


Abstract
Unbound partition coefficient (Kpuu) is important to an understanding of the asymmetric free drug distribution of a compound between cells and medium in vitro, as well as between tissue and plasma in vivo, especially for transporter-mediated processes. Kpuu was determined for a set of compounds from the SLC13A family that are inhibitors and substrates of transporters in hepatocytes and transporter-transfected cell lines. Enantioselectivity was observed, with (R)-enantiomers achieving much higher Kpuu (>4) than the (S)-enantiomers (<1) in human hepatocytes and SLC13A5-transfected human embryonic 293 cells. The intracellular free drug concentration correlated directly with in vitro pharmacological activity rather than the nominal concentration in the assay because of the high Kpuu mediated by SLC13A5 transporter uptake. Delivery of the diacid PF-06649298 directly or via hydrolysis of the ethyl ester prodrug PF-06757303 resulted in quite different Kpuu values in human hepatocytes (Kpuu of 3 for diacid versus 59 for prodrug), which was successfully modeled on the basis of passive diffusion, active uptake, and conversion rate from ester to diacid using a compartmental model. Kpuu values changed with drug concentrations; lower values were observed at higher concentrations possibly owing to a saturation of transporters. Michaelis-Menten constant (Km) of SLC13A5 was estimated to be 24 μM for PF-06649298 in human hepatocytes. In vitro Kpuu obtained from rat suspension hepatocytes supplemented with 4% fatty acid free bovine serum albumin showed good correlation with in vivo Kpuu of liver-to-plasma, illustrating the potential of this approach to predict in vivo Kpuu from in vitro systems.
Introduction
Intracellular free drug concentration plays a critical role in developing target exposure and pharmacological activity relationships for disease targets located in the intracellular domain. For compounds that reach distribution equilibrium across the cell membrane at steady state, free drug concentration in the medium might be considered the same as the intracellular free drug concentration. This is usually the case for most compounds in which the nominal concentrations can be used to correlate to pharmacological activities [e.g., concentration of an inhibitor at which the response is reduced by half (IC50)] of a compound in in vitro assays or after correction of nonspecific binding to the proteins/lipids in the assay media. However, when transporters are involved in the distribution process, the intracellular free drug concentration may be different from that in the medium owing to active processes. In addition, effects of membrane potential, lysosomal trapping, pH gradient, and mitochondria accumulation can all impact the intracellular free drug concentration relative to the medium free concentration (Fig. 1). Therefore, it is important to understand the intracellular free drug concentration and unbound distribution coefficient (Kpuu, defined as the ratio between unbound intracellular free drug concentration and unbound medium concentration and steady state is not assumed in this paper) to accurately determine the true potency of a compound, e.g., intrinsic IC50, rather than apparent IC50. Prediction of human in vivo liver-to-plasma Kpuu is of great importance in order to estimate dose, safety margin, and drug-drug interaction potentials for compounds with transporter-mediated uptake into the liver. Development of in vitro tools to predict in vivo liver-to-plasma Kpuu will be very useful for human translation in drug discovery and development, since there are no simple tools available to measure human liver concentration.

Drug disposition and trafficking in cells.
Several in vitro methods have been developed to measure the intracellular free drug concentration and Kpuu in hepatocytes or transfected cell systems expressing OATP transporters (Fig. 2). The binding method measures fraction unbound of cells (fucell), medium and total cell concentration at the steady state, from which intracellular free drug concentration and Kpuu are determined (Mateus et al., 2013). The temperature method measures the medium and total cell concentration at both 37°C and 4°C at steady state (Shitara et al., 2013). The total concentration ratio of cell to medium between 37°C and 4°C is considered to be Kpuu. This method does not consider the impact of membrane potential or metabolism. The kinetic method measures the uptake rate at multiple concentrations in the presence of the cytochrome P450 inhibitor 1-aminobenzotriazole. Kinetic parameters [maximum rate (Vmax), Michaelis-Menten constant (Km), and clearance via passive diffusion (Pdiff)] are obtained by simultaneously fitting all the data (Yabe et al., 2011). Kpuu is calculated on the basis of the kinetic parameters with the extended sequential clearance equation (Yabe et al., 2011). This approach does not consider the impact of efflux transport or metabolism on Kpuu. The permeability method measures the permeability of both neutral and ionized species and determines the Kpuu across a cell membrane on the basis of membrane potential and passive permeability (Ghosh et al., 2014). This method has not incorporated the effects of transporters or metabolism at this point. Other methods reported to determine drug concentration in the subcellular compartments (Pfeifer et al., 2013) are relevant to therapeutic targets localized in the specific subcellular organelles. Validation of the different Kpuu methods with in vitro or in vivo data are minimal in literature. We will discuss our approach for measuring Kpuu and intracellular free drug concentration, as well as validation against in vitro potency, using cell lines expressing different members of the SLC13A family, hepatocytes, and in vivo rat liver-to-plasma Kpuu data.
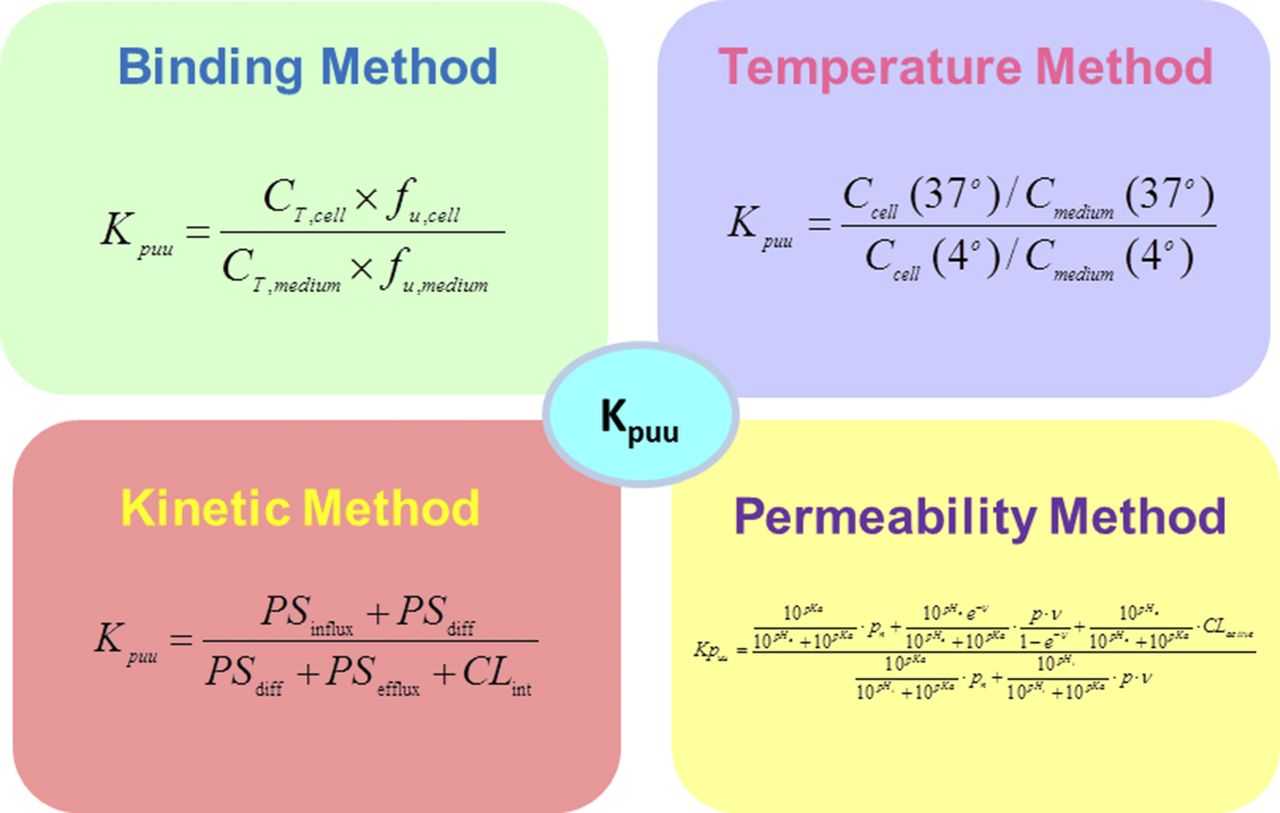
Methods for Kpuu determination.
Citrate is a key regulatory metabolic intermediate and is critical to the integration of the glycolysis and lipid synthesis pathways. Inhibition of hepatic extracellular citrate uptake through SLC13A5 has been suggested as a potential therapeutic approach to treat metabolic diseases (Huard et al., 2015, 2016; Li et al., 2016). The SLC13A family contains five family members: SLC13A1 and SLC13A4 mainly transport sulfate, and SLC13A2, SLC13A3, and SLC13A5 transport di- and tri-carboxylates such as citrate (Lee et al., 2005; Bergeron et al., 2013). SLC13A5 is highly expressed in human liver, and SLC13A2 and SLC13A3 are expressed more broadly, including in the intestine and kidney (Pajor, 2014). Therefore, selective SLC13A5 inhibitors have the potential to impact citrate flux in the liver only, whereas SLC13A2/3 inhibitors can impact citrate flux in the intestine and kidney as well. Development of specific inhibitors of SLC13A5 is proposed to be beneficial in treating metabolic disorders by reducing citrate flux into the liver, presumably exerting metabolic benefits through modification of intracellular metabolites, at the same time limiting direct effects in other tissues.
Materials and Methods
Materials
Test compounds were obtained from Pfizer Global Material Management (Groton, CT) or purchased from Sigma-Aldrich (St. Louis, MO). The syntheses of diacids PF-06649298 and PF-06649297 and monoacid PF-06757303 were previously reported (Huard et al., 2015). The syntheses of PF-06794266, PF-06793742, and PF-06761281 were also reported (Huard et al., 2016). PF-06761281 was commercially available via Sigma-Aldrich (cat. no. PZ0318). The syntheses of PF-06746350 and PF-06741415 were similar to the methods described previously (Huard et al., 2016). Other reagents were obtained from Sigma-Aldrich unless specified. The 96-well equilibrium dialysis (HTD96) device and cellulose membranes with molecular weight cut-off (MWCO) of 12–14K were purchased from HTDialysis, LLC (Gales Ferry, CT). Ninety-six-well plates, 1.2 ml deep, were from Axygen Scientific Inc. (Union City, CA), and pipette tips were obtained from Apricot Designs (Monrovia, CA).
Measurement of Fraction Unbound
Cells [hepatocytes, human embryonic kidney (HEK)293 transfected with human SLC13A transporters] at a cell density of 50–60 million cells/ml, rat and human liver tissues [1:5 dilution with phosphate buffered saline (PBS)] were homogenized at room temperature in PBS using an Omni TH tissue homogenizer (Omni International, Kennesaw, GA) with a 7-mm × 110-mm tip at high speed for 30-second pulses. Assay medium containing proteins [e.g., bovine serum albumin (BSA)] was used directly for binding measurement. The dialysis membranes were prepared prior to experimental setup. The cellulose membranes (MWCO 12–14K) were immersed in deionized water for 15 minutes, followed by 15 minutes in 30% ethanol/deionized water, then at least 15 minutes or overnight in PBS. The equilibrium dialysis device was assembled according to manufacturer’s instructions (http://htdialysis.com/page/1puq4/Operating_Instructions.html). Dimethylsulfoxide (DMSO) stock solutions of test compounds were prepared at 200 μM, added to matrices in 1:100 ratio, and mixed thoroughly with an eight-channel pipettor (Eppendorf/VWR, Radnor, PA). The final compound concentration was 2 μM containing 1% DMSO. A 150-μl aliquot of matrix (cell and tissue homogenate, assay medium, or plasma) spiked with 2 μM compound was added to one side of the membrane (donor) and 150 μl of PBS was added to the other side (receiver). The equilibrium dialysis device was covered with Breathe-Easy gas-permeable membranes (Sigma-Aldrich). Compounds were assessed in quadruplicate. Equilibrium dialysis devices were placed on an orbital shaker (VWR) at 200 rpm and incubated for 6 hours in a humidified [75% relative humidity (RH)] incubator at 37°C with 5% CO2/95% O2. At the end of the incubation, 15 μl of matrix samples from the donor wells were added to a 96-well plate containing 45 μl of PBS. Aliquots of 45 μl of dialyzed PBS from the receiver wells were added to 15 μl of blank matrix. Before and after incubation, 15 μl of matrix spiked with 2 μM compounds was added to a 96-well plate containing 45 μl of PBS. These samples were used for the recovery calculation and stability evaluation. All the samples were quenched with 200 μl of cold acetonitrile containing internal standard (IS; a cocktail of 0.5 ng/ml tolbutamide and 5 ng/ml terfenadine). The plates were sealed and mixed with a vortex mixer (VWR) for 3 minutes, then centrifuged at 3000 rpm Allegra 6R (Beckman Coulter Life Sciences, Indianapolis, IN) at room temperature for 5 minutes. The supernatant was transferred to a new 96-well plate, dried down, reconstituted, and subsequently analyzed using liquid chromatography–tandem mass spectrometry (LC-MS/MS). Sertraline was used as a quality control sample on every plate. Cell diameter was measured using Vi-CELL XR (Beckman Coulter Life Sciences) to calculate cell volume assuming spherical shape and dilution factor.
In Vitro Kpuu Measurement
Transfected Cells (SLC13A5- and SLC13A3-HEK293).
Kpuu within this paper is defined as the ratio between unbound intracellular free drug concentration and unbound medium concentration (steady state is not assumed). Protocols to generate the stable cell line overexpressing sodium citrate transporters and culture conditions were discussed previously (Huard et al., 2015). Test compounds (1 μM unless otherwise specified) were incubated with the plated cells (1–4 hours unless otherwise specified) at 37°C in a humidified (75% RH) incubator with 5% CO2/95% O2. At the end of the incubation, 100 μl of the medium were removed for concentration determination, and cold acetonitrile was added. The rest of the medium was gently removed from the cells by pipette aspiration. The cells were washed three times with cold PBS of the same starting volume. After wash, the cells were lysed by adding 80 μl of M-PER buffer (Thermo Fisher Scientific, Rockford, IL). An aliquot of the lysed cells was transferred to a new plate, matrix matched, and cold acetonitrile containing IS was added. The solutions were centrifuged at 3000 rpm for 10 minutes at room temperature. The supernatants were transferred and analyzed by LC-MS/MS against standard curves that were matrix matched for both media and cells.
Human and Rat Hepatocytes.
Cryopreserved human hepatocytes (Lot DCM) consisting of 10 donors, both males and females, were custom-pooled and prepared by BioreclamationIVT (Baltimore, MD). Wistar Han rat hepatocytes (Lot VSU) consisting 35 male donors were also purchased from BioreclamationIVT. Upon thawing, the hepatocytes were resuspended in Williams’ E medium (custom formula supplemented with 50 mM Hepes and 26 mM sodium bicarbonate; Gibco/Thermo Fisher Scientific, Waltham, MA). The cells were counted and viability was determined using the Trypan blue exclusion method. Test compounds were dissolved in DMSO at 1 mM, and 1 μl was added to 0.5 million cells/ml suspended hepatocytes in 1000 μl (final concentration 1 μM containing 0.1% DMSO). The suspension was incubated in a humidified (75% RH) incubator at 37°C with 5% CO2/95% O2. For rat hepatocytes, InVitroGRO HI medium (BioreclamationIVT) containing 0.2, 2, and 4% fatty acid–free BSA (Sigma-Aldrich) was also used in the study for in vitro–in vivo extrapolation (IVIVE) development. At appropriate time points, hepatocyte suspension was centrifuged at 500 rpm for 3 minutes and supernatant was collected and removed from the cells to determine the medium concentration. Cells were washed 3 times with cold PBS (1 ml each) and lysed by adding 80 μl of M-PER buffer (Thermo Fisher Scientific). An aliquot of the lysed cells, as well as the medium supernatant, were transferred to a new plate, matrix matched, and cold acetonitrile containing IS was added. The solutions were centrifuged at 3000 rpm for 10 minutes at room temperature. The supernatants were transferred and analyzed by LC-MS/MS against standard curves that were matrix matched for both media and cells.
Stability in Human Hepatocytes and Media
The experimental details and data processing have been reported previously (Di et al., 2012). One micromolar test compound was added to cryopreserved human or rat hepatocytes at 0.5 million cells/ml in assay medium and incubated in a humidified (75% RH) incubator at 37°C with 5% CO2/95% O2. At appropriate time points, an aliquot of the suspension was collected and quenched with cold acetonitrile containing IS. The solutions were centrifuged at 3000 rpm for 10 minutes at room temperature. The supernatants were transferred and analyzed by LC-MS/MS.
In Vivo Rat Liver-to-Plasma Kpuu Determination
The rat intravenous infusion study was conducted at BioDuro contract laboratories (Shanghai, China). Rats (Wistar Han, male, fed, n = 3) were intravenously infused via a jugular vein cannula with test compounds for 6 hours at an infusion rate of 5–9 μl/min using a Harvard 2000 programmable pump (Harvard Apparatus, Holliston, MA). The doses were 1.6 (PF-06649298), 0.21 (PF-06761281), and 5.5 (PF-06741415) mg/kg, which were estimated on the basis of intravenous bolus dose and modeled using a two-compartment model (Phoenix WinNonlin 6.3, Princeton, NJ). Compounds were formulated in 2% PEG400 (polyethylene glycol 400) and 98% of 15% sulfobutylether-β-cyclodextrin in water. Blood samples were collected from the carotid artery catheter at 0, 1, 2, 4, and 6 hours to determine time required to achieve steady state. Upon study termination, livers were collected and concentrations were determined. Free concentrations were determined by multiplying the steady state total plasma and liver concentration by fraction unbound of plasma or liver. In vivo free liver-to-plasma ratio, Kpuu, was calculated by dividing free liver concentration by free plasma concentration at steady state.
LC-MS/MS Quantification
A typical LC-MS/MS method is described here and equivalent methods were also used depending on sample characteristics. The LC mobile phases were either: (A) HPLC grade water containing 0.1% formic acid, (B) acetonitrile containing 0.1% formic acid, or (A) 95% 2 mM ammonium acetate in water and 5% 50:50 methanol/acetonitrile, (B) 90% 50:50 methanol/acetonitrile and 10% 2 mM ammonium acetate in water. A solvent gradient from 5% (B) to 95% (B) over 1.1 minutes at the flow rate of 0.5 ml/min was used to elute the compounds from the column (Waters UPLC BEH C18, 50 × 2.1 mm, 1.7 μm; Waters, Milford, MA). The total run-time was 2.5 minutes/injection. A 10-μl aliquot of sample was injected for analysis using a CTC PAL autosampler (LEAP Technologies, Carrboro, NC). The analysis was conducted with Agilent 1290 binary pump (Santa Clara, CA) connected to an AB Sciex (Foster City, CA) API 6500 triple quadrupole mass spectrometer equipped with a TurboIonSpray source using multiple-reaction monitoring mode. Analyst 1.6.1 software (Applied Biosystems, Foster City, CA) was applied to data collection processing and analysis.
Calculation Methods
Fraction unbound was calculated using eq. (1). The concentrations or area ratios of test compound to IS in receiver and donor wells were corrected to account for sampling volume differences and determined using LC-MS/MS. For diluted matrices, eqs. (2) and (3) were used for calculation, where D is the dilution factor (Kalvass and Maurer, 2002). Dilution factor for cells is calculated on the basis of medium to cell volume (calculated from cell diameter assuming spherical geometry) and cell density. Recovery and stability were calculated using eq. (4) and eq. (5), respectively. In vitro Kpuu, unbound cell concentration, and unbound medium concentration were calculated by eqs. (6) through (8). C represents concentration.
 (1)
(1) (2)
(2) (3)
(3) (4)
(4) (5)
(5) (6)
(6) (7)
(7) (8)
(8)Modeling Method for in Vitro Kpuu for Ethyl Ester Prodrug and Active Diacid
Prodrug PF-06757303 enters into hepatocytes by transcellular passive diffusion; there it can be converted to active diacid PF-06649298 in hepatocytes by esterases (Fig. 3). The Kpuu time course of PF-06649298 and PF-06757303 in media and human suspension hepatocytes was modeled using a compartmental model in NONMEM7.2 (ICON Plc, Dublin, Ireland). For the Kpuu time course of PF-06649298 (Fig. 4A), two active processes were modeled, i.e., the influx of the compound from medium to hepatocyte and the efflux of the compound from hepatocyte to medium. Since PF-06649298 was stable in human hepatocytes, no metabolism was considered. The PF-06649298 medium and hepatocyte-concentrations time course were modeled using differential eqs. (9) and (10), where A is amount (nmol); P is influx or efflux rate constant (mm/min); SA is surface area of hepatocytes (mm2); fu is fraction unbound; V is volume (μl); “8” represents PF-06649298; “in” represents the flux into hepatocytes; “out” represents the flux out of hepatocytes; “m” represents medium, and “c” represents hepatocytes. The resulting Kpuu of PF-06649298 was the ratio of the influx and efflux rate constant. Model parameters are summarized in the Supplemental Table 1S.

Conversion of ethyl ester prodrug (PF-06757303) to active diacid (PF-06649298) in hepatocytes.

Determinants of in vitro Kpuu in the computational models. (A) When PF-06649298 (active diacid) is incubated with human hepatocytes, Kpuu is determined by the influx and efflux rate constants; (B) when PF-06757303 (prodrug) is dosed in human hepatocytes, Kpuu of PF-06649298 is determined by the formation rate from prodrug (PF-06757303) as well as the influx and efflux rate constants of PF-06649298 (active).
 (9)
(9) (10)
(10)For human hepatocyte suspension incubation with ester PF-06757303 (Fig. 4B), in addition to the influx and efflux processes, the conversion of ester PF-06757303 to diacid PF-06649298 in the hepatocytes was included. PF-06757303 was found to be highly permeable and no active cellular uptake was observed. Therefore, prodrug PF-06757303 can enter into hepatocytes only by passive diffusion. Time course of medium and hepatocyte concentrations of diacid PF-06649298 and prodrug PF-06757303 was simulated using differential eqs. (11) through (14), where “3” represents PF-06757303, and kdeg is the conversion rate from PF-06757303 to PF-06649298. All the other symbols were the same as above in eqs. (9) and (10).
 (11)
(11) (12)
(12) (13)
(13) (14)
(14)Results
Enantioselectivity of Kpuu for Sodium Citrate Transporter.
The Kpuu values of a set of sodium citrate transporter inhibitors in human hepatocytes, which express high level of SLC13A5, are shown in Table 1. Kpuu within this paper is defined as the ratio between unbound intracellular free drug concentration and unbound medium concentration (steady state is not assumed). The pharmacologically active R-enantiomers all achieved moderate-to-high Kpuu values (from 4.8 to 61) in human hepatocytes, whereas the pharmacologically inactive S-enantiomers all had low Kpuu (<1). The data demonstrated that SLC13A5 was highly enantioselective toward the R-enantiomer by active influx of its substrates into the hepatocytes. The diacidic sodium citrate transporter inhibitors from this series had minimal metabolic turnover in the human hepatocytes and very low transcellular passive permeability (Huard et al., 2016), leading to high Kpuu through active uptake processes. Enantioselective transport uptake and inhibition had been reported in literature (Honjo et al., 2011; Togami et al., 2013); however, the differences in uptake are particularly striking for the two enantiomers in this case.
Enantioselectivity of Kpuu in human hepatocytes
Kpuu was measured at hepatocyte cell density of 0.5 million cells/ml, 1 μM test compound concentration, and 4-hour incubation time.
Correlation of Kpuu to In Vitro Pharmacological Activity.
Human hepatocytes and HEK293 cells expressing SLC13A transporters were used to determine the Kpuu and in vitro IC50 values for inhibition of SLC13A transporters (Huard et al., 2015, 2016) for a pair of enantiomers, PF-06649298 (R) and PF-06649297 (S) (Table 2). PF-06649298 (R) is a selective inhibitor and substrate of human SLC13A5 [mainly in liver, in contrast to SLC13A2 (intestine) and SCL13A3 (kidney)] and therefore has high Kpuu in SLC13A5-transfected HEK293 cells and human hepatocytes (29 and 4.8), but low Kpuu in SCL13A2- and SCL13A3-transfected HEK293 cells (0.1 and 0.2, respectively), which are predominately expressed in the intestine and kidney. The S-enantiomer, PF-06649297, has low Kpuu in both SLC13A5 HEK293 cells and human hepatocytes (0.6 and 0.4), suggesting it is not a substrate for the liver SLC13A5 transporter. Both the (R) and the (S) enantiomers have low Kpuu (0.1) in the parental HEK293 cells, in which expression of SLC13A5 is undetectable. Negative membrane potential, low passive permeability, and pH gradient potentially contribute to the low Kpuu of the diacids in the absence of transporter-mediated uptake in cells (e.g., in HEK293 cells). The Kpuu values are consistent with the in vitro pharmacological activity of SLC13A5 inhibition (Table 2). Comparing IC50 values derived from nominal concentration for PF-06649298, the human hepatocyte IC50 is 39-fold higher than that in the SLC13A5 HEK293 cells (Table 3). However, after correction for intracellular free drug concentration using Kpuu at each test concentration (data not shown, Kpuu values varied with concentrations), the IC50 in the two cell-based systems are comparable (Table 3), suggesting SLC13A5 is inhibited at least in part from the intracellular domain. This exemplifies the importance of measuring intracellular free drug concentration to build a correlation with pharmacological activity for intracellular targets. The high Kpuu value of PF-06761281 in SLC13A3-transfected HEK293 cells (Table 2) supports the hypothesis that active uptake of this compound by SLC13A3 in intestine may play a role in oral absorption and at least partially explains the difference in fraction absorbed (fa) compared with PF-06649298 (Huard et al., 2016). Both PF-06649298 (R) and PF-06649297 (S) have a rat fa ∼0.2, presumably through passive paracellular diffusion (Huard et al., 2016). Conversely, PF-06761281 (R), a substrate for SLC13A3, has a rat fa of 1, whereas its enantiomer PF-06761282 (S) has a rat fa of 0.07, suggesting that transport of the diacids by SLC13A3 is also enantioselective (Huard et al., 2016).
Enantioselectivity of Kpuu and SLC13 inhibition in transfected cells and human hepatocytes for an enantiomer pair, PF-06649298 (R) and PF-06649297 (S), and PF-06761281 (R)
Effect of intracellular free drug concentration on SLC13A5 inhibition for PF-06649298
Comparison between in Vitro and in Vivo Kpuu.
In vivo liver-to-plasma unbound concentration ratios at the steady state (in vivo Kpuu) were determined using rat intravenous infusion. The comparison of in vitro Kpuu in rat hepatocyte suspensions with various amounts of BSA and in vivo liver-to-plasma Kpuu is shown in Table 4. Differences in sodium concentrations are small in the system that has various amounts of BSA, and the impact on the SCL13A5 uptake is expected to be minimal. In general, the in vitro hepatocyte Kpuu with 4% BSA is in good agreement with in vivo liver-to-plasma Kpuu. PF-06649298 shows high Kpuu in vitro and in vivo (14 versus 16), whereas PF-06761281 and PF-06741415 have low/moderate in vitro and in vivo Kpuu. The differences are within assay variability from both in vitro (liver and plasma binding, Kpuu from multiple rat hepatocyte donors) and in vivo (individual differences). The data suggest that the in vitro suspension hepatocyte system could potentially be used to predict in vivo Kpuu, which is important to estimation of liver concentrations in humans (difficult to measure directly), development of pharmacokinetic/pharmacodynamic relationships, and prediction of drug-drug interaction potentials. In vitro Kpuu is sensitive to the amount of BSA in the assay medium, with 4% (plasma physiologic amount) giving the best correlation to in vivo data.
In vitro and in vivo correlation of Kpuu in WH rat and impact of BSA on Kpuu
Kpuu Measurement for Prodrug and Active Diacid.
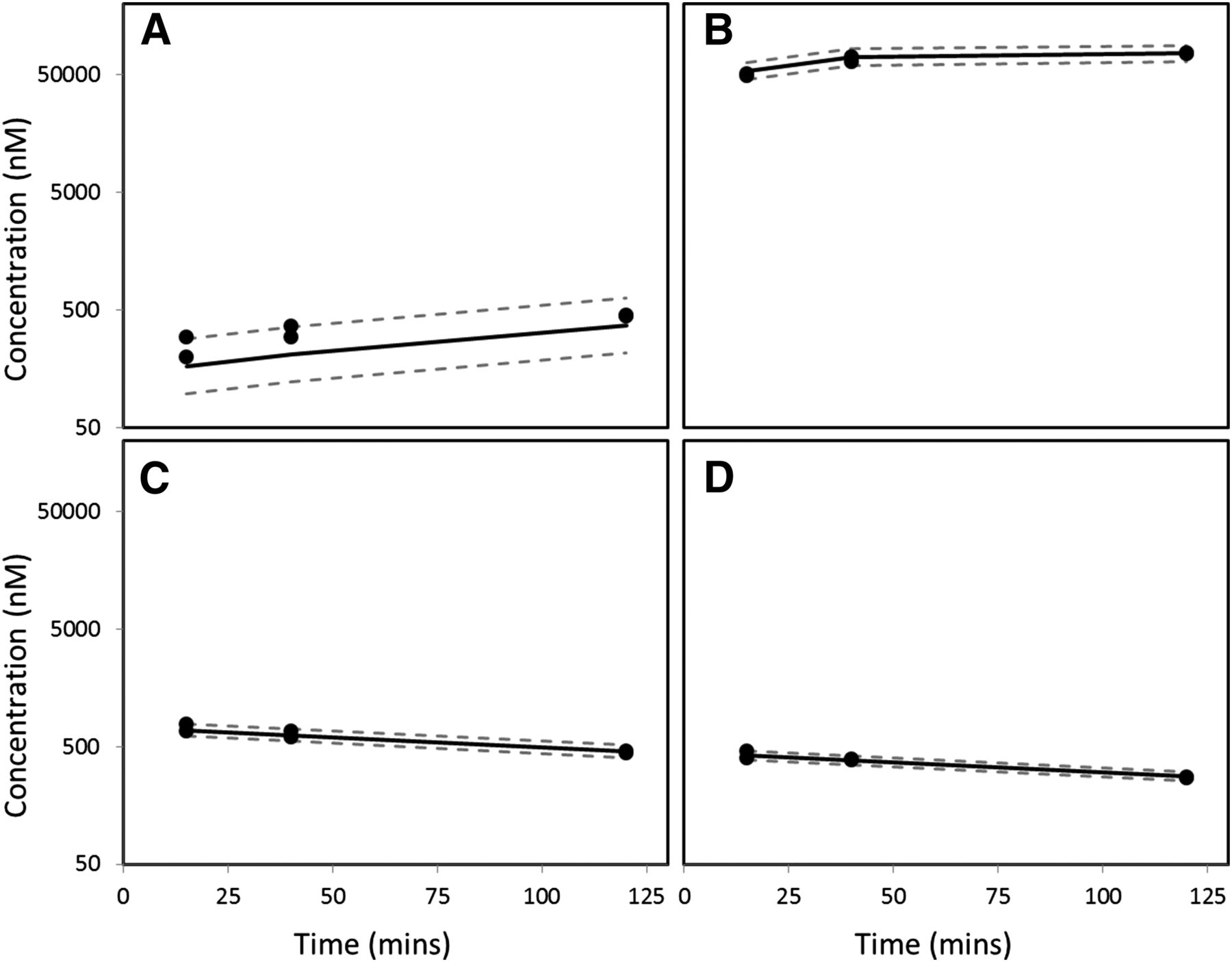
Prodrug is a strategy that can be used to enhance oral bioavailability, and the ethyl ester prodrug (PF-06757303) of diacid PF-06649298 was prepared (Fig. 3). The conversion rate from prodrug PF-06757303 to the active diacid PF-06649298 and the Kpuu in human hepatocytes are summarized in Figs. 5 and 6 and Table 5 [model parameters (Supplemental Table 1) and diagnosis plots of model fitting (Supplemental Fig. 1)]. When human hepatocytes in suspension were incubated with diacid PF-06649298, the medium concentration remained relatively unchanged at 1 μM over 2 hours (Fig. 5A, owing to the large volume difference between cells and medium), whereas the intracellular free drug concentration increased gradually to a Kpuu of 3 at 2 hours (Fig. 5B, Table 5). When human hepatocytes in suspension were incubated with ethyl ester PF-06757303, the medium and cell concentration of the ester decreased as a result of conversion to the corresponding diacid (Fig. 6, C and D), but the concentration of the diacid increased in both the medium and hepatocytes (Fig. 6, A and B). The respective Kpuu of ester PF-06757303 and diacid PF-06649298 were 0.07 and 59 at 2 hours (Table 5). The Kpuu value measured for diacid PF-06649298 was higher when the cells were treated with the prodrug rather than the diacid directly (59 versus 3). The combination of high formation rate (rapid entry of the prodrug into the hepatocytes and fast conversion to the diacid by esterases within the cells) and the slow disappearance rate (low passive diffusion, no active efflux out, and minimal metabolism) lead to a high Kpuu value for the diacid. For the prodrug, its disappearance rate (passive diffusion out of the cells and rapid conversion to the diacid) is much faster than its appearance rate (passive diffusion into the cells), leading to a Kpuu value less than one for the prodrug (0.07). Theoretically, after adequate incubation time for the ester to completely convert to the corresponding diacid, the Kpuu value of the diacid will drop to a value similar to that observed when the cells are incubated with the diacid itself.

Modeling results of human hepatocyte incubation with diacid PF-06649298, for 2 hours at 0.5 million cells/ml, 1 μM. (A) PF-06649298 concentration in medium; (B) PF-06649298 concentration in hepatocytes. LB, 95% lower prediction bound; UB, 95% upper prediction bound. Solid line represents predicted median. Dash lines represent 95% lower and upper prediction bound.

Modeling results of human hepatocyte incubation with prodrug PF-06757303, for 2 hours at 0.5 million cells/ml, 1 μM. (A) PF-06649298 concentration in medium; (B) PF-06649298 concentration in hepatocytes; (C) PF-06757303 concentration in medium; (D) PF-06757303 concentration in hepatocytes. LB, 95% lower prediction bound; UB, 95% upper prediction bound. Solid line represents predicted median. Dash lines represent 95% lower and upper prediction bound.
Kpuu of ethyl ester prodrug (PF-06757303) and its corresponding active diacid (PF-06649298) in human hepatocytes
To quantitatively understand the cellular distribution process of the ester and the diacid, a computational model was developed to evaluate the Kpuu and the half-life of both molecules in human hepatocytes (Fig. 4). The Kpuu of the diacid was estimated to be 3, and its half-life in the hepatocytes to be 40 minutes. The passive permeability of the prodrug was 60 times higher than that of diacid (0.012 versus 2 × 10−4 mm/min), consistent with negatively charged characteristics of the diacid and increased lipophilicity of the ester. The hydrolysis rate constant of the prodrug was predicted to be as high as 57 minute−1, resulting in a very short half-life of a few seconds. This disappearance rate of the prodrug was much higher than the appearance rate, leading to a low Kpuu (0.07).
Time to Steady State for Kpuu Measurement.
The ratio between unbound drug concentration in cells and unbound drug concentration in medium (Cu,cell/Cu,mdium) of PF-06649298 was determined using SLC13A5 HEK293 cells at different time points to assess how much time is required to reach steady state in this system (Fig. 7). Cu,cell/Cu,mdium increases with incubation time as molecules continue to be actively transported into the cells by SLC13A5. For PF-06649298 in SLC13A5 HEK293 cells, steady state is achieved in approximately 4 hours (estimated t1/2 ∼1 hour). The time to reach steady state for Kpuu measurement is compound- and cell system–dependent. At steady state, multiple processes (uptake, efflux, metabolism, electrochemical, and pH gradient, etc.) have reached a state where intracellular free drug concentration does not change anymore. Typically, at least two time points are taken for Kpuu measurement to ensure steady state has been achieved, meaning that Kpuu no longer changes with time.
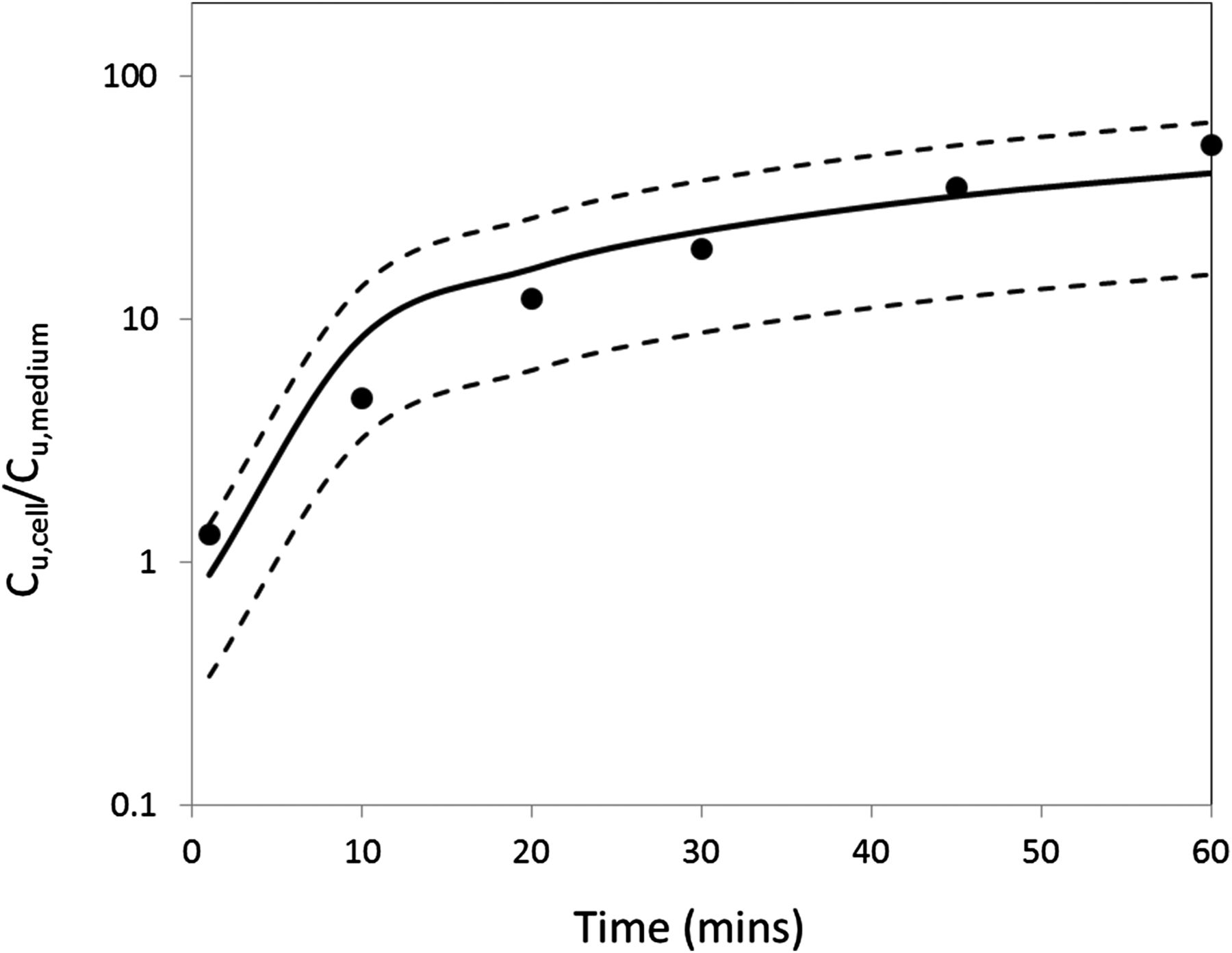
Time course of PF-06649298 in human SLC13A5 HEK293 cells as free cell concentration to free medium concentration. Kpuu was measured at cell density of 1 million cells/ml and 1 μM test compound concentration. LB, 95% lower prediction bound; UB, 95% upper prediction bound. Dash lines represent 95% lower and upper prediction bound.
Impact of Substrate Concentration on Kpuu.
Impact of substrate concentration on Kpuu was evaluated using PF-06649298 in human hepatocytes. Above a certain concentration (∼5 μM in this case), lower Kpuu values were observed in human hepatocytes with increasing test compound concentrations (Fig. 8). This is potentially attributable to saturation of the SLC13A5 transporter at high concentrations, leading to decrease in Kpuu. The estimated Km value of PF-06649298 in human hepatocytes is 24 μM (unbound Km 20 μM). Under typical experimental conditions, test compound concentrations are set below Km for Kpuu measurement.

Kpuu–concentration profile of PF-06649298 in human hepatocytes. Km is estimated to be 24 μM based upon nominal concentration in medium. LB: 95% lower prediction bound; UB: 95% upper prediction bound. Dash lines represent 95% lower and upper prediction bound.
Discussion
Drug disposition and trafficking in cells is intricate and complex (Fig. 1). Uptake transporters can increase the intracellular free drug concentration, whereas efflux transporters can reduce it. Cell membrane potential, pH gradient, binding to proteins and lipids, metabolism, lysosomal trapping, and disposition in other subcellular compartments can affect the intracellular free drug concentration and Kpuu. Intracellular free drug concentration measured by most methods represents an average free drug concentration of all the subcellular compartments. For carboxylic acids, since they have minimal lysosomal trapping and mitochondria accumulation, intracellular free drug concentration is a good surrogate for cytosolic drug concentration in hepatocytes. A number of in vitro methods have been developed to measure in vitro Kpuu (Fig. 2); however, validation of the methods with in vitro or in vivo pharmacology or pharmacokinetic data has not been thoroughly investigated. One of the validation studies used HMG-CoA reductase inhibition IC50 ratio from both rat hepatocytes and liver microsomes as a surrogate for Kpuu of statins (OATP substrates) and compared it to both the kinetic (Yabe et al., 2011) and temperature Kpuu methods (Shitara et al., 2013). The results showed that the kinetic method tended to overestimate the Kpuu, because metabolism and efflux were not considered in the method. Clearly, cross-validation of the Kpuu methods is important using various in vitro and in vivo pharmacological and pharmacokinetic approaches. In this study, a number of diacids were evaluated in the SLC13A transporter systems. These are ideal compounds for the study, as they are metabolically stable and have low passive permeability. Uptake is the main process governing the Kpuu of the compounds. In some of the studies, steady state was assumed to have been achieved on the basis of time course data. Both the in vitro SLC13A5 inhibition data and in vivo liver-to-plasma ratio data suggest that the in vitro binding Kpuu method provides a reliable measure of the intracellular free drug concentration, and good IVIVE has been established with rat suspension hepatocytes containing 4% fatty acid free BSA. This is the first time that a direct validation of the method is reported with in vitro and in vivo pharmacological and exposure/pharmacokinetic data.
A direct translation of the in vitro Kpuu to the in vivo Kpuu (measured from intravenous infusion study) is possible when a number of assumptions are met. The in vitro Kpuu can be expressed using the sequential clearance equation (Shitara et al., 2006; Watanabe et al., 2010; Yabe et al., 2011). The in vivo Kpuu between the hepatocytes and the hepatic capillary is expressed by the same equation. However, in an intravenous infusion, artery blood is sampled rather than hepatic capillaries. Since liver blood flow is only about 20% of cardiac output, the concentration difference at the two different locations will lead to different estimation of the in vivo Kpuu. However, in special cases, in vivo and in vitro Kpuu is similar. For example, if the compound has minimal metabolism by the liver and no biliary clearance, the in vivo Kpuu will be a result of drug partition between hepatocytes and hepatic capillaries, and the blood flow in (arterial blood) and out of the liver (hepatic capillary blood) will have the same drug concentrations at the steady state. Therefore, the in vivo Kpuu measured in an intravenous infusion study can be expressed using the same equation as that from the in vitro assay. Under these conditions, when the passive permeability and active uptake rates are comparable in the in vitro and in vivo settings, the Kpuu values will be similar as well. This appears to be the case for the SLC13A transport of the diacids.
A number of studies have shown that certain transporters are downregulated or internalized in cryopreserved hepatocytes (Bow et al., 2008). In vitro hepatocyte systems tend to underestimate in vivo Kpuu for OATP substrates (Morse et al., 2015), and typically, scaling factors are needed to more accurately predict in vivo outcomes (Li et al., 2014). However, in this case, the in vitro cryopreserved-suspension rat hepatocyte system appears to be able to predict in vivo rat Kpuu reasonably well. This might be owing to the unique properties of the SLC13A transporters, which may behave differently than OATPs. Further investigation of IVIVE mediated by OATP transporter uptake using hepatocyte suspension systems will provide additional insight.
Acknowledgments
The authors thank Karen Atkinson for editing the manuscript, and Larry Tremaine, Tess Wilson, and Charlotte Allerton for their leadership and support.
Authorship Contributions
Participated in research design: Riccardi, Li, Brown, Gorgoglione, Niosi, Gosset, Huard, Erion, Di.
Conducted experiments: Riccardi, Brown, Gorgoglione.
Performed data analysis: Riccardi, Li, Brown, Gorgoglione, Niosi, Gosset, Huard, Erion, Di.
Wrote or contributed to the writing of the manuscript: Riccardi, Li, Brown, Niosi, Gosset, Huard, Erion, Di.
Footnotes
- Received May 27, 2016.
- Accepted July 13, 2016.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- BSA
- bovine serum albumin
- CES
- carboxylesterase
- DMSO
- dimethylsulfoxide
- HEK
- human embryonic kidney
- HMG-CoA
- (9R,21S)-1-[(2R,3S,4R,5R)-5-(6-amino-9H-purin-9-yl)-4-hydroxy-3-(phosphonooxy)tetrahydrofuran-2-yl]-3,5,9,21-tetrahydroxy-8,8,21-trimethyl-10,14,19-trioxo-2,4,6-trioxa-18-thia-11,15-diaza-3,5-diphosphatricosan-23-oic acid 3,5-dioxide
- IC50
- concentration of an inhibitor at which the response is reduced by half
- IS
- internal standard
- IVIVE
- in vitro–in vivo extrapolation
- Km
- Michaelis-Menten constant
- Kpuu
- unbound partition coefficient, defined as the ratio between unbound intracellular free drug concentration and unbound medium concentration (steady state is not assumed) in this paper
- LC-MS/MS
- liquid chromatography–tandem mass spectrometry
- RH
- relative humidity
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}