


Abstract
PSC 833, a nonimmunosuppressive cyclosporin, is a potent inhibitor of the efflux of antitumor drugs mediated by P-glycoprotein and thus has been introduced in clinical trials as an agent to overcome multidrug resistance. The purpose of this study was to evaluate the dose-dependent pharmacokinetics of PSC 833 and its effects on the biliary excretion of endogenous substrates in rats. The major elimination route for PSC 833 is metabolism, followed by excretion into bile. The biliary clearance of PSC 833 was reduced in a dose-dependent manner, whereas no urinary excretion of PSC 833 was detectable. The tissue/blood concentration ratios for PSC 833 in the liver, kidney, intestine, and spleen were reduced in a dose-dependent manner, suggesting the presence of a saturable uptake process and/or saturable binding in these tissues. The dose-dependent increase in the tissue/blood concentration ratio in the brain suggests the presence of efflux transporters on the blood-brain barrier. PSC 833 reduced the bile flow rate by decreasing the biliary excretion of bile acids and reduced and oxidized glutathione, in a dose-dependent manner. The mechanism for the dose-dependent disposition of PSC 833 and its effects on the biliary excretion of endogenous substrates could be related to interactions with transporters.
PSC 833, a nonimmunosuppressive CsA1 derivative, is now under clinical investigation as a modulator of MDR acquired by overexpression of P-gp (an active transporter responsible for the extrusion of certain antitumor drugs from cells), because of its potent ability to inhibit P-gp function (Keller et al., 1992; Watanabe et al., 1995b). Because previous studies from this and other laboratories suggested that P-gp expressed in hepatocytes and renal tubular epithelial cells mediates the biliary and urinary excretion, respectively, of its substrates (Watanabe et al., 1992,1995a; de Lannoy et al., 1994), it is possible that the disposition of certain antitumor drugs might be altered by the concomitant administration of MDR modulators (Lum et al., 1993). In addition, we and others have indicated that modulation of the function of P-gp, which is expressed on the cerebral endothelial cells that form the blood-brain barrier, might result in enhanced brain entry of certain drugs (Kusuhara et al., 1998; Lemaire et al., 1996). Based on these findings, the effect of MDR modulators on the disposition of P-gp substrates has been the subject of extensive investigation; the effects of PSC 833 on the disposition of P-gp substrates such as vincristine, vinblastine, digoxin, Adriamycin, and colchicine have been reported (de Lannoy et al., 1994; Speeg and Maldonado, 1994).
Because the extent of the inhibitory effect of PSC 833 on P-gp function should be closely related to its blood concentrations, information on the disposition of PSC 833 is particularly important. At the present time, however, only one report is available on the disposition of PSC 833; Lush et al. (1997) analyzed the blood concentration profiles of PSC 833 after iv and oral administration to 14 subjects, at doses of 3 and 9 mg/kg, respectively, using HPLC. Extensive interindividual variation in the pharmacokinetic parameters was observed, and a total body clearance of 2.62 ± 1.90 ml/min/kg (mean ± SD) and an absolute bioavailability of 34% (range, 3–58%) were reported. The purpose of the present study was to determine the pharmacokinetics of PSC 833 in rats, with particular attention to the dose-dependent tissue distribution.
Materials and Methods
Materials.
[3H-Val]PSC 833 (54.3 or 0.344 Ci/mmol) and unlabeled PSC 833 were supplied by Novartis Pharma (Basel, Switzerland). All other reagents were of reagent grade. Female Sprague-Dawley rats (10 weeks of age, weighing 220–270 g) were purchased from Charles River Japan (Tokyo, Japan). The animals had free access to water and food. The animal experiments were performed according to the guidelines provided by the Institutional Animal Care Committee (Graduate School of Pharmaceutical Sciences, The University of Tokyo).
In Vivo Study.
The Sprague-Dawley rats underwent insertion of cannulas into the femoral vein and artery, using polyethylene tubing (polyethylene-50; i.d., 0.58 mm; o.d., 0.9655 mm; Becton Dickinson & Co., Parsippany, NJ). The bile duct was also cannulated with polyethylene tubing (polyethylene-10; i.d., 0.28 mm; o.d., 0.61 mm), as was the bladder (polyethylene-8; o.d., 2.33 mm; Hibiki Co., Tokyo, Japan), under light anesthesia with phenobarbital. The body temperature of the rats was maintained by heat from a lamp. Twenty minutes before the iv injection of PSC 833, a control bile specimen was collected. Rats received iv injections of 0, 0.1, 0.3, 3, 10, or 30 mg/kg PSC 833 dissolved in vehicle consisting of polyethylene glycol/ethanol (80:20, v/v). The injected volume of vehicle was 800 μl/kg. Aliquots of approximately 0.4 ml of blood were collected at 5, 10, 15, 30, 60, 120, and 180 min after injection of PSC 833 and were transferred to Eppendorf tubes that had been pretreated with heparin. Bile specimens were collected at 0–5, 5–10, 10–20, 20–30, 30–40, 40–60, 60–90, 90–120, 120–150, and 150–180 min. In the present study, sodium citrate (40 mM) was added to the bile specimens to prevent conversion of GSH to GSSG (Eberle et al., 1981). The volume of sodium citrate solution was adjusted to provide a final concentration of 20 mM, taking into account the bile flow rate (approximately 10 μl/min/rat), e.g. for collection at 10-min intervals, the bile specimens were collected in tubes containing 100 μl of the sodium citrate solution. Thirty microliters of the diluted bile specimens were used to determine the amounts of bile acids, total glutathione, and GSH. Urine specimens were collected at 0–30, 30–60, 60–120, and 120–180 min. Bile, urine, and blood specimens were collected on ice and stored at −50°C until analysis.
Determination of Bile Acids and Glutathione.
Total bile acids in the bile were determined by an enzymatic method, and the content of bile acids was determined by examining the absorbance of formazan at 540 nm (Mashige et al., 1981). Total glutathione and GSSG levels were determined by an enzymatic method (Adams et al., 1983; Tietze, 1969). The GSH content was determined by measuring the absorbance of 2-nitro-5-thiobenzoic acid at 412 nm. To measure GSSG, an appropriate amount ofN-ethylmaleimide was added, followed by extraction with diethyl ether to remove GSH (Adams et al., 1983). The amount of GSH was calculated as the difference between total glutathione and GSSG.
HPLC Analysis.
Adrenal glands and other tissues were homogenized with 2 and 4 ml, respectively, of distilled water, using a Polytron homogenizer (model PT 10/35; Kinematika, Luzern, Switzerland); 1 ml of homogenate was used for analysis. Two milliliters of distilled water were added to the bile and urine specimens, and 0.5 ml of the diluted specimens was used for analysis. Unlabeled PSC 833 (40 μg) was added to 100 μl of blood and the prepared specimens as an internal standard. Then 1 ml of 180 mM HCl and 5 ml of t-butyl methyl ether were added to the specimens, which were shaken for 10 min and then centrifuged at 1600g for 10 min. The t-butyl methyl ether layer was collected and evaporated under a gentle nitrogen stream at 40°C. The residue was dissolved in 300 μl of a mixture of distilled water, methanol, and acetonitrile (30:20:50, v/v/v) and then mixed after addition of 5 ml of n-heptane. The mixture was centrifuged at 1600g for 2 min, the n-heptane layer was removed, and the remaining aqueous layer was used for HPLC.
The HPLC system consisted of a pump (model PU-980; Jusco Co., Tokyo, Japan), an analytical column (5 μm, C4 bonded phase, 150 × 4.6 mm, 300 Å, YMC-PACK C4-AP; Yamamura Chemical Laboratories, Kyoto, Japan), a guard column (5 μm, C-KGC-824–30 C, 300 Å; Yamamura Chemical Laboratories), a spectrophotometer (model L-4200; Hitachi, Tokyo, Japan), a column oven (model 860-CO; Jusco Co.), and a fraction collector (model SF-2120; Advantec Tokyo Kaisha, Tokyo, Japan). The wavelength for the analysis was 210 nm. The mobile phase consisted of acetonitrile and H2O (63:37, v/v). The flow rate was 0.8 ml/min.
Determination of Kinetic Parameters.
From the time profiles for the blood concentration of [3H]PSC 833, the AUC0–180min (in fraction of dose per milliliter-minute) was calculated by the logarithmic-trapezoidal rule. The cumulative amounts of ligands excreted into bile and urine up to time t (180 min) (Xbile,tand Xurine,t, in fraction of dose) were calculated as
Statistical Method.
The results are shown as the mean ± SE of the indicated number of determinations. One-way analysis of variance was used to determine the significance of differences among groups. The Fisher t test was used to determine the significance of differences between the means of two groups, with p < 0.05 as the minimal level of significance.
Results
Disposition of PSC 833.
Fig. 1 shows the dose-normalized blood concentration-time profiles for [3H]PSC 833 after iv administration of 0.1, 0.3, 3, 10, and 30 mg/kg. Pharmacokinetic analysis indicated that the dose-normalized AUC0–180min showed dose-related increases (table1). These increases in AUC values up to 3 hr (by 50%) with increases in the administered dose of PSC 833 represent the initial phase of disposition, because the plasma half-life of PSC 833 is extremely long (approximately 7 hr) (fig.1). In the present study, the total body clearance was not determined because of the presence of the terminal phase, with a much longer half-life (fig. 1). Measurement of the total radioactivity revealed that 25–38 and 6.9% of the administered isotope was recovered in bile within 3 hr after iv administration of [3H]PSC 833 at doses of 0.1–10 and 30 mg/kg, respectively, although ≤1.1% of dose was accounted for by intact [3H]PSC 833 in all experiments (table 1). In the same way, <2% of the injected dose was recovered in urine, and no urinary excretion of intact [3H]PSC 833 was detected (table 1).
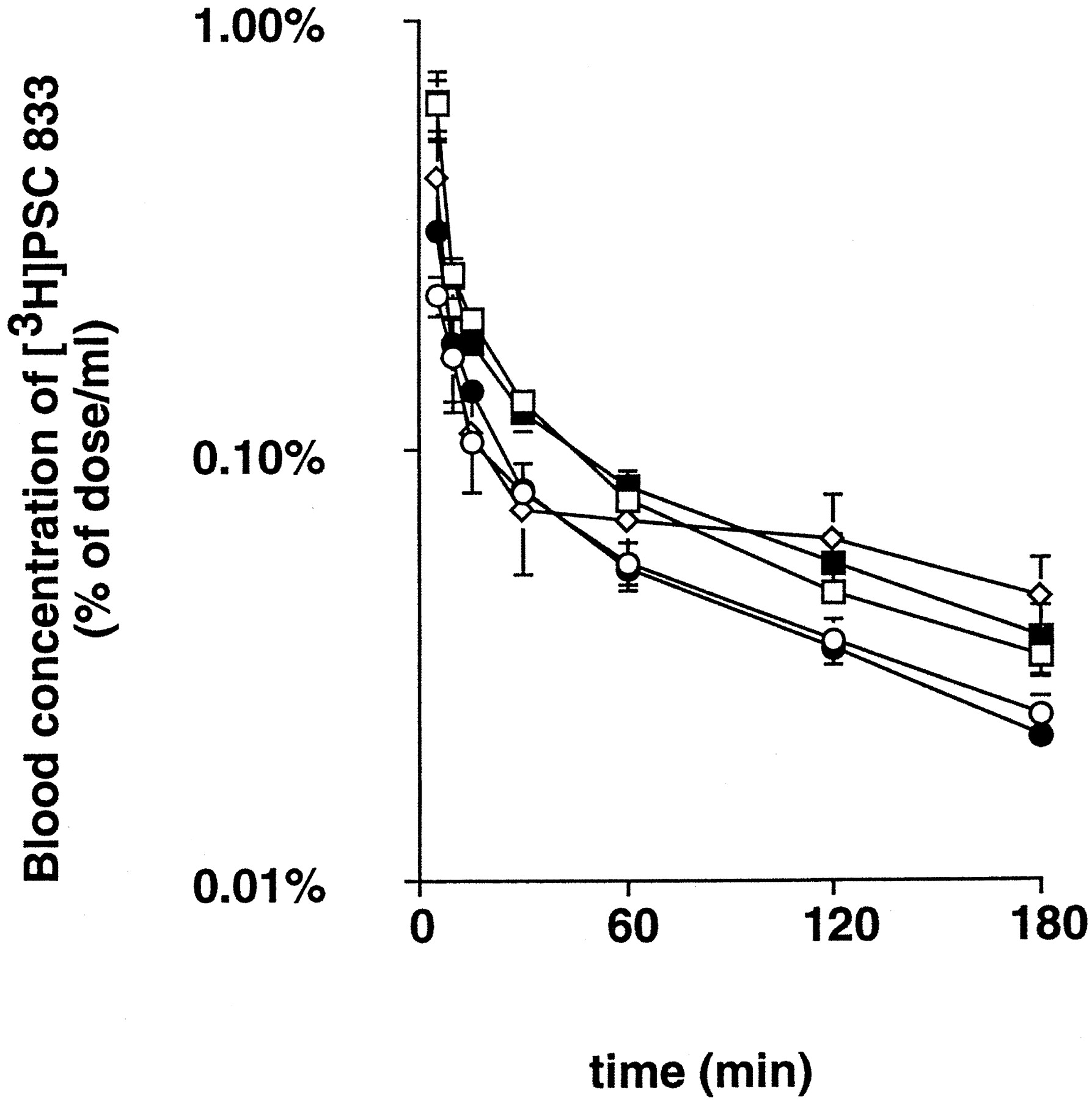
Blood concentration profiles for [3H]PSC 833 in rats.
[3H]PSC 833 was administered iv to Sprague-Dawley rats, after bile duct cannulation, at doses of 0.1 (○), 0.3 (•), 3 (■), 10 (▪), and 30 (⋄) mg/kg. Results are shown as the dose-normalized blood concentrations. All values are given as the mean ± SE of four independent experiments.
Pharmacokinetic parameters for [3H]PSC 833 after iv administration
The Kp values for [3H]PSC 833 at 3 hr after iv administration are listed in table 2. TheKp values for [3H]PSC 833 in the liver, kidney, intestine, and spleen were reduced in a dose-dependent manner (table 2). TheKp in the liver was significantly increased when the dose was increased from 10 to 30 mg/kg PSC 833. TheKp in the brain was significantly increased when the administered dose of PSC 833 was >3 mg/kg, whereas 30 mg/kg was required to increase the Kp in the lung.
Blood and tissue concentrations and tissue/blood partition coefficient for [3H]PSC 833
Dose-Dependent Effects of PSC 833 on the Bile Flow Rate and the Excretion of Bile Acids and Glutathione.
The bile flow rate was temporarily reduced after iv administration of PSC 833 at doses of 10 and 30 mg/kg (fig.2). The biliary excretion rate for bile acids was also reduced, in a reversible manner, at doses of 10 and 30 mg/kg (fig. 3). Similarly, the rates for GSSG and GSH were significantly reduced after administration of the same doses (figs. 4 and5); the rates measured after the dose of 30 mg/kg did not recover to the control levels before 180 min.

Effect of PSC 833 on the bile flow rate in rats.
[3H]PSC 833 was given iv to Sprague-Dawley rats, after bile duct cannulation, at doses of 0 (○), 0.1 (•), 0.3 (■), 3 (▪), 10 (⋄), and 30 (♦) mg/kg. All values are given as the mean ± SE of four to seven independent experiments. *,p < 0.05; **, p < 0.01, significantly different from control by Fisher t test.

Effect of PSC 833 on the biliary excretion of bile acids in rats.
[3H]PSC 833 was given iv to Sprague-Dawley rats, after bile duct cannulation, at doses of 0, 0.1, 0.3, 3, 10, and 30 mg/kg. All values are given as the mean ± SE of four to seven independent experiments. *, p < 0.05; **,p < 0.01, significantly different from control by Fisher t test.

Effect of PSC 833 on the biliary excretion of GSSG in rats.
[3H]PSC 833 was given iv to Sprague-Dawley rats, after bile duct cannulation, at doses of 0, 0.1, 0.3, 3, 10, and 30 mg/kg. All values are given as the mean ± SE of three or four independent experiments. *, p < 0.05; **,p < 0.01, significantly different from control by Fisher t test.

Effect of PSC 833 on the biliary excretion of GSH in rats.
[3H]PSC 833 was given iv to Sprague-Dawley rats, after bile duct cannulation, at doses of 0, 0.1, 0.3, 3, 10, and 30 mg/kg. All values are given as the mean ± SE of three or four independent experiments. *, p < 0.05; **,p < 0.01, significantly different from control by Fisher t test.
Discussion
In the present study, the disposition of PSC 833 was examined in rats. It is suggested that metabolism is the predominant pathway for the elimination of PSC 833, because a minimal amount of the radioactivity excreted in bile and urine was associated with intact [3H]PSC 833, although approximately 30% of the administered radioactivity was excreted in bile and urine by 3 hr after iv administration of 0.1 mg/kg [3H]PSC 833 (table 1). Extensive metabolism of [3H]CsA in rats has also been reported; Wagner et al. (1987) found that 47% of the administered radioactivity was recovered in bile by 24 hr after iv administration to rats, with minimal excretion of intact [3H]CsA. Although the major elimination route for PSC 833 has been shown to be metabolism, little is known about the metabolites in rats. In humans, it has been reported that P450 3A is responsible for the extensive metabolism of PSC 833 as well as CsA;Lush et al. (1997) reported that the predominant metabolite PSC M9, a metabolite of PSC 833 hydroxylated at the γ-position of L-methyl-leucine whose formation is catalyzed by P450 3A, accounts for approximately 20% of the parent compound in human blood. Unfortunately, no report is available on the effect of PSC M9 on the transport mediated by P-gp or other transporters, although Toffoliet al. (1997) reported that the hydroxylated and demethylated metabolites of CsA (M1, M17, andM21) did not affect the transport activity of P-gp at concentrations detected in the whole blood of patients (0.5–2.2 μM). In contrast, the metabolites of PSC 833 in rats and the interactions with transporters have not yet been determined. If we consider the previous belief that a substrate or inhibitor for P-gp can be a substrate for P450 3A (Wacher et al., 1995), it is possible that PSC 833, in turn, inhibits the metabolism of concomitantly administered drugs whose metabolism is mediated by the same enzyme. Because it has been reported that certain antitumor drugs are recognized by P-gp and P450 3A (Kivisto et al., 1995; Wacheret al., 1995), it is possible that the altered disposition of these drugs after administration of PSC 833 may be accounted for by the inhibition of both metabolism and efflux. CsA may preferentially inhibit drug transfer, rather than drug metabolism, in rats, however; the IC50 of CsA for α-hydroxylation of midazolam mediated by P450 3A in rat microsomes was found to be approximately 6 μM (Li et al., 1990), which is >15 times higher than the Ki value associated with the transport of daunorubicin mediated by P-gp located on rat bile canalicular membranes (Böhme et al., 1993). The kinetic parameters for the metabolism of PSC 833 by rat P450 3A enzymes have not been reported. Although AUC0–180min was increased by 50% by increases in the administered dose of PSC 833, the increase in AUC represents the initial phase of disposition, because the plasma half-life of PSC 833 is extremely long (approximately 7 hr) (fig. 1).
In addition, we determined the dose-dependent tissue disposition of PSC 833. The dose-dependent reduction in Kp values in the liver, kidney, intestine, and spleen (table 2) can be accounted for by saturable uptake and/or tissue binding. Although Table2 shows the total radioactivity in the liver, it is difficult to discuss the saturation of the metabolism of PSC 833 from these data because no information is available on the metabolic pathways or the hepatic elimination of metabolites in rats. Because the plasma half-life of PSC 833 is extremely long (approximately 7 hr) (fig. 1), the increase in the amount of PSC 833 associated with the liver at higher PSC 833 concentrations can be accounted for by saturation of biliary excretion across the bile canalicular membranes, although we cannot exclude possible saturation of metabolism. The dose-dependent increase in Kp in the brain (table 2) may result from saturation of the efflux transporter located on the cerebral endothelial cells that form the blood-brain barrier. Our result is consistent with the previous report by Lemaire et al. (1996). They determined the blood and brain levels of total radioactivity 2 hr after iv administration of [3H]PSC 833, and they found that theKp in the brain was markedly increased if a dose of >3 mg/kg was administered; theKp values in the brain were 0.17, 0.93, and 1.4 after iv administration of 0.1, 3, and 10 mg/kg, respectively. Previously, we examined the dose-dependent effects of PSC 833 on the disposition of [3H]digoxin, another substrate for P-gp, at 6 hr after iv administration to rats and found that the same dose of PSC 833 (3 mg/kg) was required to increase theKp of digoxin in the brain (Song SH, Suzuki H, Kawai R and Sugiyama Y, submitted for publication). Our previous finding that, after the iv administration of 10 mg/kg, theKp of [3H]quinidine in the brain in wild-type mice was significantly increased, to a level comparable to that inmdr1a-knockout mice, supports the hypothesis that 10 mg/kg PSC 833 can completely inhibit the function of P-gp located on cerebral endothelial cells (Kusuhara et al., 1998). One of the most plausible explanations for the dose-dependent disposition of PSC 833 in the brain assumes that PSC 833 acts as a substrate for P-gp located on the luminal membranes of cerebral endothelial cells. However, the contribution of P-gp to the disposition of PSC 833 in vitrois controversial. Archinal-Mattheis et al. (1995) and Naitoet al. (1996) provided evidence that PSC 833 may be a poor substrate for P-gp. Archinal-Mattheis et al. (1995) examined the effect of verapamil on the disposition of PSC 833 by MDA-435 (a human breast adenocarcinoma cell line sensitive to chemotherapy) and its derived resistant cell lines exhibiting extensive P-gp expression. They found that the influx and efflux of PSC 833 did not differ between sensitive and resistant cell lines, and they observed no significant effect of verapamil on the transport of PSC 833 (Archinal-Mattheiset al., 1995). The disposition of PSC 833 was in marked contrast to that of CsA and vincristine, in that the authors observed verapamil-sensitive reduced influx and increased efflux in the resistant cell lines for these two P-gp substrates. The same conclusion was obtained by Naito et al. (1996), using the K562 human myelogenous leukemia line and its subline resistant to Adriamycin; the authors found no difference in the accumulation of PSC 833 between the two cell lines, whereas there was reduced accumulation of CsA in the resistant cells that was modulated by MRK 16, a monoclonal antibody against human P-gp. However, it is possible that, in some cell lines, the P-gp-mediated penetration of PSC 833 may be difficult to detect because of significant passive diffusion. Therefore, we cannot establish whether the dose-dependent increase in theKp of PSC 833 in the brain may be accounted for by saturation of P-gp. It is also possible that efflux transporters other than P-gp may be responsible for our observations. Although the total radioactivity associated with the brain is shown in table 2, it is difficult to assess the saturation of PSC 833 metabolism in the brain and the efflux of its metabolites across the blood-brain barrier from these data, because several metabolites (some of which represent sequential metabolites) have been found and the properties of metabolite transport across the blood-brain barrier have not been determined. In addition, it is possible that PSC 833 and/or its metabolite(s) can be extruded into the intestinal lumen.
Another finding suggests a possible interaction of PSC 833 with transporters; administration of >10 mg/kg PSC 833 temporarily reduced the bile flow rate (fig. 2), which was associated with reduced biliary excretion of bile acids (fig. 3). These results are consistent with the previous report by Böhme et al. (1994), who found a 50% reduction in bile flow rate 10 min after iv administration of PSC 833 (25 mg/kg); the rate then recovered to a normal level. Those authors also found reduced biliary excretion of [14C]taurocholic acid administered iv 10 min after injection of PSC 833 (Böhme et al., 1994). Because PSC 833 competitively inhibits the transport of taurocholic acid in in vitro experiments with isolated CMV (Böhmeet al., 1993), it is possible that the inhibition by PSC 833 of this export carrier results in a reduced bile acid-dependent bile flow rate.
In addition, it has been reported that approximately 50% of the bile flow rate is dependent on the excretion of conjugated metabolites (such as glutathione, glucuronide, and sulfate conjugates) (Erlinger, 1996). Because the biliary excretion of certain conjugates is mediated by the cMOAT, which was recently cloned in this and other laboratories (Paulusma et al., 1996; Büchler et al., 1996; Ito et al., 1996, 1997), and because Böhmeet al. (1993), using CMV, reported that PSC 833 inhibited cMOAT function, it is also possible that PSC 833 inhibits the excretion of these cMOAT substrates and thus reduces the bile acid-independent bile flow rate. In this study, we found that the biliary excretion of GSSG, a physiological substrate for cMOAT (Fernandez-Checa et al., 1992), was markedly reduced by PSC 833 up to 3 hr after administration (fig. 4). It was also found that the biliary excretion of GSH was reduced by administration of PSC 833 (fig. 5). Although the mechanism by which GSH is excreted into bile has not been clarified,Fernandez-Checa et al. (1992) proposed that GSH is transported across the bile canalicular membrane in exchange for GSSG. The decreased GSH excretion into bile may thus result from inhibition of the biliary excretion of GSSG mediated by cMOAT. Hyperbilirubinemia, which is one of the adverse effects of PSC 833 (Boote et al., 1996), may result from inhibition of cMOAT function, because bilirubin glucuronide is one of the physiological substrates for cMOAT (Oude Elferink et al., 1995; Yamazaki et al., 1996). Indeed, Boote et al. (1996) reported a positive correlation between the peak blood concentration of bilirubin and the mean blood concentration of PSC 833 in human subjects.
The present in vivo results should be discussed in relation to the previous in vitro data reported by Böhmeet al. (1993). Those authors determined theKi values of PSC 833 for the transport mediated by P-gp, the bile acid transporter, and cMOAT and obtained values of 0.3, 0.6, and 29 μM, respectively, by examining the inhibitory effects of PSC 833 on the ATP-dependent uptake of daunorubicin, taurocholic acid, and leukotriene C4 into CMV. With the assumption that the biliary excretion of PSC 833 is mediated by P-gp, their in vitrodata agree semiquantitatively with the present in vivoobservations; 3 mg/kg PSC 833 was required to significantly reduce the biliary excretion of both PSC 833 and taurocholic acid, whereas higher doses (>10 mg/kg) were required to inhibit the transport mediated by cMOAT (figs. 3-5).
In conclusion, PSC 833 is eliminated predominantly by metabolism. Possible inhibition of transporters other than P-gp was also demonstrated. The information on the pharmacokinetics of PSC 833 and its interaction with transporters for endogenous substrates is important for the proper modulation of MDR and for the reduction of antitumor drug toxicity possibly induced by the concomitant administration of PSC 833.
Footnotes
-
Send reprint requests to: Dr. Yuichi Sugiyama, Graduate School of Pharmaceutical Sciences, The University of Tokyo, 7–3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan.
-
This work was supported in part by a grant-in-aid from the Ministry of Education, Science, Sports, and Culture of Japan and by the Core Research for Evolutional Sciences and Technology of Japan Science and Technology Corp.
- Abbreviations used are::
- CsA
- cyclosporin A
- MDR
- multidrug resistance
- P-gp
- P-glycoprotein
- P450
- cytochrome P450
- cMOAT
- canalicular multispecific organic anion transporter
- Kp
- tissue/blood concentration ratio
- GSH
- reduced glutathione
- GSSG
- oxidized glutathione
- CMV
- canalicular membrane vesicles
- Received January 30, 1998.
- Accepted July 2, 1998.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}