
Abstract
There is a recognized need for new treatment options for type 2 diabetes mellitus (T2DM). Recovery of glucose from the glomerular filtrate represents an important mechanism in maintaining glucose homeostasis and represents a novel target for the management of T2DM. Recovery of glucose from the glomerular filtrate is executed principally by the type 2 sodium-glucose cotransporter (SGLT2). Inhibition of SGLT2 promotes glucose excretion and normalizes glycemia in animal models. First reports of specifically designed SGLT2 inhibitors began to appear in the second half of the 1990s. Several candidate SGLT2 inhibitors are currently under development, with four in the later stages of clinical testing. The safety profile of SGLT2 inhibitors is expected to be good, as their target is a highly specific membrane transporter expressed almost exclusively within the renal tubules. One safety concern is that of glycosuria, which could predispose patients to increased urinary tract infections. So far the reported safety profile of SGLT2 inhibitors in clinical studies appears to confirm that the class is well tolerated. Where SGLT2 inhibitors will fit in the current cascade of treatments for T2DM has yet to be established. The expected favorable safety profile and insulin-independent mechanism of action appear to support their use in combination with other antidiabetic drugs. Promotion of glucose excretion introduces the opportunity to clear calories (80–90 g [300–400 calories] of glucose per day) in patients that are generally overweight, and is expected to work synergistically with weight reduction programs. Experience will most likely lead to better understanding of which patients are likely to respond best to SGLT2 inhibitors, and under what circumstances.
Similar content being viewed by others

Introduction
Type 2 diabetes mellitus (T2DM) is a growing epidemic: in the United States of America alone, nearly 25.8 million people (approx 8.3% of the total population) have the disease.1 Estimates have placed the global prevalence of the disease at around 217 million.2 The consequence to the individual of prolonged exposure to hyperglycemia is a marked increase in the risk of mortality and morbidity, with an associated reduction in life expectancy of around 12–13 years.3 Diagnosis usually occurs some time after development of the disease and they have often already experienced occult pathology by the time patients receive a diagnosis. Early adoption of an aggressive approach to disease management improves patient outcome, with marked reductions in morbidity and mortality.4–6
The first line approach to treatment is through lifestyle modification.7 However, T2DM is progressive in nature, and lifestyle changes sufficient to halt the disease are difficult to achieve. Intense efforts by patients and the healthcare team frequently fail. Similarly, most conventional antidiabetic drugs (eg, metformin, sulfonylureas etc.) often fail to slow the progression of T2DM, despite the availability of a broad range of agents employing different mechanisms of action.7–9 The progressive nature of the disease and how it overwhelms available therapies was highlighted in the UKPDS study; suggesting that fewer than half of all patients actually achieve adequate levels of disease control.4,5
There is a recognized need for new treatment options for T2DM. Characterization of the mechanisms facilitating glucose resorption by the kidney has raised the possibility of a novel treatment for diabetes: inhibition of the type 2 sodium-glucose transporter (SGLT2), a 672 amino-acid, high capacity, low affinity transmembrane protein that promotes reabsorption of glucose as the glomerular filtrate passes down the nephrons.10 Several candidate molecules are currently in development and may soon be available for use in the treatment of diabetes. We provide a brief review of SGLT2 inhibitors and their possible role in the treatment of T2DM.
Role of Sodium Glucose Transporter in Renal Glucose Excretion
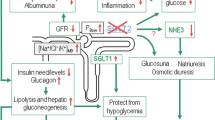
Most (>99%) of the plasma glucose entering the kidney filters into the nephrons though the glomeruli. Under normal circumstances, the reabsorptive capacity of the early part of the nephron, the proximal tubule, is sufficient to clear the filtered glucose load from the luminal fluid before it enters the Loop of Henlé. In normal individuals, around 180 g of glucose passes into the proximal tubules each day, from where it is almost completely reabsorbed.11,12 As plasma glucose concentrations increase, the filtered glucose load increases in a linear manner. When the rate of glucose entering the nephron rises above 260–350 mg/min/1.73 m2, for example in patients with diabetes, the excess glucose outstrips resorptive capacity and appears in the urine (glycosuria).13 In a healthy adult, this equates to a blood glucose concentration of approximately 11 mmol/L (200 mg/dL).14 As much as 90% of the filtered glucose load is extracted in the S1 segment, and the remaining 10% is removed in the distal straight tubules (S2 and S3 segments) (Figure 1). Until recently, the mechanisms behind glucose reabsorption were poorly understood, although it was proposed as early as 1960 that glucose transmembrane flux could be achieved through the coupling of glucose transport with that of sodium.15

Summary of glucose reabsorption from the lumen of the nephrons. Glucose concentration in the glomerular filtrate reflects plasma concentration. In most normal healthy subjects, the majority of glucose reabsorption is believed to occur in the early part of the proximal tubule.
Since the start of the 20th century, phlorizin, a toxic 2′-glucoside of phloretin, has been known to increase glycosuria, and has been used in the study of renal function.16,17 During the 1930s, phlorizin was used in non-invasive human experiments that revealed some of the fundamental mechanisms of renal hemodynamics and metabolic transport.18 In the 1950s, studies delineated phlorizin’s mechanism of action on inhibition of glucose transport in the kidney and small intestine at the cellular and molecular levels. Renal micropuncture studies conducted with phlorizin in the 1970s showed that the transporter was located in the brush border of the proximal tubule, and that sodium was required for the renal absorption of glucose.11,19,20 Studies performed since then confirmed that phlorizin is a competitive inhibitor of glucose transport, with a binding affinity for the transporter that is 1000- to 3000-fold greater than that of glucose.21
The rabbit homolog of the human type 1 sodium-glucose transporter (SGLT1), which is coded by the SLC5A gene, was the first mammalian cotransporter carrier protein to be identified, cloned, and sequenced.22 A family of SLC5A gene sodium-dependent transporters has since been sequenced and identified in a broad range of tissues.23,24 SGLT1 and SGLT2 are, perhaps, the SLC5A family members that have received greatest coverage within the literature. The high affinity, low capacity SLGT1 is the main gastrointestinal glucose transporter. However, SLGT1 accounts for only a small proportion of renal tubular glucose reabsortion. The relatively widespread distribution of SGLT1 is contrasted by the almost exclusive expression on the luminal surface of proximal tubules (mainly in the renal cortex) of the low glucose affinity, high capacity SGLT2, responsible for most renal tubular glucose reabsorption.22–26 Cellular glucose and sodium uptake occurs in a 1:1 ratio (Figure 2). The sodium:potassium adenosine triphosphatase (ATP) pump transports sodium across the basolateral surface into the intracellular fluid, maintaining the physiological levels of sodium in the cell. The inward sodium concentration gradient drives the ‘uphill’ glucose reabsorption (a secondary active transport mechanism). Cellular glucose concentrations are maintained by facilitative glucose outflow through transporters in the basolateral membrane of the cell. After binding intracellular glucose the transporters undergo a conformational change that subsequently moderates the movement of glucose (down its concentration gradient) back into the blood (Figure 3).

Representation of the 1:1 transport of sodium and glucose across the luminal membrane of the epithelial cells of the early part of the proximal tubule facilitated by SGLT2

Summary of the glucose transport cascade in the brush border epithelial cells of the proximal tubule demonstrating how the process is driven by the sodium gradient maintained by the Na+-K+ ATPase of the basal membrane
SGLT2 Inhibitors
The antidiabetic properties of phlorizin were investigated in the 1980s. In partially pancreatectomized rats, phlorizin increased glucose secretion in urine and this was associated with a normalizing of plasma glucose, without inducing hypoglycemia.17 Despite its promising in vitro properties, phlorizin does not fit the profile that we have come to expect from a modern therapeutic agent. Phlorizin is hydrolyzed to phloretin in the gut, resulting in poor oral bioavailability. Phlorizin is also potentially toxic and is non-selective, inhibiting both SGLT1 and SGLT2 transporters. In the last decade, several alternative candidate molecules, targeted to specifically inhibit SGLT2, have been investigated in both pre-clinical and clinical settings.27 The aim has been to take advantage of the potential for ‘turning-off’ glucose reabsorption as a new therapeutic target for the treatment of T2DM.
First reports of devised SGLT2 inhibitors started to emerge in the scientific literature in the second half of the 1990s. Developed with a view to overcoming the shortcomings of phlorizin, SGLT2 inhibitors represented a new mechanism to manage hyperglycemia that acted independently of insulin and irrespective of patients’ glycemic status. First indications suggest that the mechanism of action, which is independent of insulin, further reduces glycemia when used in combination with traditional antidiabetic treatments.
Results with early compounds were promising in terms of specificity for the transporter: the compound T-1095 has inhibitory capacity for SGLT2 that is 4-fold greater than for SGLT1.25 Pharmacodynamic studies demonstrated attenuated hyperinsulinemia and hypertriglyceridemia in KK rats following oral administration of T-1095.26 Lowering of insulin resistance and HbA1c levels along with normalized hepatic glucose production and glucose utilization rate were also observed in streptozotocin-induced diabetic rats26,28 and Zucker diabetic fatty rats28,29 following oral administration of T-1095. Long-term administration of T-1095 restored impaired insulin secretion from pancreatic β-cells in Goto-Kakizaki (GK) rats30 and suppressed diabetic complications in both C57BL/KsJ-db/db mice and GK rats.31,32 However, retained co-inhibition of SGLT1 by T-1095 led to development of the compound being discontinued in 2003, having reached phase II clinical trials.
Various SGLT2 inhibitors based on the glucoside structure of phlorizin have since been proposed (Table 1), and narratives of the discovery pathway of the different inhibitors have recently been published.27,33 The glucoside moiety of phlorizin binds to SGLT2 transporters and the ‘O’-linked phenolic distal ring is responsible for its inhibitory properties.34 Structure-activity analysis of the parent molecule shows that addition of lipophilic groups to the distal ring augments the inhibition of the SGLT2 transporter, and increases selectivity for SGLT2 over SGLT1.25 However, the O-linkage is a metabolic target for β-glucosidase enzymes that can curtail the activity of SGLT2 inhibitors in vivo. To address this possible limitation to therapeutic utility, candidate SGLT2 inhibitors have been synthesized that employ a C-glucoside linkage (Table 1). Both the O- and C-glucosides appear to bind to a single site on the SGLT2 transporter. The aromatic and heteroaromatic C-glucosides are metabolically more stable than O-glucosides, due to their relative resistance to hydrolysis. Alternative candidate SGLT2 inhibitors that have also been considered include modified sugar rings, N-glucosides and, more recently, a bridged ketal ring.35

Candidate SGLT2 inhibitors
Manufacturers: Johnson & Johnson/Tanabe Seiyaku Co. Ltd ( Japan) (T-1095); Sanofi-Aventis (AVE-2268); GlaxoSmithKline (remogliflozin/KGT 1681/sergliflozin); Wyeth (WAY-123783); Astellas Pharma Inc (ASP 1941/YM-543); Boehringer Ingelheim GmbH (BI-10773/BI-44847); Boehringer Ingelheim, GmbH/Ajinomoto (canagliflozin); Johnson & Johnson/Tanabe Mitsubishi (TA-7284/JNJ 28431754); Bristol-Myers Squibb Co/AstraZeneca (dapagliflozin); Lexicon (LX 4211); Isis Pharmaceuticals (ISIS 388626); Roche/Chugai (R7201/CSG452); Pfizer (PF-04971729). Candidate molecules have also been registered by Kissei, Taisho, Theracos, and Daiichi Sankyo. NA; not applicable.
Another approach uses antisense oligonucleotides to inhibit expression of SGLT2. Administration of synthesized strands of nucleic acid targeted to specifically bind to SGLT2 messenger RNA blocks the transporter’s translation, protein production, and expression in the cells of the proximal tubule.
A summary of the status of inhibitor development is provided in Table 2.36–54
Clinical Expectations for SGLT-2 Inhibitors
As the above discussion suggests, there are several hypothetical reasons why the SGLT2 transporter represents an opportune target for managing blood glucose. However, the challenge is to establish therapeutic utility while demonstrating an acceptable safety profile. A detailed summary of clinical findings has recently been published.55
Efficacy
The mechanism of action of SGLT2 inhibitors predicts a beneficial effect, but the long-term glucose lowering capacity in a clinical setting may not impart significant reductions in HbA1c. Modest HbA1c lowering in the region of 0.5%–0.9%, that may be predicted from early clinical studies, would be comparable to that achieved with other currently marketed oral agents.55 It remains to be seen whether promoting glucose excretion will result in long-term benefits for the patient in terms of returning metabolic balance, or even weight loss. Clearly, blocking glucose reabsorption permits the clearance of glucose from the body, and thus must eventually serve to reduce levels of plasma glucose. The amount of glucose available for excretion is dependent on the amount entering the nephrons, which, in turn, depends on blood glucose concentration at the glomerulus. Thus, the amount of glucose excreted is greater when the blood plasma glucose concentrations are highest. In effect, glucose ‘removal’ might be expected only to be greatest at times when it is most needed (when glucose levels are highest), such as during post-prandial hyperglycemia.
The benefit to those patients in whom treatment has provided mild-to-moderate glycemic control might be questioned, as the potential for glucose excretion would be relatively low. Nevertheless, patients who achieve moderate glycemic control may be exposed to clinically relevant post-prandial glucose excursions that can impart disproportionate effects on HbA1c and possibly the morbidity and mortality associated with T2DM.56 In such a patient population, SGLT2 inhibitors might attenuate the impact of post-prandial glucose spikes. Nevertheless, clinical experience with agents, such as the meglitinides, that target post-prandial glucose control, suggest that the clinical benefit of this approach is disappointing. Treatments targeting post-prandial glucose levels provide little more than modest improvements in HbA1c with little evidence of long-term outcome benefits for patients.57
As SGLT2 may be responsible for as much as 90% of glucose reabsorption by the kidney, there is the clinical potential for as much as 160 g of glucose to be excreted each day following effective SGLT2 inhibition.23 However, it appears that the actual glucose loss achieved in clinical studies is only about half that predicted.38 It is not clear whether this is a consequence of compensating mechanisms undertaking tubular reabsorption or incomplete inhibition of the transporter.
Safety
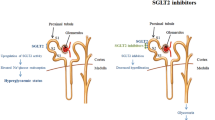
Thus far, the safety profile of SGLT2 inhibitors reported from clinical studies appears to fulfill expectations.33,34,55,40,58 SGLT2 inhibitors are ‘designed’ to target a highly specific membrane transporter that is almost exclusively expressed within the renal tubules. Clearly, compared with less specific molecules, the potential for cross-reaction should be low. It is also unlikely that SGLT2 inhibitors will induce hypoglycemia, since when plasma glucose levels are low the amount of glucose excreted will also be low.59 This prediction appears to be confirmed by clinical studies reported thus far, which show no apparent increases in hypoglycemic episodes with SGLT2 inhibitors.50,60 Even when SGLT2 is blocked completely, a degree of renal glucose recovery is maintained via the relatively unhindered SGLT1 transporter.
One aspect of SGLT2 inhibition that has been raised as a potential issue of safety concern is that of glycosuria, which could predispose patients to increased urinary tract infections (UTI). The extent to which increases in infection will occur has yet to be established. There have been some reports of infection in clinical studies.60,61 However, a study that reviewed risk factors for developing UTIs in women with diabetes observed that glucosuria was not a significant contributing factor.62
Interestingly, there is a rare group of individuals who do not express the SGLT2 transporter or in which its functionality has been partially or completely lost due to a genetic mutation for which both an autosomal recessive and dominant pattern of inheritance has been reported. These people do not appear to suffer any ill consequences, suggesting that blockade of the transporter per se in T2DM patients would offer no immediate risk. Patients expressing these mutations have decreased renal tubular reabsortion of glucose from the lumen in the absence of hyperglycemia, or any other signs of tubular dysfunction. It is not clear whether familial renal glucosuria (due to SGLT2 mutation) protects against T2DM; although SGLT2 deletion in animal models appears to improve glucose homeostasis and preserve pancreatic β-cell function.63 We did not find any recorded evidence of an increased disposition to urinary tract or vulvovaginal infections, although identification and study of these subjects is difficult due to the rarity of the disease. Clearly, clinical development programs will need to address the concern of a possible increased risk of UTI.
Increased glucose content in the urine following SGLT2 inhibition will likely serve to increase urinary flow as a consequence of the osmotic diuretic effect in the lumen of the nephron. This could result in modest, possibly beneficial, reductions in blood pressure, but raises additional safety concerns associated with possible loss of fluid and solutes. This may be of particular concern in elderly patients or those who do not have the capacity to maintain their fluid balance. However, it should be noted that the effect is considerably lower than that seen with frequently used loop diuretics and there is no apparent change in glomerular filtration rate that would be indicative of a direct effect on renal function. Simple instructions on maintaining a state of hydration with regular drinks may serve to overcome the concerns over both urinary infection and fluid imbalance.
Positioning the SGLT2 Inhibitors
The question arises as to where SGLT2 inhibitors might fit in the current cascade of treatments for the management of T2DM. While treatment of T2DM follows prescribed guidelines, there are many approaches and permutations to their application in clinical practice. Initial treatment strategies involve lifestyle interventions to promote weight loss and improve glycemic control. Although the SGLT2 inhibitors’ mechanism of action would make them suitable for initial monotherapy in patients with early stage T2DM, it is unlikely they would be considered at this stage. Most treatments are currently initiated with metformin, which is relatively inexpensive, has a good historical safety profile, and is efficacious. Clearly, when they are first launched, the SGLT2 inhibitors will not be able to compete with metformin, purely on the issue of cost. If considered as second line treatment, SGLT2 inhibitors may offer a synergistic effect. However, as explained earlier, the advantage of this may be minimal in patients achieving a degree of glycemic control.
By promoting an ‘escape’ mechanism for glucose, SGLT2 inhibitors introduce a new mode to the control of T2DM. With the exception of α-glucosidase inhibitors, which block glucose uptake from the gut, all currently available antidiabetic therapies directly or indirectly modulate insulin to manipulate endogenous glucose utilization. Despite the modest effect on HbA1c predicted for SGLT2 inhibitors, the introduction of a novel means of reducing hyperglycemia increases the treatment options available to physicians for a disease that frequently requires the use of multiple agents to achieve control targets.57 The expected favorable safety profile and insulin-independent mechanism of action appear to support the use of SGLT2 inhibitors in combination with other antidiabetic drugs. Insulin-dependent therapies become less effective with the development of insulin resistance and/or deterioration of β-cell function; particularly in patients with low insulin resistance (high glucose) or poorly controlled disease. The insulin-independent action of SGLT2 inhibitors suggests potential for a synergistic effect in such scenarios. The insulin-independent action of SGLT2 inhibitors also means that they may be of use in type 1 diabetes, perhaps as a means of moderating post-prandial glucose excursions.
By increasing excretion of glucose, SGLT2 inhibitors offer an opportunity to increase calorie loss in T2DM patients, most of whom are overweight. The continual loss of 80–90 g of glucose per day (300–400 calories) is a significant loss of calories that should work synergistically with weight reduction programs.64 Short-term studies in both animals and man appear to confirm the predicted weight reducing property. This contrasts with several drug therapies, including sulfonylureas, insulin, and thiazolidinediones, which are generally associated with weight gain.65,66 But there are currently no data to confirm whether the rate of calorie loss continues with chronic therapy.
In a clinical setting it would be easy to overcome any ‘benefits’ that patients may derive from SGLT2 inhibition if they perceived the drug as a means of ‘escaping’ strict adherence to lifestyle modifications. In addition, it has yet to be established whether patients ‘acclimatized’ to higher levels of plasma glucose would experience a marked change in appetite with substantial loss of glucose/calories.67 One final issue is that for these drugs to work, they need to be delivered to the epithelial luminal surface of the nephron, requiring them to be cleared by the glomerulus. Consequently, their efficacy may be affected in those instances when the functioning of the kidney is impaired, for example in diabetic nephropathy. However, this has yet to be confirmed clinically or in any animal model.
Conclusion
Most patients with T2DM eventually succumb to the progressive nature of the disease and will, at some point, require multiple therapies to attain treatment targets. Only half the patients with T2DM achieve the HbA1c target of less than 7%, despite the many different treatment options currently available. With SGLT2 inhibitors introducing an alternative means of managing glucose, we gain a further treatment option that may increase our ability to control T2DM. Experience will lead to better understanding of which patients are likely to respond best, and under what circumstances.
References
American Diabetes Association website. Available at: http://www.diabetes.org/diabetes-basics/diabetes-statistics/?utm_source=www&utm_medium=DropDownDB&utm_content=Statistics&utm_campaign=CON. Accessed 14 June 2011.
Smyth S, Heron A. Diabetes and obesity: the twin epidemics. Nat Med. 2006;12:75–80.
Manuel DG, Schultz SE. Health-related quality of life and health-adjusted life expectancy of people with diabetes in Ontario, Canada, 1996–1997. Diabetes Care. 2004;27:407–414.
American Diabetes Association. Implications of the United Kingdom Prospective Diabetes Study. Diabetes Care. 2003;26(Suppl.):S 28–32.
UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet. 1998;352:854–865.
The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329:977–986.
Nathan DM, Buse JB, Davidson MB, et al. Management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy. A consensus statement from the American Diabetes Association and the European Association for the Study of Diabetes. Diabetologia. 2006;49:1711–1721.
AACE Diabetes Mellitus Clinical Practice Guidelines Task Force. American Association of Clinical Endocrinologists Medical Guidelines for Clinical Practice for the Management of Diabetes Mellitus. Endocr Pract. 2007;13(Suppl. 1):4–68.
American Diabetes Association. Standards of Medical Care in Diabetes 2007. Diabetes Care. 2007;30:S4–S41.
Kanai Y, Lee W-S, You G Y, Brown D, Hediger MA. The human kidney low affinity Na+ / glucose cotransporter SGLT2. J Clin Invest. 1994;93:397–404.
Bakris GL, Fonseca VA, Sharma K, Wright EM. Renal sodium-glucose transport: role in diabetes mellitus and potential clinical implications. Kidney Int. 2009;75:1272–1277.
Wright EM. Renal Na+-glucose cotransporters. Am J Physiol Renal Physiol. 2001;280:F10–18.
Zelikovic I. Aminoaciduria and glycosuria. In: Avner ED, Harmon WE and Niaudet P, eds. Pediatric Nephrology. 5th edition. Philadelphia: Lippincott Williams & Wilkins: 2004;701–728.
Moe OW, Wright SH and Palacín M. Renal handling of organic solutes. In: Brenner BM, ed. Brenner and Rector’s The Kidney. 8th edition. Philadelphia: Saunders Elsevier: 2008;214–247.
Miller D and Bihler I. The restrictions on possible mechanisms of intestinal transport of sugars. In: Kleinzeller A and Kotyk A, eds. Membrane Transport and Metabolism. Proceedings of a Symposium held in Prague. Prague: Czech Academy of Sciences & Academic Press: 1960;439–49.
Rossetti L, Shulman GI, Zawalich W, DeFronzo RA. Effect of chronic hyperglycemia on in vivo insulin secretion in partially pancreatectomized rats. J Clin Invest. 1987;80:1037–1044.
Ehrenkranz RRL, Lewis NG, Kahn CR, Roth J. Phlorizin: a review. Diabetes Metab Res Rev. 2005;21:31–38.
Smith HW. The evolution of the kidney. In: Smith HW ed. Lectures on the Kidney. Kansas: University of Kansas Press: 1943;3.
Vick HD, Deidrich DF, Baumann K. Re-evaluation of renal tubular glucose transport inhibition by phlorizin analogs. Am J Physiol. 1973;224:552–557.
Silverman M. Glucose transport in the kidney. Biochim Biophys Acta. 1976;457:303–351.
Bishop JH, Elegbe R, Green R, Thomas S. Effects of phlorizin on glucose, water and sodium handling by the rat kidney. J Physiol. 1978;275:467–480.
Hediger MA, Coady MJ, Ikeda TS, Wright EM. Expression cloning and cDNA sequencing of the Na+/glucose co-transporter. Nature. 1987;330:379–381.
Wright EM, Hirayama BA and Loo DF. Active sugar transport in health and disease. J Intern Med. 2007;261:32–43.
Wright EM and Turk E. The sodium/glucose cotransport family SLC5. Eur J Physiol. 2004;447:510–518.
Tsujihara K, Hongu M, Saito K, et al. Na+-glucose cotransporter (SGLT) inhibitors as antidiabetic agents. 4. Synthesis and pharmacological properties of 4′-dihydroxyphlorizin derivative substituted on the B ring. J Med Chem. 1999;42:5311–5324.
Oku A, Ueta K, Nawano M, et al. Antidiabetic effect of T-1095, an inhibitor of Na+-glucose cotransporter, in neonatally streptozotocin-treated rats. Eur J Pharmacol. 2000;391:183–192.
Washburn WN. Evolution of sodium glucose cotransporter 2 inhibitors as anti-diabetic agents. Expert Opin Ther Patents. 2009;19:1485–1499.
Nawano M, Oku A, Ueta K, et al. Hyperglycemia contributes insulin resistance in hepatic and adipose tissue but not skeletal muscle of ZDF rats. Am J Physiol Endocrinol Metab. 2000;278:E535–543.
Fujimoto Y, Torres TP, Donahue EP, Shiota M. Glucose toxicity is responsible for the development of impaired regulation of endogenous glucose production and hepatic glucokinase in Zucker diabetic fatty rats. Diabetes 2006;55:2479–2490.
Nunoi K, Yasuda K, Adachi T, et al. Beneficial effect of T-1095, a selective inhibitor of renal Na+-glucose cotransporters, on metabolic index and insulin secretion in spontaneously diabetic GK rats. Clin Exp Pharmacol Physiol. 2002;29:386–390.
Arakawa K, Ishihara T, Oku A, et al. Improved diabetic syndrome in C57BL/KsJdb/ db mice by oral administration of the Na+-glucose cotransporter inhibitor T-1095. Br J Pharmacol. 2001;132:578–586.
Ueta K, Ishihara T, Matsumoto Y, et al. Long-term treatment with the Na+-glucose cotransporter inhibitor T-1095 causes sustained improvement in hyperglycemia and prevents diabetic neuropathy in Goto-Kakizaki rats. Life Sci. 2005;76:2655–2668.
Washburn WN. Development of the renal glucose reabsorption inhibitors: a new mechanism for the pharmacotherapy of diabetes mellitus type 2. J Med Chem. 2009;52:1785–1794.
Katsuno K, Fujimori Y, Takemura Y, et al. Sergliflozin, a novel selective inhibitor of low-affinity sodium glucose cotransporter (SGLT2), validates the critical role of SGLT2 in renal glucose reabsorption and modulates plasma glucose level. J Pharmacol Exp Ther. 2007;320:323–330.
Mascitti V, Maurer TS, Robinson RP, et al. Discovery of a clinical candidate from the structurally unique dioxa-bicyclo[3.2.1]octane class of sodium-dependent glucose cotransporter 2 Inhibitors. J Med Chem. 2011;54:2952–2960.
Kasichayanula S, Chang M, Hasegawa M, et al. Pharmacokinetics and pharmacodynamics of dapagliflozin, a novel selective inhibitor of sodium-glucose co-transporter type 2, in Japanese subjects without and with type 2 diabetes mellitus. Diab Ob Metab. 2011;13:357–365.
Kasichayanula S, Liu X, Shyu WC, et al. Lack of pharmacokinetic interaction between dapagliflozin, a novel sodium-glucose transporter 2 inhibitor, and metformin, pioglitazone, glimepiride or sitagliptin in healthy subjects. Diab Ob Metab. 2011;13:47–54.
Wilding JPH, Woo V, Pahor A, Sugg J, Langkilde A, Parikh S. Effect of dapagliflozin, a novel insulin-independent treatment, over 48 weeks in patients with type 2 diabetes poorly controlled with insulin. Diabetologia. 2010;53(Suppl. 1):S348.
Ng CM, Zhang L, List J, Pfister M. Mechanismbased disease model to describe the plasma/urine glucose-time profiles in placebo-or dapagliflozin-treated normal and type 2 diabetes mellitus (T2DM) subjects. Clin Pharm Ther. 2010;87(Suppl. 1):S17–S18.
Komoroski B, Vachharajani N, Boulton D, et al. Dapagliflozin, a novel SGLT2 inhibitor, induces dose-dependent glucosuria in healthy subjects. Clin Pharm Ther. 2009;85:520–526.
Komoroski B, Vachharajani N, Feng Y, Li L, Kornhauser D, Pfister M. Dapagliflozin, a novel, selective SGLT2 inhibitor, improved glycemic control over 2 weeks in patients with type 2 diabetes mellitus. Clin Pharm Ther. 2009;85:513–519.
Feng Y, Kasichayanula S, Leslie B, List J, Zhang L, Pfister M. Dapagliflozin mechanism of action in type 2 diabetes mellitus: characterization of biomarker response. Clin Pharm Ther. 2009;85(Suppl. 1):S15.
Skee D, Shalayda K, Vandebosch A, Usiskin K, Tickel-Logan R, Devineni D. The effects of multiple doses of canagliflozin on the pharmacokinetics and safety of single doses of an oral contraceptive containing ethinyl estradiol and levonorgestrel. Clin Pharm Ther. 2010;87(Suppl. 1):S35.
Rosenstock J, Polidori D, Zhao Y, et al. Canagliflozin, an inhibitor of sodium glucose cotransporter 2, improves glycaemic control, lowers body weight, and improves beta cell function in subjects with type 2 diabetes on background metformin. Diabetologia. 2010;53(Suppl. 1):S349.
Sarich T, Devineni D, Ghosh A, et al. Canagliflozin, a novel inhibitor of sodium glucose co-transporter 2, increases 24-hour urinary glucose excretion and reduces body weight in obese subjects over 2 weeks of treatment. Diabetologia. 2010;53(Suppl. 1):S349–S350.
Rothenberg PL, Devineni D, Ghosh A, et al. Canagliflozin, a novel inhibitor of sodium glucose co-transporter 2, improved glucose control in subjects with type 2 diabetes: Results of a phase 1b study. Diabetologia. 2010;53(Suppl. 1):S350–S351.
Polidori D, Sha S, Sarich T, Devineni D, Rothenberg PL. Canagliflozin lowers the renal threshold for glucose excretion in lean, obese and type 2 diabetic subjects. Diabetologia. 2010;53(Suppl. 1):S350.
Zhang W, Townsend R, Abeyratne A, Smulders R. Lack of pharmacokinetic interactions between ASP1941, a selective sodium glucose co-transporter 2 (SGLT2) inhibitor, and sitagliptin in healthy subjects. Clin Pharm Ther. 2011;89(Suppl. 1):S81–S82.
Zhang W, Townsend R, Abeyratne A, Smulders R. Lack of pharmacokinetic interactions between ASP1941, a selective sodium glucose co-transporter 2 (SGLT2) inhibitor, and pioglitazone in healthy subjects. Clin Pharm Ther. 2011;89(Suppl. 1):S82.
Hussey EK, Clark RV, Amin DM, et al. Single-dose pharmacokinetics and pharmacodynamics of sergliflozin etabonate, a novel inhibitor of glucose reabsorption, in healthy volunteers and patients with type 2 diabetes mellitus. J Clin Pharmacol. 2010;50:623–635.
Hussey EK, Dobbins RL, Stoltz RR, et al. Multiple-dose pharmacokinetics and pharmacodynamics of sergliflozin etabonate, a novel inhibitor of glucose reabsorption, in healthy overweight and obese subjects: a randomized double-blind study. J Clin Pharmacol. 2010;50:636–646.
Hussey EK, Kapur A, O’Connor-Semmes RL, Tao W, Poo JL, Dobbins RL. Safety, pharmacokinetics and pharmacodynamics of Remogliflozin etabonate (SGLT2 Inhibitor) and metformin when co-administered in type 2 diabetes mellitus (T2DM) patients. Diabetes. 2009;58(Suppl. 1A):582P.
Kapur A, O’Connor-Semmes RL, Hussey EK, et al. First human dose escalation study with remogliflozin etabonate (RE) in healthy subjects and in subjects with type 2 diabetes mellitus (T2DM). Diabetes. 2009;58(Suppl. 1A):509P.
Dobbins RL, Kapur A, Kapitza C, O’Connor-Semmes RL, Tao W, Hussey EK. Remogliflozin etabonate, a selective inhibitor of the sodiumglucose transporter 2 (SGLT2) reduces serum glucose in type 2 diabetes mellitus (T2DM) patients. Diabetes. 2009;58(Suppl. 1A):573P.
Chao EC, Henry RR. SGLT2 inhibition — a novel strategy for diabetes treatment. Nat Rev Drug Disc. 2010;9:551–559.
Landgraf R. The relationship of postprandial glucose to HbA1c. Diabetes Metab Res Rev. 2004;20:S9–S12.
Shomali M. Add-on therapies to metformin for type 2 diabetes. Expert Opin Pharmacother. 2011;12:47–62.
List JF, Woo V, Morales E, Tang W, Fiedorek FT. Sodium-glucose cotransport inhibition with dapagliflozin in type 2 diabetes. Diabetes Care. 2009;32:650–657.
Meng W, Elsworth BA, Nirschl AA, et al. Discovery of dapagliflozin: a potent, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes. J Med Chem. 2008;51:1145–1149.
Wilding JP, Norwood P, T’joen C, Bastien A, List JF, Fiedorek FT. A study of dapagliflozin in patients with type 2 diabetes receiving high doses of insulin plus insulin sensitizers: applicability of a novel insulin-independent treatment. Diabetes Care. 2009;32:1656–1662.
Bailey CJ, Gross JL, Pieters A, Bastien A, List JF. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with metformin: a randomised, double-blind, placebo-controlled trial. Lancet. 2010;375:2223–2233.
Geerlings SE, Stolk RP, Camps, Netten PM, Collet TJ, Hoepelman AIM, on behalf of the Diabetes Women Asymptomatic Bacteriuria Utrecht Study Group. Risk factors for symptomatic urinary tract infection in women with diabetes. Diabetes Care. 2000;23:1737–1741.
Jurczak MJ, Lee H-Y, Birkenfeld AL, et al. SGLT2 deletion improves glucose homeostasis and preserves pancreatic (beta)-cell function. Diabetes. 2011;60:890–898.
Hermansen K, Mortensen LS. Bodyweight changes associated with antihyperglycaemic agents in type 2 diabetes mellitus. Drug Saf. 2007;30:1127–1142.
Goudswaard AN, Furlong NJ, Rutten GE, Valk GD, Stolk RP, Rutten GEHM. Insulin monotherapy versus combinations of insulin with oral hypoglycaemic agents in patients with type 2 diabetes mellitus. Cochrane Database Syst Rev. 2004:CD003418.
Nesto RW, Bell D, Bonow RO, et al. Thiazolidinedione use, fluid retention, and congestive heart failure: a consensus statement from the American Heart Association and American Diabetes Association. Diabetes Care. 2004;27:256–263.
Ryan M, Livingstone MBE, Ritz P. Insulin treatment and weight gain in type 2 diabetes: is our knowledge complete? Current Nutrition and Food Science. 2006;2:51–58.
Acknowledgments
The authors declare that they have no competing interests. The authors are not currently engaged, retained, or acting in any capacity for any drug company working on SGLT2 inhibitors. Simon Dubrey provided the clinical perspective of the review, which was conceived and the first draft was prepared by Tim Hardman. Tim Hardman is the guarantor for this article, and takes responsibility for the integrity of the work as a whole.
Open Access. This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Additional information
To view enhanced content go to www.diabetestherapy-open.com
This article is published with open access at Springerlink.com
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Hardman, T.C., Dubrey, S.W. Development and potential role of type-2 sodium-glucose transporter inhibitors for management of type 2 diabetes. Diabetes Ther 2, 133–145 (2011). https://doi.org/10.1007/s13300-011-0004-1
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13300-011-0004-1