


Abstract
Overdose of isoniazid (INH), an antituberculosis drug, can be life-threatening because of neurotoxicity. In clinical practice for management of INH overdose and acute toxicity, the potential of INH-induced hepatotoxicity is also considered. However, the biochemical basis of acute INH toxicity in the liver remains elusive. In the current study, we used an untargeted metabolomic approach to explore the acute effects of INH on endobiotic homeostasis in mouse liver. We found that overdose of INH resulted in accumulation of oleoyl-l-carnitine and linoleoyl-l-carnitine in the liver, indicating mitochondrial dysfunction. We also revealed the interactions between INH and fatty acyl-CoAs by identifying INH-fatty acid amides. In addition, we found that overdose of INH led to the accumulation of heme and oxidized NAD in the liver. We also identified an INH and NAD adduct in the liver. In this adduct, the nicotinamide moiety in NAD was replaced by INH. Furthermore, we illustrated that overdose of INH depleted vitamin B6 in the liver and blocked vitamin B6–dependent cystathionine degradation. These data suggest that INH interacts with multiple biochemical pathways in the liver during acute poisoning caused by INH overdose.
Introduction
Tuberculosis (TB) is a public health problem (Orcau et al., 2011). In 2012, there were ∼8.6 million incident cases of TB and ∼1.3 million TB-associated deaths worldwide (World Health Organization, 2013). Isoniazid (INH), also known as isonicotinylhydrazide, is a first-line drug for TB treatment and prevention. With the numbers of patients receiving INH remaining high, the number of acute poisoning cases is expected to be significant (Maw and Aitken, 2003). Accidental or intentional overdose of INH causes seizures, metabolic acidosis, coma, and even death (Temmerman et al., 1999; Khoharo et al., 2009). In adults, the regular dose of INH is 300 mg/d (∼5 mg/kg/d). Doses over 30 mg/kg/d often produce neurotoxicity and seizures. It has been estimated that INH is responsible for ∼5% of all cases of seizures associated with drug intoxications (Olson et al., 1993). When ingested in amounts of ≥80–150 mg/kg/d, INH can rapidly be fatal because of acute neurotoxicity (Romero and Kuczler, 1998).
Besides neurotoxicity, INH poisoning causes hepatotoxicity. In a 2-year-old girl who accidentally received 2 g of INH, liver function test results were found to be abnormal (Caksen et al., 2003). In a 26-year-old man who ingested 8 g of INH, the elevation of activities of serum aspartate aminotransferase, alanine aminotransferase, γ-glutamyl transpeptidase, and prolonged prothrombin time were observed, suggesting the occurrence of INH-induced acute hepatitis (Tai et al., 1996). A higher dose of INH results in more significant liver injury, as shown by 20-fold increases of both serum aspartate aminotransferase and lactate dehydrogenase activities in a 21-year-old woman who ingested 12 g of INH (Bear et al., 1976). These clinical observations are supported by a preclinical study showing that a high dose of INH causes the accumulation of reactive oxygen species and reactive nitrogen species in mouse liver (Shuhendler et al., 2014). Therefore, the monitoring of liver functions and prothrombin time is recommended in clinical practice for the management of INH poisoning (Romero and Kuczler, 1998).
Understanding the acute effects of INH on the liver will be helpful for the management of INH-induced liver injury during INH poisoning (Tai et al., 1996; Romero and Kuczler, 1998; Temmerman et al., 1999). However, limited information is available in this regard (Isoniazid, 2016). Metabolomics, a methodology for exploring metabolite profiles in biologic matrices, has been considered a powerful tool for illustrating the biochemical basis of drug toxicity and disease (O'Connell and Watkins, 2010; Beyoğlu and Idle, 2013). In the current study, we used a metabolomic approach to investigate endobiotic homeostasis in the liver of mice receiving a high dose of INH. We found that INH interacts with multiple biochemical pathways in the liver, including heme, fatty acids, oxidized NAD, vitamin B6, and cystathionine.
Materials and Methods
Chemicals and Reagents.
INH, hemin, cystathionine, NAD, pyridoxal (PL), pyridoxal phosphate (PLP), heptafluorobutyric acid (HFBA), and porcine brain NAD-glycohydrolase (NADase) were purchased from Sigma-Aldrich (St. Louis, MO). Oleoyl-l-carnitine and linoleoyl-l-carnitine were purchased from INDOFINE Chemical Company (Hillsborough, NJ). Deuterium-labeled INH (d4-INH) was purchased from Toronto Research Chemicals (Toronto, ON, Canada). All solvents for metabolite analysis were of the highest grade commercially available.
Animals and Treatments.
Mouse is a commonly used model for the studies on INH (Metushi et al., 2012). In the current study, wild-type mice (FVB/NJ, male, 8 weeks old) were treated orally with INH at 0, 50, or 200 mg/kg. These doses were chosen according to the dosage conversion factor between humans and mice (Reagan-Shaw et al., 2008). Because the regular dose of INH is 5 mg/kg/d in humans (Isoniazid, 2016), 50 mg/kg INH in mice mimics the pharmacological dose in humans, and 200 mg/kg INH in mice mimics the toxic dose in humans. We did not choose the doses over 200 mg/kg, because the LD50 of INH (by mouth) in mice is 176 mg/kg (Isoniazid, 2016). To explore the effect of INH on liver metabolome, the mice were sacrificed at 30 minutes after INH treatment. Moribundity was not observed at this time point. Blood and liver samples were collected for metabolomic analysis. Serum alanine transaminase and aspartate transaminase were also analyzed. We chose 30 minutes for sample collection, because most mice will die between 45 and 90 minutes after treatment with 200 mg/kg INH. To determine the time-dependent effect of INH on vitamin B6 in the liver, the mice were sacrificed at 15 and 45 minutes after INH treatment. All procedures involving mouse care and handling were in performed in accordance with study protocols approved by the Institutional Animal Care and Use Committee from the University of Kansas Medical Center and University of Pittsburgh.
Sample Preparation.
Liver tissues samples were homogenized in water (100 mg of liver tissue in 300 µl of water). Subsequently, a 400-µl aliquot of ice-cold MeOH was added to 200 µl of the resulting mixture, followed by vortexing and centrifugation at 18,000g for 20 minutes. The supernatant was transferred to a new Eppendorf tube for a second centrifugation (18,000g for 10 minutes). Each supernatant was transferred to an autosampler vial, and 5.0 µl was injected into an ultraperformance liquid chromatography (UPLC) coupled with quadrupole time-of-flight mass spectrometry (QTOFMS) for metabolite analysis.
UPLC-QTOFMS Analysis.
The same approach has been used in our previous studies on the metabolism of atazanavir and tipranavir (Li et al., 2010, 2011a). In brief, a 100 × 2.1 mm (1.7 µm) UPLC BEH C-18 column (Acquity; Waters, Milford, MA) was used for metabolite separation. The flow rate of the mobile phase was set as 0.3 ml/min. The gradient ranged from 2% to 98% acetonitrile containing 0.1% formic acid in the first 8.5 minutes, 98% acetonitrile for 8.5–18 minutes, then equilibration at 2% acetonitrile for 2 minutes. QTOFMS (Waters) was operated in a positive mode with electrospray ionization. The source temperature and desolvation temperature were set at 120°C and 350°C, respectively. Nitrogen was used as the cone gas and desolvation gas. Argon was applied as the collision gas. QTOFMS was calibrated with sodium formate and monitored by the intermittent injection of lock mass leucine enkephalin in real time. The capillary voltage and cone voltage were set at 3.5 kV and 35 V, respectively. Screening and identification of major metabolites were performed by using MarkerLynx software (Waters) based on accurate mass measurement (mass errors less than 10 ppm). Tandem mass spectrometry fragmentation was conducted with a collision energy ramp ranging from 10 to 40 V.
Inhibitory Effect of INH on NADase Activity.
The inhibitory effect of INH (0–10 mM) on NADase activity was performed according to the findings of a previous report (Escande et al., 2013). Briefly, NADase activity was measured using recombinant human CD38 and expressed as arbitrary fluorescent units per minute.
Formation of INH-NAD Adduct in S9 Fraction of Mouse Liver and NADase.
The metabolite VII was proposed as an INH-NAD adduct. To verify the structure of the INH-NAD adduct and to determine the role of NADase in the formation of this adduct, we incubated INH or d4-INH with NAD in S9 fraction of mouse liver and porcine NADase, respectively. Briefly, the reactions were conducted in an incubation system containing liver S9 fractions or NADase, 2 mM INH or d4-INH, and 2 mM NAD at 37°C for 2 hours. The reactions were terminated by adding a twofold volume of ice-cold MeOH, followed by vortexing and centrifugation at 10,000g for 10 minutes. The supernatant was analyzed by UPLC-QTOFMS.
Bioanalysis of PL and PLP.
PL is one of the three natural forms of vitamin B6, and the active form of vitamin B6 is PLP. PL and PLP were analyzed according to a published method with a slight modification (van der Ham et al., 2012). In brief, an aliquot of 100 µl of liver homogenate was mixed with 20 µl of trifluoroacetic acid, followed by vortexing and centrifugation at 15,000g for 10 minutes. Two microliters of the supernatant was injected into the UPLC-QTOFMS system for analysis of PL and PLP. An Acquity HSS T3 column (100 × 2.1 mm, 1.7 µm) was used for chromatographic separation. A 4-minute gradient at a flow rate of 0.4 mL/min was used with mobile phase A (0.65 M acetic acid with 0.01% HFBA in water) and mobile phase B (0.65 M acetic acid with 0.01% HFBA in acetonitrile). The QTOFMS was operated in positive mode.
Synthesis of INH-PL Adduct.
We expected the formation of an INH-PL adduct. To verify its structure, we synthesized this adduct. Briefly, 200 µl of PL (400 µM in H2O) and 200 µl of INH (400 µM) were added to an Eppendorf tube. The resulting mixture was placed on a shaker at 37°C for 30 minutes. The final product was analyzed by UPLC-QTOFMS. High-resolution mass spectrometry (electrospray ionization) calculated for C14H15N4O3 [M+H]+ 287.1144, found 287.1146.
Metabolomic Analysis.
An untargeted metabolomic approach was used in the current study. Briefly, mass chromatograms and spectra were acquired by MassLynx software in centroid format from a mass-to-charge ratio (m/z) of 50 to 1000. Following deconvolution by MarkerLynx software, a multivariate data matrix containing information on sample identity, ion identity (retention time and m/z), and ion abundance was produced through deisotoping, filtering, peak recognition, and integration. We excluded INH and known INH metabolites (Li et al., 2011b) from the data matrix to focus on endobiotics in the liver. Principal component analysis (PCA) and orthogonal projection to latent structures-discriminant analysis were further conducted on Pareto-scaled data to explore INH-mediated changes in liver metabolome. We next focused on the endogenous metabolites that were significantly altered after INH treatment.
Statistics.
All quantified data are expressed as the mean ± S.E.M. Statistical analysis was conducted using ANOVA. A p value of <0.05 was considered to be statistically significant.
Results and Discussion
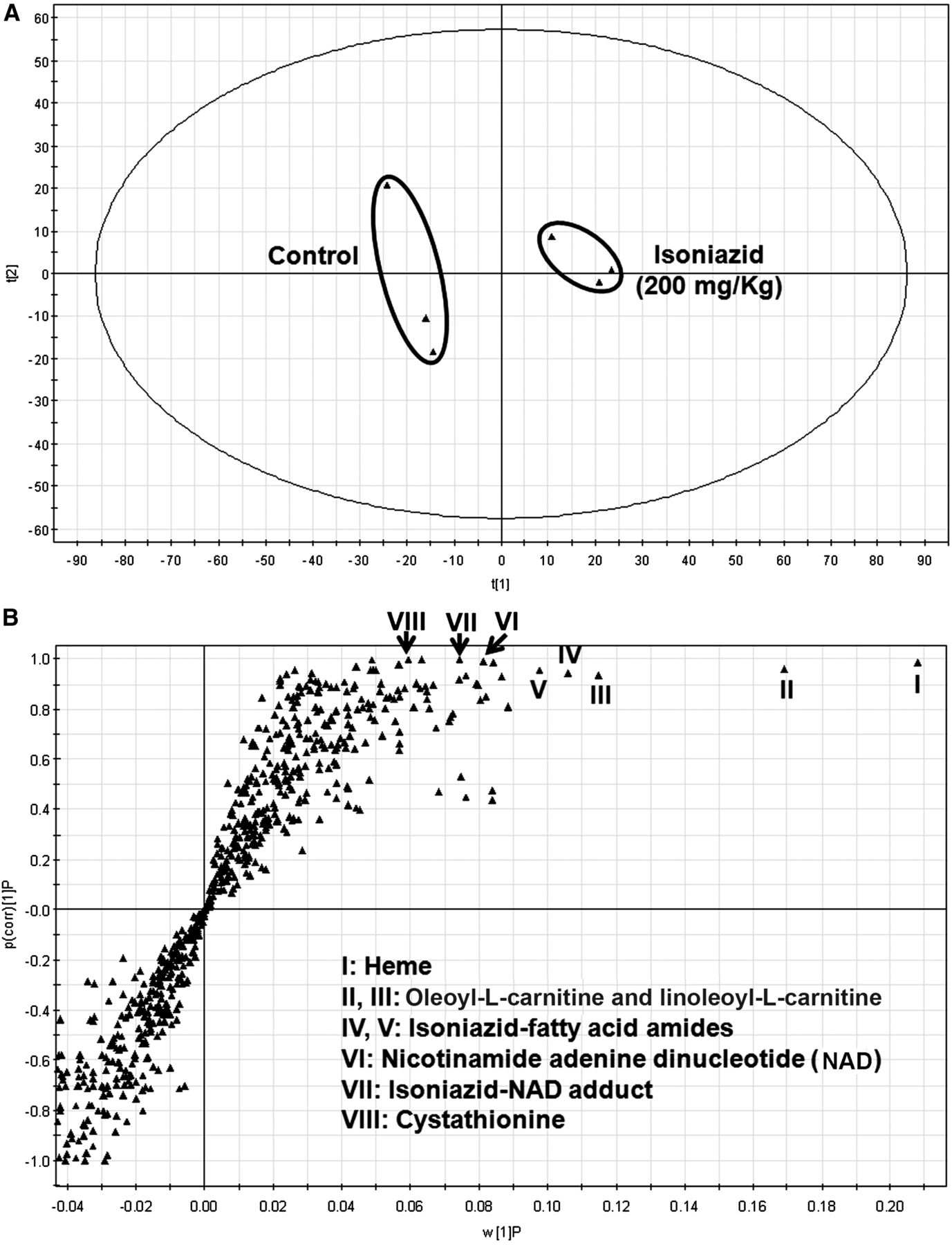
Metabolomics has revealed an elevation of bile acids, carnitine, and carnitine derivatives in the urine and bile of mice treated in the long term with a pharmacological dose of INH (Cheng et al., 2013). In addition, disturbance of the heme synthesis pathway has been found in pregnane X receptor–humanized mice cotreated in the long term with rifampicin and INH (Li et al., 2013). However, limited information is available on the acute effects of INH on endobiotic homeostasis in the liver. In the current study, an untargeted metabolomic approach was used to address this topic in mice. The PCA analysis revealed two clusters corresponding to the control and 200 mg/kg INH-treated groups in a score plot (Fig. 1A). The corresponding S-plot displays the ion contribution to the group separation (Fig. 1B). The top ranking ions were identified as heme (I), oleoyl-l-carnitine (II), and linoleoyl-l-carnitine (III), INH (IV)-fatty acid amides (V), NAD (VI), INH-NAD adduct (VII), and cystathionine (VIII). The serum metabolites were also profiled using the same method. However, the biomarkers identified in the liver cannot be found in the serum of mice treated with INH (Supplemental Table 1 and Supplemental Fig. 1). In addition, treatment with INH had no significant effect on serum alanine transaminase and aspartate transaminase activities (Supplemental Fig. 2).

Metabolomic analysis of mouse liver from the control and INH-treated groups. Wild-type mice were treated with vehicle or 200 mg/kg INH (by mouth). Liver samples were collected 30 minutes after treatment. All samples were analyzed by UPLC-QTOFMS. (A) Separation of control and INH-treated groups in a PCA score plot. The t[1] and t[2] values represent the score of each sample in principal components 1 and 2, respectively. (B) Loading S-plot generated by orthogonal projection to latent structures discriminant analysis. w[1]P, measure of the relative abundance of ions; p(corr)[1]P, measure of the correlation of each ion to the model.
INH-Mediated Accumulation of Heme (I) in the Liver.
Metabolite I was eluted at 6.03 minutes (Fig. 2A). Tandem mass spectrometry (MS/MS) of metabolite I revealed the major product ions at m/z values of 616, 557, and 498 (Fig. 2B), which exactly matched the fragments of heme. The structure of metabolite I was further confirmed by comparing the retention time and MS/MS results to the authentic standard of heme. The INH-mediated accumulation of heme in mouse liver is dose dependent. Compared with the control group, the levels of heme elevated 4- and 10-fold, respectively, in the 50 and 200 mg/kg groups (Fig. 2C).

Accumulation of heme (I) in the livers of mice treated with INH. (A) The extracted chromatogram of heme. (B) MS/MS findings of heme. (C) Relative abundance of heme. The data are expressed as the mean ± SEM (n = 3). ***p < 0.001 versus control group.
Heme is a functional component of a variety of critical cellular proteins and is involved in numerous physiologic processes, such as oxygen transport, signal transduction, and cell differentiation and proliferation (Beri and Chandra, 1993; Ponka, 1999). However, an excessive amount of free heme can cause membrane lipid peroxidation and the formation of reactive oxygen species, which eventually lead to the impairment of lipid bilayers and organelles, such as nuclei and mitochondria (Ryter and Tyrrell, 2000). Therefore, a high level of free heme will result in oxidative stress and tissue damage (Wagener et al., 2001, 2003). We found that the levels of free heme increased in mouse liver shortly after INH treatment, suggesting that heme may contribute in part to liver damage during acute poisoning with INH.
Heme homeostasis is tightly controlled. The biosynthetic pathway of heme includes eight enzymatic reactions that take place partly in the mitochondria and partly in the cytoplasm, whereas heme degradation is mediated by heme oxygenase 1 (Tenhunen et al., 1968, 1969). The possible mechanisms of INH-mediated heme accumulation include: (1) the induction of heme biosynthesis, and (2) inhibition of heme oxygenase 1–mediated heme degradation. Because the levels of heme in the liver increased shortly after INH treatment, it is more likely that INH inhibits heme degradation rather than the induction of heme biosynthesis. Further studies are needed to determine the mechanism by which INH disturbs heme homeostasis during acute poisoning with INH.
INH-Mediated Accumulation of Oleoyl-l-Carnitine and Linoleoyl-l-Carnitine in the Liver.
Metabolite II was eluted at 7.48 minutes (Fig. 3A). The MS/MS of metabolite II produced the major ions at m/z values of 426, 367, 144, and 85 (Fig. 3B). Metabolite III was eluted at 7.15 minutes (Fig. 3A), with a molecule [M+H]+ at an m/z value of 424. The major fragmental ions of III included m/z values of 365, 144, and 85 (Fig. 3C). Both metabolites II and III were identified as acylcarnitines because of their typical MS fragments at 85 and 144. Their structures were further determined as oleoyl-l-carnitine and linoleoyl-l-carnitine, respectively, by comparing the retention time and MS/MS findings to the authentic standards. The INH-mediated accumulation of oleoyl-l-carnitine and linoleoyl-l-carnitine was dose dependent (Fig. 3D).

Accumulation of oleoyl-l-carnitine (II) and linoleoyl-l-carnitine (metabolite III) in the livers of mice treated with INH. (A) Extracted chromatograms of acylcarnitines II and III. (B) MS/MS findings of oleoyl-l-carnitine (II). (C) MS/MS findings of linoleoyl-l-carnitine (III). (D) Relative abundance of acylcarnitines II and III in the liver. The data are expressed as the mean ± SEM (n = 3). **p < 0.01, ***p < 0.001 versus control group.
Mitochondrial dysfunction is one of the major mechanisms of drug-induced hepatotoxicity (Labbe et al., 2008; Pessayre et al., 2010, 2012). Long-chain fatty acid acylcarnitines are intermediates in fatty acid β-oxidation that occurs in mitochondria. Therefore, the accumulation of oleoyl-l-carnitine and linoleoyl-l-carnitine suggests mitochondrial dysfunction (Makowski et al., 2009; Sampey et al., 2012). Studies have found that an overdose of acetaminophen, a well-known hepatotoxin targeting mitochondria, leads to acylcarnitine accumulation in mice (Chen et al., 2009; McGill et al., 2014). In the current study, we found that overdose of INH resulted in acylcarnitine accumulation in the liver, indicating that a high dose of INH causes mitochondrial dysfunction. This finding is supported by the fact that hydrazine, an INH metabolite, suppresses mitochondria complex II (Lee et al., 2013; Boelsterli and Lee, 2014).
Formation of INH-Fatty Acid Amides (Metabolites IV and V).
Metabolites IV and V were eluted at 8.85 and 8.31 minutes, respectively (Fig. 4A). The mol. wt. difference of metabolites IV and V is 2 Da, but both of them have the major fragments of INH at m/z values of 138 and 121 (Fig. 4, B and C), suggesting conjugation with INH. We further proved that metabolites IV and V are amides formed through interactions between INH and fatty acids. With the increase of INH dosage, the formation of INH-fatty acid amides IV and V increased significantly (Fig. 4D).

Formation of INH-fatty acid amides (IV and V). (A) Extracted chromatograms of amides IV and V. (B) MS/MS findings of amide IV. (C) MS/MS findings of amide V. (D) Relative abundance of amides IV and V. The data are expressed as the mean ± SEM (n = 3). **p < 0.01 versus control group. (E) The scheme of the formation of INH-fatty acid amides. N.D., not detected.
The following possible mechanism for the formation of INH-fatty acid amides (metabolites IV and V) was proposed: (1) fatty acyl-CoA synthetase catalyzes fatty acids to form fatty acyl-CoAs; and (2) INH attacks fatty acyl-CoAs to form amides (Fig. 4E). The formation of fatty-acyl-CoAs is an important step in fatty acid β-oxidation. Therefore, the interaction between INH and fatty-acyl-CoAs would reduce fatty acid β-oxidation. Together with the INH-mediated accumulation of oleoyl-l-carnitine and linoleoyl-l-carnitine (Fig. 3), our results suggest that an overdose of INH suppresses fatty acid metabolism, which may explain in part the mechanism of INH-induced fatty liver in mice (Richards et al., 2004; Church et al., 2014).
INH-Mediated NAD (Metabolite VI) Accumulation in the Liver.
Metabolite VI was eluted at 0.87 minute and had a protonated molecule [M+H]+ at an m/z value of 664. The MS/MS of metabolite VI produced the major ions at m/z values of 542, 524, 428, 348, 232, and 136, which exactly matched the fragments of NAD (Fig. 5A). The structure of metabolite VI was further confirmed by comparing the retention time and MS/MS findings to the authentic standard of NAD. The INH-mediated NAD accumulation in mouse liver is dose dependent (Fig. 5B).
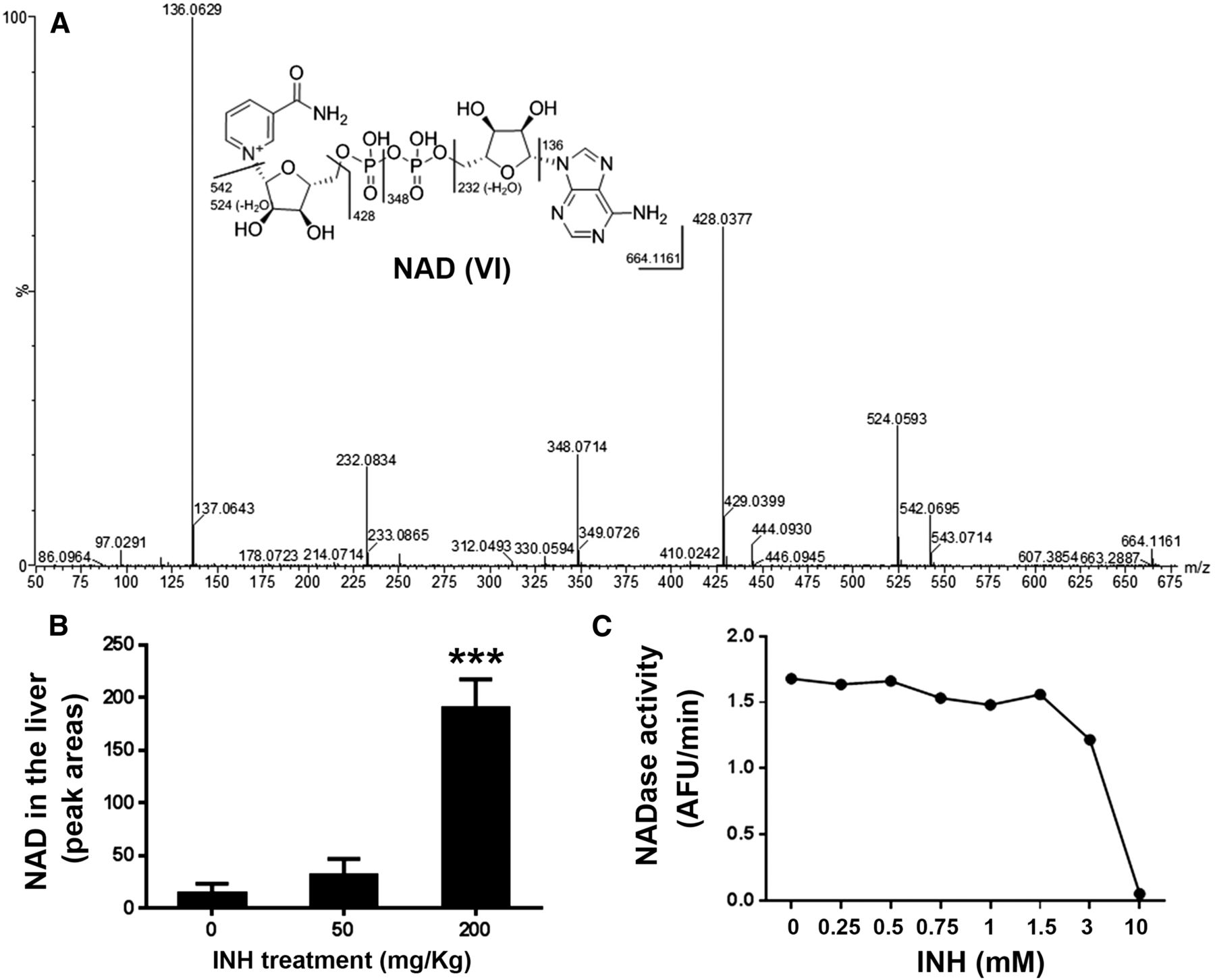
Accumulation of NAD (VI) in the livers of mice treated with INH. (A) MS/MS findings of NAD. (B) Relative abundance of NAD in the liver. The data are expressed as the mean ± SEM (n = 3). ***p < 0.001 versus control group. (C) The inhibitory effect of INH on NADase activity.
NAD, a coenzyme in redox reactions, serves as an electron carrier in fatty acid β-oxidation, glycolysis, and the Krebs cycle (Pollak et al., 2007). In addition to redox reactions, NAD participates in post-translational modifications of proteins, including NAD-dependent deacetylation, and mono- and poly-ADP-ribosylation (Dolle et al., 2013). NAD-dependent deacetylation is a key mechanism to control the activities of many mitochondrial enzymes that are involved in the Krebs cycle, fatty acid metabolism, antioxidant response, oxidative phosphorylation, and amino acid catabolism (Dolle et al., 2013; Nikiforov et al., 2015). Therefore, INH-mediated elevation of NAD in the liver may disrupt hepatic energy metabolism.
NAD is synthesized either in a de novo pathway from amino acids or in salvage pathways by recycling preformed components, such as nicotinamide, back to NAD (Nikiforov et al., 2015). However, the synthetic pathways may not be important in the INH-mediated NAD accumulation in the liver, because: (1) the synthesis of NAD may require a certain time to build up a high level of NAD, and (2) NAD accumulation occurs sharply after INH treatment. Based on the role of NAD in redox reactions, protein deacetylation, and ADP-ribosylation (Pollak et al., 2007; Dolle et al., 2013), it is possible that overdose of INH inhibits the functions of the NAD-dependent enzymes. In addition, NAD can be hydrolyzed to ADP-ribose and nicotinamide in mitochondria by NADase (Nakazawa et al., 1968; Zhang et al., 1995). We found that INH dose dependently inhibits NADase activity (Fig. 5C), suggesting that the INH-mediated accumulation of NAD is in part caused by INH-mediated NADase inhibition. Further studies are needed to determine the detailed mechanism by which an overdose of INH leads to NAD accumulation in the liver.
Formation of INH-NAD Adduct (Metabolite VII).
Metabolite VII had a protonated molecule [M+H]+ at an m/z value of 679. MS/MS of metabolite VII produced the primary ions at m/z values of 542, 524, 428, and 136 (Fig. 6A), which are exactly the same as for the fragments of NAD (Fig. 5A). However, the mol. wt. of metabolite VII is 15 Da larger than NAD. We proposed the metabolite VII as an INH and NAD adduct in which the nicotinamide moiety in NAD was replaced by INH, because the mol. wt. difference of INH and nicotinamide is 15 Da. The structure of the INH-NAD adduct was further confirmed by the studies using d4-INH (Fig. 6B). In addition, the structure of the INH-NAD adduct reported in the current study was different from that of the previously reported INH-NAD adduct generated by the mycobacterial catalase-peroxidase enzyme KatG or host INH activation (Zhang et al., 1992; Mahapatra et al., 2012).

Formation of INH-NAD adduct (VII) in the livers of mice treated with INH. (A) MS/MS findings of INH-NAD adduct. (B) MS/MS findings of d4-INH-NAD adduct. (C) Relative abundance of INH-NAD adduct in the liver. (D) The role of NADase in the formation of INH-NAD adduct. The quantified data are expressed as the mean ± SEM (n = 3). N.D., not detected. ***p < 0.001 versus control group.
Formation of an INH-NAD adduct is dose dependent (Fig. 6C). In the control group, no INH-NAD was detected. However, a considerable abundance of INH-NAD was detected in the livers of mice treated with 50 mg/kg INH, and it came to be much more abundant in the group treated with 200 mg/kg (Fig. 6C). We proposed that the formation of the INH-NAD adduct is mediated by NADase. In addition to the activity of hydrolysis, NADase catalyzes the base-exchange reaction, where it transfers the ADP-ribose moiety of NAD to pyridine derivatives, such as nicotinamide and thionicotinamide (Nakazawa et al., 1968; Chini, 2009). INH contains a pyridine ring and its structure is similar to nicotinamide. Therefore, NADase may incorporate INH and ADP-ribose to form the INH-NAD adduct. We recaptured the formation of INH-NAD adduct using the purified NADase when it was coincubated with equimolar amounts of INH and NAD (Fig. 6D). Further studies are needed to characterize the functions of INH-NAD adduct and its association with INH-induced liver damage.
INH-Mediated Accumulation of Cystathionine (Metabolite VIII).
Metabolite VIII was eluted at 0.81 minute (Fig. 7A). Metabolite VIII had a protonated molecule [M+H]+ at an m/z value of 223, with major fragments at m/z values 134 and 88, which exactly matched that of cystathionine (Fig. 7B). The structure of cystathionine was further confirmed by comparing the retention time and MS/MS findings to the authentic standard of cystathionine. INH-mediated accumulation of cystathionine is dose dependent (Fig. 7C).

Accumulation of cystathionine (VIII) in the livers of mice treated with INH. (A) The extracted chromatogram of cystathionine from the liver. (B) MS/MS findings of cystathionine. (C) Relative abundance of cystathionine in the liver. (D) MS/MS findings of INH-PL adduct. (E) Relative abundance of PLP in the liver. All quantified data are expressed as the mean ± SEM (n = 3). **p < 0.01, ***p < 0.001 versus control group. (F) The proposed mechanism by which INH disturbs cystathionine homeostasis. CSE, cystathionine gamma-lyase.
Cystathionine is an important intermediate in the transsulfuration pathway (Stipanuk, 1986). Cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE) are the enzymes for the synthesizing and cleaving of cystathionine, respectively. Both CBS and CSE require the active form of vitamin B6 (PLP) as a cofactor (Binkley et al., 1952). INH can cause vitamin B6 deficiency in part due to the formation of the INH and vitamin B6 adduct (Mandel, 1959). In the current study, an INH-PL adduct was detected in the serum of mice treated with INH (Fig. 7D). In addition, the concentrations of PLP in the liver were dramatically decreased after INH treatment (Fig. 7E). Although PLP is needed for both cystathionine synthesis and degradation, PLP deficiency affects CSE activity more than it affects CBS activity (Binkley et al., 1952; Hope, 1964). Therefore, we propose that the accumulation of cystathionine in the liver after INH overdose is due to PLP deficiency (Fig. 7F).
In the current study, we used an untargeted metabolomic approach to explore the INH-mediated global changes of endobiotic metabolites in mouse liver. We found that overdose of INH resulted in the accumulation of oleoyl-l-carnitine and linoleoyl-l-carnitine in the liver, indicating mitochondrial dysfunction. We also revealed the interactions between INH and fatty acyl-CoAs by identifying INH-fatty acid amides. In addition, we found that overdose of INH led to the accumulation of heme, NAD, and INH-NAD adduct in the liver. Furthermore, we illustrated that overdose of INH depleted vitamin B6 in the liver and blocked vitamin B6-dependent cystathionine degradation. All of these hepatic biomarkers of INH presented in a dose-dependent manner, suggesting that a pharmacological dosage of INH can also interrupt the metabolism of endobiotics, especially for the slow metabolizers of N-acetyltransferase 2, in which INH metabolism is suppressed (Chen et al., 2006; Zabost et al., 2013). In summary, our data suggest that INH can disturb multiple endogenous pathways in the liver (Fig. 8), which are potentially associated with INH-induced liver toxicity.
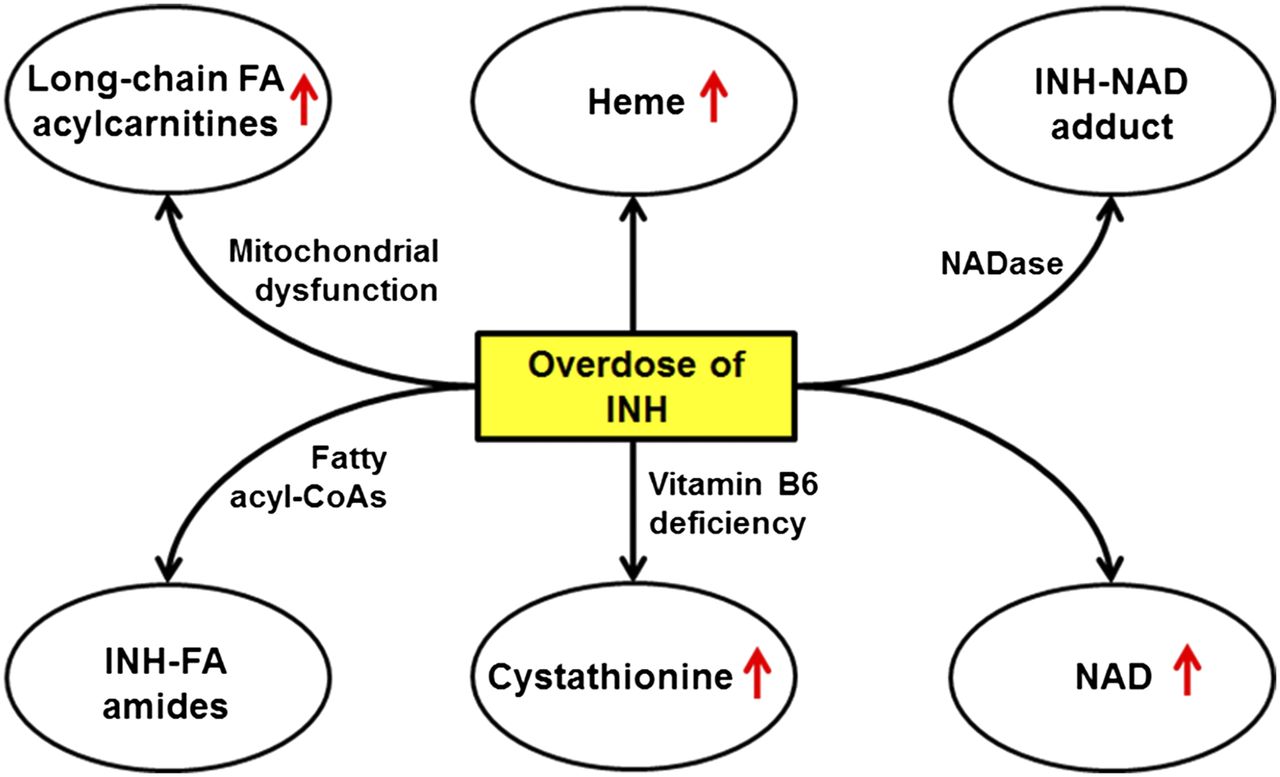
The scheme of disturbance of endogenous pathways in mouse liver by a high dose of INH. FA, fatty acid.
Authorship Contributions
Participated in research design: Li, Wang, and Ma
Conducted experiments: Li, Wang, Liu, Tarrago, and Lu
Contributed the new reagents or analytic tools: Li, Wang, Liu, Tarrago, and Chini
Performed data analysis: Li, Wang, Tarrago, and Ma
Wrote or contributed to the writing of manuscript: Li, Wang, and Ma
Footnotes
- Received April 9, 2016.
- Accepted August 15, 2016.
F.L. and P.W. contributed equally to this work.
This work was supported in part by the National Institute of Diabetes and Digestive and Kidney Diseases [Grant number DK090305]. The authors declare that there are no conflicts of interest.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- CBS
- cystathionine b-synthase; CSE, cystathionine g-lyase
- HFBA
- heptafluorobutyric acid
- INH
- isoniazid
- MS/MS
- tandem mass spectrometry
- m/z
- mass-to-charge ratio
- NAD
- nicotinamide adenine dinucleotide
- PCA
- principal component analysis
- PL
- pyridoxal
- PLP
- pyridoxal phosphate
- QTOFMS
- quadrupole time-of-flight mass spectrometry
- TB
- tuberculosis
- UPLC
- ultraperformance liquid chromatography
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}