


Abstract
Animals are not commonly used to assess drug-drug interactions due to poor clinical translatability arising from species differences that may exist in drug-metabolizing enzymes and transporters, and their regulation pathways. In this study, a transgenic mouse model expressing human pregnane X receptor (PXR), constitutive androstane receptor (CAR), CYP3A4/CYP3A7, and CYP2D6 (Tg-composite) was used to investigate the effect of induction mediated by rifampin on the pharmacokinetics of tamoxifen and its metabolites. In humans, tamoxifen is metabolized primarily by CYP3A4 and CYP2D6, and multiple-day treatment with rifampin decreased tamoxifen exposure by 6.2-fold. Interestingly, exposure of tamoxifen metabolites 4-hydroxytamoxifen (4OHT), N-desmethyltamoxifen (NDM), and endoxifen also decreased. In the Tg-composite model, pretreatment with rifampin decreased tamoxifen area under the time-concentration curve between 0 and 8 hours (AUC0-8) from 0.82 to 0.20 µM*h, whereas AUC0-8 of 4OHT, NDM, and endoxifen decreased by 3.4-, 4.7-, and 1.3-fold, respectively, mirroring the clinic observations. In the humanized PXR-CAR (hPXR-CAR) model, rifampin decreased AUC0-8 of tamoxifen and its metabolites by approximately 2-fold. In contrast, no significant modulation by rifampin was observed in the nonhumanized C57BL/6 (wild-type) animals. In vitro kinetics determined in microsomes prepared from livers of the Tg-composite animals showed that, although Km values were not different between vehicle- and rifampin-treated groups, rifampin increased the Vmax for the CYP3A4-mediated pathways. These data demonstrate that, although the hPXR-CAR model is responsive to rifampin, the extent of the clinical rifampin-tamoxifen interaction is better represented by the Tg-composite model. Consequently, the Tg-composite model may be a suitable tool to examine the extent of rifampin-mediated induction for other compounds whose metabolism is mediated by CYP3A4 and/or CYP2D6.
Introduction
As the pursuit of discovering innovative medicines continues, it is important to recognize that patients are increasingly being prescribed multiple drugs to treat various medical conditions. Despite the therapeutic advantages, there are several factors to consider for combination drug therapies. In particular, unwanted drug-drug interactions (DDIs) can have serious clinical consequences. Inhibition of drug-metabolizing enzymes may lead to adverse effects due to increased exposure to parent drug, whereas induction may also cause toxicity due to enhanced formation of harmful metabolites, and may result in loss of efficacy because drug exposure decreases. To assess the potential DDI liability early in drug discovery, in vitro and in silico tools are used (Bjornsson et al., 2003; Rostami-Hodjegan and Tucker, 2004; Fahmi and Ripp, 2010). However, in vivo nonclinical models are not typically used to characterize the DDI liability due to their poor translatability to in vivo in humans. This disconnect can be partly attributed to species differences in the expression and activity of drug-metabolizing enzymes and drug transporters (Shimada et al., 1997; Nelson et al., 2004; Chu et al., 2013). Indeed, these differences can complicate data interpretation, as exemplified by the observation that human CYP3A4-specific probes midazolam and triazolam undergo biotransformation via endogenously expressed mouse Cyp2c in the Cyp3a knockout mice (van Waterschoot et al., 2008, 2009). In addition to drug-metabolizing enzymes, species differences also exist in these enzyme's regulation pathways. For example, one of the mechanisms associated with the induction of CYP3A involves the activation of pregnane X receptor (PXR). Although the DNA binding domain of PXR is highly conserved, the sequence homology of the ligand binding domain between human and mouse is only 76% (Lehmann et al., 1998). As a result, extensive species differences in CYP3A4 induction have been reported, such as with rifampin, which exhibits inductive effects in humans but not in rats (Xie et al., 2000; Lu and Li, 2001).
Genetically modified animal models expressing human orthologs have been developed in an effort to bridge the gap between nonclinical animals and humans. For example, intrinsic clearance values determined in humanized CYP3A4 mice were shown to exhibit reasonable correlation to humans (Mitsui et al., 2014), and studies in these models provided further insight into the mechanism of cobimetinib disposition (Choo et al., 2015). Moreover, transgenic animals expressing multiple human proteins, such as PXR and CYP3A4, were able to capture the magnitude of rifampin-amprenavir interaction (Ma et al., 2008). However, although CYP3A4 is an important enzyme responsible for the metabolism of many drugs, there are limitations in the utility of models expressing only one cytochrome P450 (P450) isoform, since the metabolism of many drugs is mediated by multiple P450 isoforms.
Tamoxifen is a selective estrogen receptor modulator for the treatment of breast cancer. The main route of elimination is metabolism through several biotransformation reactions. In particular, as illustrated in Fig. 1, tamoxifen is metabolized extensively to N-desmethyltamoxifen (NDM) and to minor metabolite 4-hydroxytamoxifen (4OHT), which are catalyzed primarily by CYP3A4 and CYP2D6, respectively. NDM and 4OHT are further metabolized to form an active metabolite, endoxifen, by CYP2D6 and CYP3A4, respectively (Crewe et al., 2002; Desta et al., 2004). In a randomized crossover clinical trial with patients with breast cancer, 15-day oral administration of rifampin resulted in a 6.2-fold decrease in the exposure of tamoxifen. Unexpectedly, instead of yielding higher metabolite concentrations, the decrease in tamoxifen levels was accompanied by a 3.2-, 3.7-, and 3.2-fold decrease in exposures of NDM, 4OHT, and endoxifen, respectively (Binkhorst et al., 2012). The current work examined the impact of multiple-day administration of rifampin on the pharmacokinetics of tamoxifen and its metabolites in a composite transgenic mouse model expressing human PXR, constitutive androstane receptor (CAR), CYP3A4/7, and CYP2D6 (Tg-composite). Previous studies in the humanized animal models have demonstrated that CYP3A4 and CYP2D6 are expressed in both the liver and the intestines, and that treatment with rifampin for 3 days increased the protein expression of CYP3A4 in both tissues but not that of CYP2D6 (Hasegawa et al., 2011; Scheer et al., 2015). Effect of rifampin treatment was also examined in a transgenic mouse model expressing only the human PXR and CAR (hPXR-CAR) and in the nonhumanized, wild-type (WT) animals. The aims were to investigate the translatability of the Tg-composite model to the observed rifampin-tamoxifen DDI in humans and to gain further insights into the mechanism of rifampin-mediated induction involving tamoxifen metabolism pathways.

Illustration describing the metabolic pathways for tamoxifen and its metabolites.
Materials and Methods
Materials.
Tamoxifen, NDM, 4OHT (or afimoxifene), endoxifen, rifampin, potassium phosphate, sucrose EDTA, dithiothreitol, leupeptin, potassium chloride, and phenylmethylsulfonylfluoride were purchased from Sigma-Aldrich (St. Louis, MO). 1× Complete protease inhibitor was purchased from Roche Applied Sciences (Indianapolis, IN). All other chemicals and reagents were of analytical grade and were purchased from Sigma-Aldrich.
Pharmacokinetic Study Design.
Female transgenic mouse expressing human orthologs of PXR, CAR, CYP3A4/7, and CYP2D6 (Tg-composite); female transgenic mouse expressing only the human orthologs of PXR and CAR (hPXR-CAR; Taconic Farms/Artemis, Cologne, Germany); and female background WT C57BL/6 (Charles River Laboratories, Hollister, CA) were housed at a controlled temperature and humidity in an alternating 12-hour light/dark cycle with access to food and water ad libitum. All in vivo studies were performed in accordance with Institutional Animal Care and Use Committee guidelines at Genentech, Inc., and in harmony with the Guide for Laboratory Animal Care and Use (National Research Council, 2011). Four animals from each mouse model were pretreated with either vehicle (35% polyethylene glycol 400/65% saline) or 50 mg/kg rifampin orally for 3 days. On the fourth day, all animals were given a single 20-mg/kg oral dose of tamoxifen (35% polyethylene glycol 400/65% saline). Each animal was serially bled via tail nick at 0.25, 0.5, 1, 3, 6, 8, and 24 hours postdose. Fifteen microliters of blood was collected at each time point and diluted with 60 µl of water containing 1.7 mg/ml of EDTA. All blood samples were stored at approximately −80°C until thawed for quid chromatography–mass spectrometry/mass spectrometry (LC-MS/MS) analysis. At approximately 24 hours postdose, livers from these animals were harvested and stored at −80°C until microsome preparation.
Preparing Microsomes.
Liver microsomes were prepared from livers collected from the Tg-composite animals that were in the pharmacokinetic study. Approximately 1 g of tissue was thawed in 1 ml of homogenization buffer [0.1 M potassium phosphate buffer (pH 7.4) with 250 mM sucrose, 1 mM EDTA, 0.1 mM dithiothreitol, 2 μg/ml leupeptin, 150 mM potassium chloride, 1× complete protease inhibitor, and freshly added 1 mM phenylmethylsulfonylfluoride], and the samples were homogenized on ice with a Dounce glass A homogenizer (Kontes, Seattle, WA). The suspension was centrifuged at 9000g for 20 minutes at 4°C. The resulting supernatant was centrifuged again in an ultracentrifuge at 105,000g for 60 minutes at 4°C, and the pellet was resuspended in 0.1 M potassium phosphate buffer (pH 7.4) containing 250 mM sucrose and stored at −80°C. Total microsome protein concentrations were determined using a BCA assay kit from Pierce (Rockford, IL).
Microsomal Incubations.
A mixture containing 0.05 mg/ml liver microsomes prepared from vehicle- and rifampin-treated Tg-composite mice and 1 mM MgCl2 were added to 0.1 M potassium phosphate buffer (pH 7.4). The mixture was added to vials containing tamoxifen (0.25–40 µM), NDM (0.05–25 µM), or 4OHT (0.25–50 µM) dissolved in dimethylsulfoxide (0.1% w/v). The mixture was briefly vortexed, and following preincubation at 37°C for approximately 15 minutes, the reactions were initiated by adding 1 mM NADPH. The total reaction volume for all experiments was 200 μl. The incubation was allowed to continue at 37°C, and after 10 minutes, the reactions were terminated with the addition of equal volumes of acetonitrile containing 100 nM cortisone as the internal standard. The concentration of the microsomes and the time point were optimized to ensure that the in vitro experiment was being conducted to yield linear metabolite formation for 1) tamoxifen to 4OHT, 2) tamoxifen to NDM, 3) 4OHT to endoxifen, and 4) NDM to endoxifen. The microsomal protein concentrations ranged between 0.05 and 0.5 mg/ml, and the reaction was monitored for 45 minutes. Based on these experiments, a 0.05-mg/ml protein concentration at 10 minutes was an optimal in vitro condition, yielding linear metabolite formation for all metabolic pathways (data not shown). Each sample was centrifuged, and the supernatant was injected onto LC-MS/MS. Formation of NDM, 4OHT, and endoxifen was quantitated using a standard curve prepared in a similar manner as stated earlier for the in vitro incubation reaction. All experiments were done in triplicate for each animal in the study.
Sample Analysis.
Concentrations of tamoxifen, NDM, 4OHT, and endoxifen in the blood were determined by LC-MS/MS. The LC-MS/MS system consisted of a Nexera LC system (Shimadzu, Columbia, MD) and an API-5500 Qtrap mass spectrometer (Sciex, Redwood City, CA). The separation was achieved on a Kinetex PF column (50 × 2 mm, 2.6-μm particle size; Phenomenex, Torrance, CA) at a flow rate of 0.6 ml/min with gradient elution using mobile phases of water with 0.1% formic acid (A) and acetonitrile with 0.1% formic acid (B). High-performance liquid chromatography gradient was 40% B for 0.33 minute, increased to 60% B at 2.4 minutes, and to 95% at 2.6 minutes. The gradient was maintained at 95% for 1.8 minutes before returning to the initial composition of 40% B within 0.01 minute. The system was allowed to equilibrate for 2.4 minutes before the next injection. The total run time was 5 minutes. The concentrations of tamoxifen and its metabolites were determined using multiple reaction monitoring in the positive ion mode. The following multiple reaction monitoring transitions were monitored: m/z 372.1 to 129 for tamoxifen, m/z 358.2 to 128 for NDM, m/z 388.1 to 129.2 for 4OHT, and m/z 374 to 128.9 for endoxifen. The standard curve ranged from 0.003 to 50 µM, and the lower limit of quantification was 0.006 µM for tamoxifen and its metabolites.
Data Analysis.
Concentrations for tamoxifen and its metabolites were below the limit of quantitation at 24 hours, which was observed in previous studies (Reid et al., 2014). Therefore, area under the blood concentration-time curve was determined by noncompartmental analysis using Phoenix WinNonlin, version 6.3 (Pharsight Corporation, Mountain View, CA) for 0–8 hours (AUC0-8). Metabolite-to-parent ratios (MPRs) were calculated as AUC0-8metabolite/AUC0-8parent. Analysis compared tamoxifen, NDM, 4OHT, or endoxifen AUC0-8 between the vehicle- and rifampin-treated groups. Statistical analysis was conducted using the 2-tailed, heteroscedastic t test function in Excel (Microsoft, Redmond, WA). Mean (± S.D.) and P < 0.05 were reported.
Km and Vmax from the in vitro microsome experiments were determined using the Michaelis-Menten function in a nonlinear regression curve fit with Prism 5 (GraphPad, San Diego, CA). The absolute sum of squares and R2 generated by Prism were used to assess the goodness of the fit.
Results
Pharmacokinetics of Tamoxifen, NDM, 4OHT, and Endoxifen in Tg-Composite, hPXR-CAR, and WT Animals.
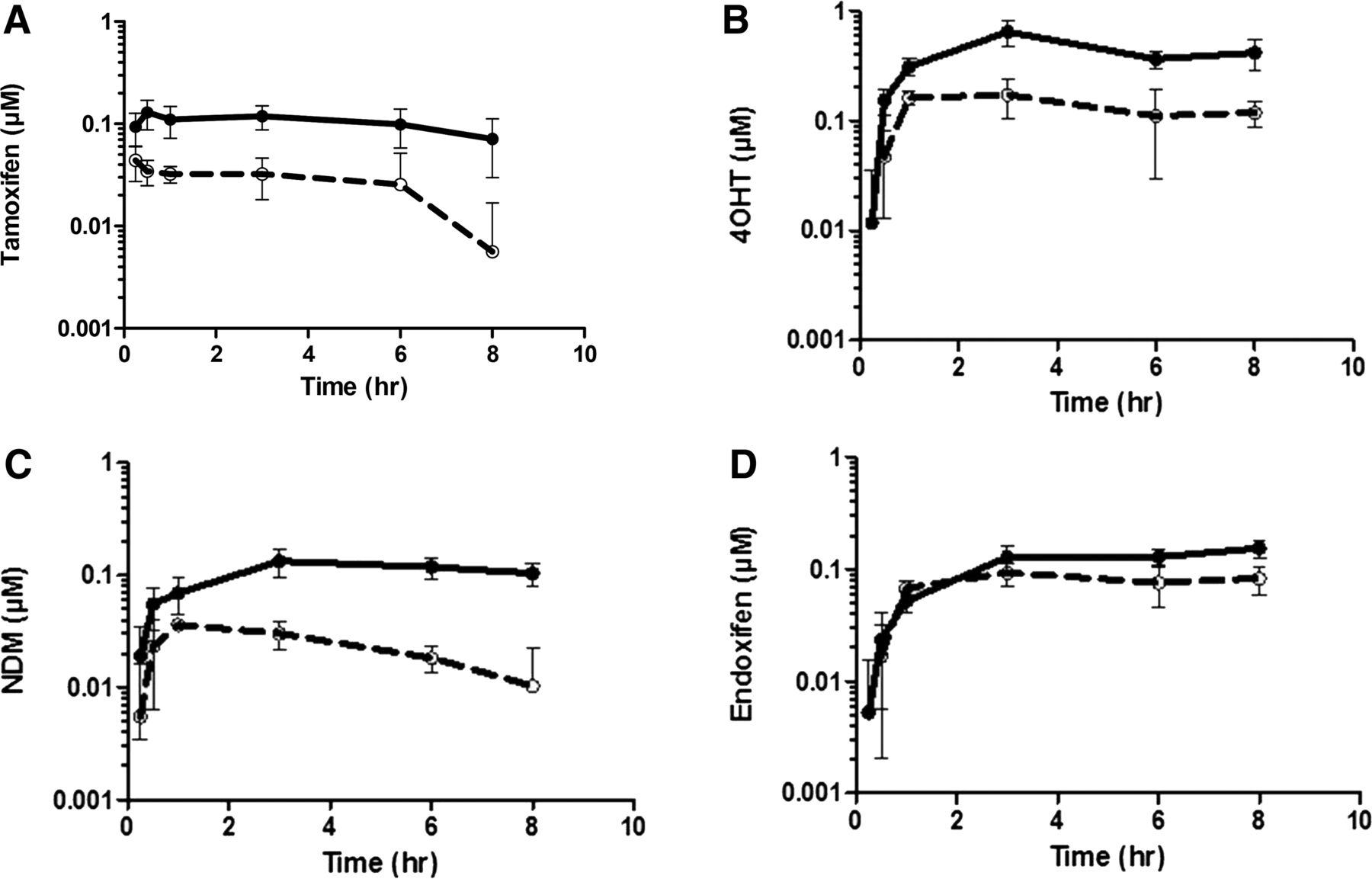
Tamoxifen was extensively metabolized to form NDM, 4OHT, and endoxifen following 20-mg/kg oral administration of tamoxifen to the Tg-composite, hPXR-CAR, and WT animals. Figure 2 shows that the shapes of the concentration-time profiles between the vehicle- and rifampin-treated groups for tamoxifen, NDM, 4OHT, and endoxifen in the Tg-composite animals were similar. However, the concentrations of tamoxifen and its metabolites in the rifampin-treated groups were significantly lower overall. Table 1 summarizes the AUC0-8 and Cmax of tamoxifen, NDM, 4OHT, and endoxifen determined in the Tg-composite, hPXR-CAR, and WT animals when tamoxifen was orally administered following pretreatment with vehicle or rifampin. In the Tg-composite group, AUC0-8 and Cmax of tamoxifen in the vehicle-treated animals were 0.82 µM*h and 0.15 µM, respectively, which are within a reasonable range of the exposures reported in humans. When pretreated with rifampin, the AUC0-8 of tamoxifen decreased 4.1-fold to 0.20 µM*h, and Cmax decreased 3.1-fold to 0.048 µM. Along with the decrease in tamoxifen AUC0-8 and Cmax, pretreatment with rifampin also decreased the AUC0-8 and Cmax of tamoxifen metabolites. AUC0-8 of NDM was decreased 4.7-fold to 0.18 µM*h, 4OHT was decreased 3.4-fold to 1.0 µM*h, and endoxifen was decreased 1.3-fold to 0.74 µM*h. Cmax of NDM was decreased 3.5-fold to 0.037 µM*h, 4OHT was decreased 3.4-fold to 0.19 µM*h, and endoxifen was decreased 1.6-fold to 0.11 µM*h. The decreases in both AUC0-8 and Cmax for tamoxifen, NDM, and 4OHT were comparable with what was observed in vivo in humans (Binkhorst et al., 2012). In the hPXR-CAR group, tamoxifen AUC0-8 and Cmax in the vehicle-treated animals were comparable to the Tg-composite animals at 1.0 µM*h and 0.33 μM, respectively. Pretreatment with rifampin also decreased tamoxifen and its metabolites in the hPXR-CAR animals. However, the magnitude of change was smaller, as rifampin decreased the AUC0-8 of tamoxifen, NDM, 4OHT, and endoxifen by 2.4-, 1.8-, 1.9-, and 1.9-fold, respectively. Similarly, the Cmax of tamoxifen, NDM, 4OHT, and endoxifen decreased by 1.9-, 1.4-, 1.4-, and 1.2-fold, respectively. In contrast to the Tg-composite and hPXR-CAR animals, rifampin had no effect on the exposure of tamoxifen and its metabolites in the WT animals.

Summary of blood concentration-time profile of tamoxifen (A), 4OHT (B), NDM (C), and endoxifen (D) following 20-mg/kg oral administration of tamoxifen in transgenic animals expressing human PXR, CAR, CYP3A4/7, and CYP2D6 (Tg-composite). In each panel, solid symbols with solid lines represent transgenic animals treated with vehicle, and open symbols with broken lines represent transgenic animals treated with rifampin. Concentrations for tamoxifen and its metabolites were below the limit of quantitation at 24 hours. Data are presented as the mean (± S.D.) of four animals.
Summary of mean (± S.D.) AUC0-8 and Cmax of tamoxifen, NDM, 4OHT, and endoxifen when administered 20 mg/kg tamoxifen orally following vehicle or rifampin treatment for 3 days (N = 4)
AUC0-8 values were used to calculate the MPR and are summarized in Table 2. In the TG-composite animals, MPR values in the vehicle-treated group were 4.1, 1.0, and 1.1 for 4OHT, NDM, and endoxifen, respectively, relative to tamoxifen AUC0-8. In contrast, MPR values for all of the metabolites were below unity in the hPXR-CAR and WT animals. Differences in the MPR values suggest that the metabolism in the Tg-composite animals to form NDM, 4OHT, and endoxifen is not the same as in the hPXR-CAR and WT animals, which do not express the human orthologs of CYP3A4/7 and CYP2D6. For all models, pretreatment with rifampin did not significantly alter the MPRs for any of the metabolites.
Summary of mean (± S.D.) MPR calculated from AUC0-8 (N = 4)
In Vitro Kinetics in Liver Microsomes Prepared from Tg-Composite Animals Treated with Either Vehicle or Rifampin.
Microsomes were prepared from livers harvested from Tg-composite animals treated with either vehicle or rifampin to further characterize the kinetics of tamoxifen and its metabolites in this model. Tamoxifen was incubated, and the formation of 4OHT and NDM was monitored. In microsomes prepared from the vehicle-treated groups, the apparent Km (Km,app) values for the formation of NDM and 4OHT were 3.8 and 7.9 µM, respectively. Km,app values did not change when using microsomes prepared from the rifampin-treated groups. However, whereas the Vmax values were comparable between the vehicle- and rifampin-treated groups for the formation of 4OHT, the Vmax of NDM formation was enhanced by 3.3-fold to 2.6 nmol/min/mg protein in the rifampin-treated group.
In addition to tamoxifen, NDM and 4OHT were incubated in separate microsomal reactions to determine the formation kinetics of endoxifen. In microsomes prepared from the vehicle-treated groups, Km,app was 5.9 and 13 µM when incubating NDM and 4OHT, respectively. As with tamoxifen, there was no effect on Km,app with rifampin pretreatment. However, Vmax of endoxifen formation from 4OHT increased 7.4-fold to 2.6 nmol/min/mg protein in the rifampin-treated animals, whereas the Vmax of endoxifen formation from NDM was not changed.
Discussion
Estimating the extent of P450 induction in vivo in humans is one of the hurdles in drug discovery due to its complex mechanism (Lin, 2006; Shou et al., 2008; Fahmi and Ripp, 2010). The gold standard for assessing P450 induction is with human hepatocytes, but their ability to quantitatively predict DDI is limited because of the need to scale in vivo. Unfortunately, due to poor translatability to humans resulting from species differences in drug-metabolizing enzymes, drug transporters, and their regulation pathways, nonclinical animals are not good models for DDI risk assessment. However, transgenic mouse models whose endogenous genes are replaced by human orthologs may be a useful tool to address these limitations. The objectives of this work were to investigate the translatability of the Tg-composite model expressing human orthologs of PXR, CAR, CYP3A4/7, and CYP2D6 to the clinically observed rifampin-tamoxifen DDI, and to gain further insights into the mechanism of rifampin-mediated induction involving tamoxifen metabolism pathways.
In the current study, tamoxifen was used as a probe substrate with a specific intention, as it is mainly metabolized in humans by CYP3A4 and CYP2D6, and because it exhibits an interesting but complex DDI profile. In particular, metabolism of tamoxifen yields at least two active metabolites in 4OHT (Jordan et al., 1977) and endoxifen (Johnson et al., 2004). Moreover, although it is normally expected that P450 induction would result in decreases of drug concentration accompanied by increases of metabolite levels, pretreatment with rifampin decreased both tamoxifen and its metabolite levels in humans (Binkhorst et al., 2012).
Rifampin has been characterized as a strong activator of human PXR, but not that of rodents (Jones et al., 2000). Indeed, rifampin had no effect on the disposition of tamoxifen or its metabolites in the WT animals (Table 1). Contrarily, in the Tg-composite model where the human ortholog of PXR is expressed, rifampin decreased the AUC0-8 of tamoxifen, NDM, 4OHT, and endoxifen by 4.1-, 4.7-, 3.4-, and 1.3-fold, respectively. Comparable decreases were observed with Cmax. The extent of decrease for both AUC0-8 and Cmax was comparable to the effect that was observed in vivo in humans. Not surprisingly, hPXR-CAR animals were also susceptible to rifampin pretreatment, as the AUC0-8 of tamoxifen, NDM, 4OHT, and endoxifen decreased by 2.4-, 1.8-, 1.9-, and 1.9-fold, respectively. However, the magnitude of change observed in the hPXR-CAR animals was lower than what was observed in vivo in humans, especially for tamoxifen and the major human metabolite NDM. Therefore, although both Tg-composite and hPXR-CAR animals were able to capture the complex rifampin-tamoxifen interaction, Tg-composite animals were more sensitive. In addition, these data suggest that the expression of additional human orthologs of CYP3A4/7 and CYP2D6 in the Tg-composite model in predicting the magnitude of tamoxifen induction may go beyond a qualitative assessment, but may also be used to quantitatively capture the extent of change.
MPR is useful to evaluate if a particular metabolic pathway has been altered. When metabolism is induced, MPR should typically increase. However, ratios for NDM/tamoxifen and 4OHT/tamoxifen in both Tg-composite and hPXR-CAR animals did not change with rifampin. This finding does not necessarily imply that induction of their metabolic pathways did not occur, but instead suggests that the extent of induction in both the formation and elimination of NDM and 4OHT is similar. In fact, NDM and 4OHT undergo further oxidative metabolism to endoxifen by CYP2D6 and CYP3A4, respectively. In addition, CYP3A4 can also metabolize NDM to other oxidative metabolites (Coller et al., 2004). This hypothesis corresponds to the findings of studies with atorvastatin and glyburide, where the MPR did not change with induction because the parent and the metabolite were comparable substrates for the same enzymes and transporters (Lau et al., 2007; Zheng et al., 2009). In contrast, the endoxifen/tamoxifen ratio increased 3.4-fold in the Tg-composite animals. Since MPR is proportional to the formation of metabolite and is inversely proportional to the clearance of the metabolite, these data indicate that the impact of rifampin-mediated induction was greater on the formation of endoxifen than its elimination, and that the elimination of endoxifen may be different than with NDM and 4OHT. Indeed, it has been shown that glucuronidation and sulfation are responsible for endoxifen metabolism (Poon et al., 1993; Kisanga et al., 2005). Interestingly, the endoxifen/tamoxifen ratio did not change in the hPXR-CAR animals. However, this differential effect of rifampin between Tg-composite and hPXR-CAR animals is not surprising given the likelihood that the extent and rate of tamoxifen, NDM, and 4OHT elimination are different between the two models. This assumption is supported by the fact that MPR values for NDM and 4OHT from the WT and hPXR-CAR animals were much reduced compared with the Tg-composite animals.
In vitro metabolism pathways for tamoxifen, NDM, and 4OHT were further studied in microsomes prepared from livers harvested from vehicle- and rifampin-treated Tg-composite animals. Table 3 shows that the Km,app was not markedly different between the two treatment groups. However, whereas Vmax for the tamoxifen-to-4OHT and NDM-to-endoxifen metabolic pathways was similar, Vmax was enhanced in the rifampin pretreated group for the tamoxifen-to-NDM and 4OHT-to-endoxifen pathways by 3.3- and 7.4-fold, respectively. These observations are consistent with the expectation that rifampin-mediated induction via PXR activation would not alter the binding affinity, but only increase the activity of the enzymes through upregulation of transcription. However, Vmax did not increase for all metabolic pathways that were examined, but only in two pathways. In particular, metabolic pathways where the primary catalyst was CYP3A4 were enhanced, whereas there was no modulation in the CYP2D6 pathways. These findings corroborate previous findings which show that CYP2D6 is not susceptible to induction by rifampin (Rodriguez-Antona et al., 2000; Rae et al., 2001), but that its activity is affected predominantly by genetic polymorphisms (Dilger et al., 1999; Giessmann et al., 2004). Therefore, these studies give support to the fact that the human PXR, CYP3A4, and CYP2D6 function properly in the TG-composite animals.
RESIZESummary of mean Km,app and Vmax (± S.D.) determined from microsomes prepared from livers of the Tg-composite animals treated with vehicle or rifampin (N = 4)
Previous studies have shown that hydroxylation and N-oxide formation in rodents are much greater than in humans (Robinson et al., 1991; Lim et al., 1994). Consequently, although formation of the major human metabolite NDM was enhanced compared with the WT and hPXR-CAR animals, the major circulating metabolite was 4OHT in the Tg-composite animals. It has been reported that endogenous Cyp2c12 and Cyp1a2 in mice may contribute to the enhanced hydroxylation (Mani et al., 1993), and therefore, the high formation of 4OHT can be attributed to endogenous mouse metabolizing enzymes. Therefore, despite the effort to circumvent species differences in metabolism, the transgenic mice were not able to completely recapitulate the human metabolite profile, resulting in differences in MPR between humans and the Tg-composite animals. This observation highlights one of the drawbacks of the utility of the humanized transgenic model in that it cannot completely mirror the human metabolism machinery. Drug-metabolizing enzymes exhibit a wide range of substrate specificities, and drugs frequently exploit multiple metabolism pathways for elimination. In addition, a specific biotransformation pathway that a particular drug favors may shift depending on the changing circumstances, such as with DDI and various disease states. To complicate this matter, there are unknown metabolizing enzymes and elimination pathways, and as such, it may be difficult to develop a complete humanized model. For example, studies have shown that the classic human CYP3A4 probe midazolam can also undergo metabolism via the endogenous mouse Cyp2c (van Waterschoot et al., 2008). Models where the mouse livers are replenished with human hepatocytes are available, but they also come with their own disadvantages, such as the animals being immune-compromised and the fact that the mouse livers cannot be repopulated with 100% human hepatocytes.
In summary, Tg-composite animals were able to mirror the magnitude of rifampin-tamoxifen DDI that is observed in humans. This is one of the first studies in which the transgenic model was able to demonstrate a complex DDI where both the parent and metabolites were reduced following induction. Humanized PXR animals have already been used to assess the impact of rifampin-mediated induction on victim compounds (Ma et al., 2008). However, as discussed previously, evaluating a compound as a victim of induction may be complex, and it is important to coexpress human enzymes. Therefore, despite the limitations, one potential application of the Tg-composite model and the work presented here is to investigate the likelihood that a new molecular entity is an inducer of CYP3A4 and/or CYP2D6 using tamoxifen as a victim substrate. In addition, this model may be suitable to study compounds exhibiting a more complicated DDI profile, such as protease inhibitors, which are both inducers and time-dependent inhibitors of P450 isoforms (Yakiwchuk et al., 2008). The hope is that these transgenic animal models would not necessarily replace existing methodologies to assess DDI, but that they could complement the in vitro and in silico assessments to provide additional confidence during early assessment of DDI liabilities.
*P < 0.05 relative to vehicle.
Authorship Contributions
Participated in research design: Chang, Ly.
Conducted experiments: Chang, Chen, Liu, Messick, Ly.
Performed data analysis: Chang, Ly.
Wrote or contributed to the writing of the manuscript: Chang, Chen, Ly.
Footnotes
- Received June 17, 2016.
- Accepted August 17, 2016.
Abbreviations
- AUC0-8
- area under the time-concentration curve between 0 and 8 hours
- CAR
- constitutive androstane receptor
- DDI
- drug-drug interaction
- hPXR-CAR
- humanized PXR-CAR
- LC-MS/MS
- liquid chromatography–mass spectrometry/mass spectrometry
- NDM
- N-desmethyltamoxifen
- 4OHT
- 4-hydroxytamoxifen
- P450
- cytochrome P450
- PXR
- pregnane X receptor
- Tg
- transgenic
- WT
- wild type
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}