently reported DLTs across all arms included grade 3 febrile neutropenia and increased ALT/AST; AST/ALT elevations were reversible with dose holds, reductions, or delays.
The safety profile of pevonedistat plus docetaxel or carboplatin plus paclitaxel was generally favorable. Common AEs reported in this study (fatigue, nausea, anemia, constipation, and diarrhea) were similar to those previously observed in patients receiving standard-of-care chemotherapies [27,28,29]. With dose reductions or delays, repeat dosing of pevonedistat plus docetaxel or pevonedistat in combination with carboplatin plus paclitaxel was feasible in patients with solid tumors, and at the time of reporting, two patients (arm 2) remained on study receiving single-agent pevonedistat 20 mg/m2 (currently cycle 43+). Pevonedistat treatment did not appear to result in additional toxicity when added to standard-of-care chemotherapy, and there was no cumulative toxicity from pevonedistat despite prolonged treatment.
Evaluation of concentration-time data from 58 patients for whom blood samples were available revealed that the pharmacokinetic profile of pevonedistat was unchanged in the presence of docetaxel or gemcitabine compared with historical single-agent pevonedistat pharmacokinetic data [7,8,9, 11]. There was a trend toward higher pevonedistat plasma concentrations when given with carboplatin plus paclitaxel, compared with single-agent administration. This apparent drug-drug interaction effect, which cannot be explained at this time, warrants further investigation. Although some overlap exists in the metabolizing enzymes and/or transporter proteins involved in the drug clearance of pevonedistat and the standard-of-care agents, none are known inhibitors or inducers. A radiolabeled mass balance study is ongoing to characterize the clearance pathways of pevonedistat in humans (ClinicalTrials.gov identifier NCT03057366). Additionally, because of the limited pharmacokinetic sampling scheme used in this study, individual concentration-time data will be pooled with other study data and analyzed using a population modeling approach to quantify the observed effects of carboplatin and paclitaxel on pevonedistat pharmacokinetics.
The combination of pevonedistat with carboplatin plus paclitaxel (arm 2) showed the most promising broad-based antitumor activity in pretreated patients (≥1 prior therapies). Notably, all but one of the eight responders in arm 2 and one responder in arm 2a had previously received platinum, taxanes, or both. The ORR in arm 2 was 35%, including two patients with CR (bladder cancer and endometrial carcinoma). These two patients discontinued carboplatin plus paclitaxel chemotherapy at cycle 9 and 13, respectively, and went on to maintenance therapy with single-agent pevonedistat; both patients are currently continuing treatment in cycle 43 without evidence of disease. Consistent with preclinical studies reporting synergy between pevonedistat and standard-of-care chemotherapy [13, 24], the long treatment durations in this study and objective responses in patients resistant to prior platinum/taxane therapy suggest the potential reversal of resistance by the addition of pevonedistat. Further exploration of pevonedistat and carboplatin plus paclitaxel in the platinum- and/or taxane-resistant setting is warranted.
Pevonedistat was shown in model systems to synergize with platinum-containing agents by interfering with NER and ICR pathways [13]. Previous studies reported that low ERCC1 expression is associated with clinical benefit in advanced cancer patients receiving platinum-based chemotherapy [19,20,21]. Interestingly, in our study, patients receiving pevonedistat plus paclitaxel and carboplatin, who had high ERCC1 expression appeared to show greater clinical benefit and remained on study longer than those with low ERCC1 expression. Further investigations are warranted to provide insight into this observation, which suggests the possibility that pevonedistat might re-sensitize patients to platinum-based chemotherapy and provide a potential new approach to therapy in these patients.
The observed clinical benefit in patients treated in arm 2, especially in patients with prior taxane and/or platinum exposure, supports further investigation of pevonedistat with carboplatin plus paclitaxel in phase II trials.
References
Ciechanover A (2005) Intracellular protein degradation: from a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Cell Death Differ 12(9):1178–1190
Soucy TA, Smith PG, Rolfe M (2009) Targeting NEDD8-activated cullin-RING ligases for the treatment of cancer. Clin Cancer Res 15(12):3912–3916
Zhao Y, Sun Y (2013) Cullin-RING ligases as attractive anti-cancer targets. Curr Pharm Des 19(18):3215–3225
Brownell JE, Sintchak MD, Gavin JM, Liao H, Bruzzese FJ, Bump NJ, Soucy TA, Milhollen MA, Yang X, Burkhardt AL, Ma J, Loke HK, Lingaraj T, Wu D, Hamman KB, Spelman JJ, Cullis CA, Langston SP, Vyskocil S, Sells TB, Mallender WD, Visiers I, Li P, Claiborne CF, Rolfe M, Bolen JB, Dick LR (2010) Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol Cell 37(1):102–111
Luo Z, Yu G, Lee HW, Li L, Wang L, Yang D, Pan Y, Ding C, Qian J, Wu L, Chu Y, Yi J, Wang X, Sun Y, Jeong LS, Liu J, Jia L (2012) The Nedd8-activating enzyme inhibitor MLN4924 induces autophagy and apoptosis to suppress liver cancer cell growth. Cancer Res 72(13):3360–3371
Li L, Wang M, Yu G et al (2014) Overactivated neddylation pathway as a therapeutic target in lung cancer. J Natl Cancer Inst 106(6):dju083
Sarantopoulos J, Shapiro GI, Cohen RB, Clark JW, Kauh JS, Weiss GJ, Cleary JM, Mahalingam D, Pickard MD, Faessel HM, Berger AJ, Burke K, Mulligan G, Dezube BJ, Harvey RD (2016) Phase I study of the investigational NEDD8-activating enzyme inhibitor Pevonedistat (TAK-924/MLN4924) in patients with advanced solid tumors. Clin Cancer Res 22(4):847–857
Bhatia S, Pavlick AC, Boasberg P, Thompson JA, Mulligan G, Pickard MD, Faessel H, Dezube BJ, Hamid O (2016) A phase I study of the investigational NEDD8-activating enzyme inhibitor pevonedistat (TAK-924/MLN4924) in patients with metastatic melanoma. Investig New Drugs 34(4):439–449
Shah JJ, Jakubowiak AJ, O'Connor OA, Orlowski RZ, Harvey RD, Smith MR, Lebovic D, Diefenbach C, Kelly K, Hua Z, Berger AJ, Mulligan G, Faessel HM, Tirrell S, Dezube BJ, Lonial S (2016) Phase I study of the novel investigational NEDD8-activating enzyme inhibitor Pevonedistat (MLN4924) in patients with relapsed/refractory multiple myeloma or lymphoma. Clin Cancer Res 22(1):34–43
Faessel HM, Mould DR, Dezube BJ, Santillana S, Venkatakrishnan K (2015) Abstract B148: Population pharmacokinetics (PK) of the investigational NEDD8-activating enzyme (NAE) inhibitor pevonedistat (TAK-924/MLN4924) administered alone or in combination with azacitidine in patients (pts) with solid or hematologic malignancies. Mol Cancer Ther 14(12 Supplement 2):B148-B148
Swords RT, Erba HP, DeAngelo DJ et al (2015) Pevonedistat (MLN4924), a first-in-class NEDD8-activating enzyme inhibitor, in patients with acute myeloid leukaemia and myelodysplastic syndromes: a phase 1 study. Br J Haematol 169(4):534–543
Swords RT, Watts J, Erba HP, Altman JK, Maris M, Anwer F, Hua Z, Stein H, Faessel H, Sedarati F, Dezube BJ, Giles FJ, Medeiros BC, DeAngelo DJ (2017) Expanded safety analysis of pevonedistat, a first-in-class NEDD8-activating enzyme inhibitor, in patients with acute myeloid leukemia and myelodysplastic syndromes. Blood Cancer J 7(2):e520
Bouck DC, Garcia K, Blank JL et al (2015) Abstract C55: Nedd8-activating enzyme inhibitor pevonedistat synergizes with cisplatin and carboplatin through interference with nucleotide excision repair and interstrand cross-link repair mechanisms in non-small cell lung cancer. Mol Cancer Ther 14(12 Supplement 2):C55-C55
Kirschner K, Melton DW (2010) Multiple roles of the ERCC1-XPF endonuclease in DNA repair and resistance to anticancer drugs. Anticancer Res 30(9):3223–3232
Iyama T, Wilson DM 3rd (2013) DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (Amst) 12(8):620–636
Olaussen KA, Dunant A, Fouret P, Brambilla E, André F, Haddad V, Taranchon E, Filipits M, Pirker R, Popper HH, Stahel R, Sabatier L, Pignon JP, Tursz T, le Chevalier T, Soria JC, IALT Bio Investigators (2006) DNA repair by ERCC1 in non-small-cell lung cancer and cisplatin-based adjuvant chemotherapy. N Engl J Med 355(10):983–991
Friboulet L, Olaussen KA, Pignon JP, Shepherd FA, Tsao MS, Graziano S, Kratzke R, Douillard JY, Seymour L, Pirker R, Filipits M, André F, Solary E, Ponsonnailles F, Robin A, Stoclin A, Dorvault N, Commo F, Adam J, Vanhecke E, Saulnier P, Thomale J, le Chevalier T, Dunant A, Rousseau V, le Teuff G, Brambilla E, Soria JC (2013) ERCC1 isoform expression and DNA repair in non-small-cell lung cancer. N Engl J Med 368(12):1101–1110
Smith DH, Fiehn AM, Fogh L et al (2014) Measuring ERCC1 protein expression in cancer specimens: validation of a novel antibody. Sci Rep 4:4313
Handra-Luca A, Hernandez J, Mountzios G, Taranchon E, Lacau-St-Guily J, Soria JC, Fouret P (2007) Excision repair cross complementation group 1 immunohistochemical expression predicts objective response and cancer-specific survival in patients treated by cisplatin-based induction chemotherapy for locally advanced head and neck squamous cell carcinoma. Clin Cancer Res 13(13):3855–3859
Chen S, Zhang J, Wang R, Luo X, Chen H (2010) The platinum-based treatments for advanced non-small cell lung cancer, is low/negative ERCC1 expression better than high/positive ERCC1 expression? A meta-analysis. Lung Cancer 70(1):63–70
Li S, Wu J, Chen Y, Tang W, Peng Q, Deng Y, Xie L, Wang J, Huang S, Li R, Qin X, Zhao J (2014) ERCC1 expression levels predict the outcome of platinum-based chemotherapies in advanced bladder cancer: a meta-analysis. Anti-Cancer Drugs 25(1):106–114
Jazaeri AA, Shibata E, Park J, Bryant JL, Conaway MR, Modesitt SC, Smith PG, Milhollen MA, Berger AJ, Dutta A (2013) Overcoming platinum resistance in preclinical models of ovarian cancer using the neddylation inhibitor MLN4924. Mol Cancer Ther 12(10):1958–1967
Nawrocki ST, Kelly KR, Smith PG, Espitia CM, Possemato A, Beausoleil SA, Milhollen M, Blakemore S, Thomas M, Berger A, Carew JS (2013) Disrupting protein NEDDylation with MLN4924 is a novel strategy to target cisplatin resistance in ovarian cancer. Clin Cancer Res 19(13):3577–3590
Garcia K, Blank JL, Bouck DC, Liu XJ, Sappal DS, Hather G, Cosmopoulos K, Thomas MP, Kuranda M, Pickard MD, Liu R, Bandi S, Smith PG, Lightcap ES (2014) Nedd8-activating enzyme inhibitor MLN4924 provides synergy with mitomycin C through interactions with ATR, BRCA1/BRCA2, and chromatin dynamics pathways. Mol Cancer Ther 13(6):1625–1635
Swords RT, Coutre S, Maris MB et al (2016) Results of a Clinical Study of Pevonedistat (Pev), a First-in-Class NEDD8-Activating Enzyme (NAE) Inhibitor, Combined with Azacitidine (Aza) in Older Patients (Pts) with Acute Myeloid Leukemia (AML). Paper presented at the ASH 58th Annual Meeting and Exposition, San Diego, California, December 3–6, 2016
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45(2):228–247
Tabernero J, Climent MA, Lluch A, Albanell J, Vermorken JB, Barnadas A, Antón A, Laurent C, Mayordomo JI, Estaun N, Losa I, Guillem V, Garcia-Conde J, Tisaire JL, Baselga J (2004) A multicentre, randomised phase II study of weekly or 3-weekly docetaxel in patients with metastatic breast cancer. Ann Oncol 15(9):1358–1365
Chen YM, Perng RP, Lee YC, Shih JF, Lee CS, Tsai CM, Whang-Peng J (2002) Paclitaxel plus carboplatin, compared with paclitaxel plus gemcitabine, shows similar efficacy while more cost-effective: a randomized phase II study of combination chemotherapy against inoperable non-small-cell lung cancer previously untreated. Ann Oncol 13(1):108–115
Smorenburg CH, Bontenbal M, Seynaeve C, Zuylen C, Heus G, Verweij J, Wit R (2001) Phase II study of weekly gemcitabine in patients with metastatic breast cancer relapsing or failing both an anthracycline and a taxane. Breast Cancer Res Treat 66(1):83–87
Acknowledgements
This study was funded by Millennium Pharmaceuticals, Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. The authors thank the study participants, as well as their families and staff at the study centers. The authors also acknowledge Yosef Mansour, PhD, and Laila Cancian, PhD, (FireKite, an Ashfield Company, part of UDG Healthcare plc) for medical writing support in compliance with Good Publication Practice 3 ethical guidelines (Battisti et al., Ann Intern Med 2015;163:461–4), which was funded by Millennium Pharmaceuticals, Inc., and Janice Y. Ahn, PhD (Millennium Pharmaceuticals, Inc.) for editorial support.
Funding
Research was sponsored by Millennium Pharmaceuticals, Inc., Cambridge, MA, USA, a wholly owned subsidiary of Takeda Pharmaceutical Company Limited.
Author information
Authors and Affiliations
Contributions
Study conception and design: CA, FS, BJD, AD, DH, and HF Collection and assembly of data: CA, FS, BJD, AD, ABD, ACL, RBC, CBL, and TMB Data analysis and interpretation: CA, FS, BJD, AD, ABD, ACL, RBC, DH, HF, DVF and TKN TMB, CBL, DH and AD provided the study materials or patients. All authors contributed to the writing of the manuscript and critically revised the manuscript; all authors reviewed and approved the final version of the manuscript and agreed to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Conflicts of interest
ACL has no conflicts of interest to disclose. CA reports honoraria and advisory role fees from Genentech, BMS, and Eli Lilly, and research funding from Macrogenics to the Perelman School of Medicine at the University of Pennsylvania, Philadelphia, PA, USA. TMB reports research funding from Daiichi Sankyo, Medpachto Inc., Incyte, Mirati Therapeutics, MedImmune, Abbvie, AstraZeneca, Leap Therapeutics, MabVax, Stemline Therapeutics, Merck, Eli Lilly, GSK, Novartis, Pfizer, Principa Biopharma, Genentech/Roche, Deciphera, Merrimack, Immunogen, Takeda, Ignyta, Calithera Biosciences, Kolltan Pharmaceuticals, Principa Biopharma, Peleton, Immunocore, Roche, Aileron Therapeutics, BMS, and Amgen to the Sarah Cannon Research Institute/Tennessee Oncology PLLC, Nashville, TN, USA. CBL reports travel/accommodation fees or expenses from Novartis and research funding form Takeda, Novartis, and Genentech to the University of North Carolina at Chapel Hill, Chapel Hill, NC, USA. DH reports research funding from Takeda. R.B.C. reports consulting or advisory roles for Takeda, BMS, Kolltan, Cerulean Pharma Inc., and Constellation Pharmaceuticals, and research funding from Takeda. FS, HF, and ABD report stock ownership in and employment by Millennium Pharmaceuticals Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. FS reports travel/accommodation fees or expenses from Takeda. DVF reports employment by Millennium Pharmaceuticals Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited, and patents or intellectual property interests with Phoenicia BioSciences. TKN and BJD are former employees of Millennium Pharmaceuticals Inc., a wholly owned subsidiary of Takeda Pharmaceutical Company Limited. A.D. reports consulting or advisory role fees from Abbvie and AstraZeneca, and research funding from Abbvie, EMD Serono, Genentech, Eli Lilly, Piqur, Taino, Vertex, and Takeda.
Ethical approval
All procedures performed in this study involving human participants were in accordance with the ethical standards of the participating institutions and the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Written informed consent was obtained from all individual participants included in the study.
Electronic supplementary material
ESM 1
(DOCX 45 kb)
Supplementary Fig. 1
(PDF 1335 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Lockhart, A.C., Bauer, T.M., Aggarwal, C. et al. Phase Ib study of pevonedistat, a NEDD8-activating enzyme inhibitor, in combination with docetaxel, carboplatin and paclitaxel, or gemcitabine, in patients with advanced solid tumors. Invest New Drugs 37, 87–97 (2019). https://doi.org/10.1007/s10637-018-0610-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-018-0610-0