


Abstract
(S,S)-3-[3-(Methylsulfonyl)phenyl]-1-propylpiperidine hydrochloride [(−)-OSU6162] is a weak dopamine D2 receptor modulator that possesses potential for the treatment of levodopa (l-DOPA)-induced dyskinesias in patients with Parkinson's disease. In this report, incubations with human liver microsomes revealed that (−)-OSU6162 is selectively metabolized viaN-dealkylation to yield N-depropyl (−)-OSU6162. Kinetics evidence is presented that theN-depropylation of (−)-OSU6162 in human hepatic microsomes is mediated by multiple cytochrome P450 (P450) enzymes, in particular CYP2D6. This hypothesis is borne out by several lines of in vitro evidence; 1) incubations of (−)-OSU6162 (5 μM) with hepatic microsomes from a panel of human donors showed that (−)-OSU6162N-depropylase activity correlated well with CYP2D6-catalyzed dextromethorphan O-demethylase activity but not with other P450 enzyme-specific activities; 2) quinidine, a CYP2D6-specific inhibitor, inhibited (−)-OSU6162N-depropylation, whereas other P450 enzyme-specific substrates/inhibitors did not significantly inhibit this activity; 3) CYP2D6 possessed highest intrinsic (−)-OSU6162N-depropylase activity when compared with a battery of recombinant heterologously expressed human P450 enzymes. In addition, the selectivity of (−)-OSU6162 to inhibit six human P450 enzymes (CYP1A2, CYP2C9, CYP2C19, CYP2E1, CYP2D6 and CYP3A4) was evaluated using an in vitro inhibition screen. Of the enzymes examined, only the activity of CYP2D6 was inhibited by coincubation with (−)-OSU6162. Thus, it is concluded that (−)-OSU6162 is metabolized by several P450 enzymes and that CYP2D6 accounts for the majority of the observed P450N-depropylase activity in vitro.
(S,S)-3-[3-(methylsulfonyl)phenyl]-1-propylpiperidine hydrochloride [(−)-OSU61621] is a substituted (S)-3-phenylpiperidine derivative, which exhibits some affinity to the dopamine D2 receptor family (Tedroff et al., 1998). In vivo, the compound displays a unique normalizing profile on psychomotor activity by an intriguing mixture of stimulatory and inhibitory properties (Ekesbo et al., 1997; Tahar et al., 2001). As a consequence, (−)-OSU6162 is under clinical development for the treatment of levodopa (l-DOPA)-induced dyskinesias in disease patients with Parkinson's disease.
As (−)-OSU6162 represents a potentially useful therapeutic agent in the treatment of Parkinson's disease, knowledge concerning the enzyme(s) involved in the metabolism of this drug in humans is of key interest (Peck et al., 1993). To this end, we set out to identify the hepatic enzymes responsible for (−)-OSU6162 metabolism and to examine the effect of (−)-OSU6162 upon hepatic cytochrome P450 activities. After characterizing the metabolic profile of (−)-OSU6162 in vitro, a combination of four different in vitro strategies was then used to identify the enzyme(s) responsible for (−)-OSU6162 metabolism, namely 1) kinetic analysis in human microsomal preparations, 2) correlation analysis using a human liver bank, 3) chemical inhibition, and 4) metabolism by recombinant human P450 enzymes. Moreover, additional experiments were conducted to examine the potential inhibitory effects of (−)-OSU6162 on various human cytochrome P450 enzymes using select human P450 activity marker substrates.
Experimental Procedures
Chemicals.
(−)-OSU6162 (Fig. 1) synthesized with a uniform carbon-14 radiolabel (specific activity = 0.142 mCi/mg) was obtained from Pharmacia Corporation (Kalamazoo, MI). The radiochemical purity of [14C](−)-OSU6162 was 99.8% as determined by high performance liquid chromatography (HPLC) with radiochemical detection. Other chemicals, including nonlabeled (−)-OSU6162, N-despropyl (−)-OSU6162, bropirimine (BROP), 3-cyclopentyloxy-N-(3,5-dichloro-4-pyridyl)-4-methoxybenzamide (CPMB), and [14C]delavirdine were obtained from Pharmacia Corporation. [14C](S)-mephenytoin, [14C]diclofenac and [14C]chlorzoxazone were purchased from Amersham Biosciences Inc. (Piscataway, NJ); [14C]testosterone was obtained from PerkinElmer Life Sciences (Boston, MA); [14C]para-nitrophenol, coumarin (COUM), sulfaphenazole (SULF), para-nitrophenol (NTRO), quinidine (QUIN), ketoconazole (KETO), and NADPH were purchased from Sigma-Aldrich (St. Louis, MO). (S)-mephenytoin (MEPH) was a gift from Dr. W. F. Trager, Department of Medicinal Chemistry, University of Washington (Seattle, WA). UltimaFlo M liquid scintillant was purchased from PerkinElmer Life Sciences. All other reagents and solvents were of analytical grade.

Typical HPLC radiochemical chromatographic profile for (−)-OSU6162 and major metabolite after incubation with human liver microsomes.
∗, radiochemical impurity.
Microsomes.
Human livers were acquired from the International Institute for the Advancement of Medicine (IIAM, Exton, PA). Liver microsomal protein isolation and the specific catalytic activity of individual enzymes of P450 were determined as previously described (Wienkers et al., 1996). Microsomes from a baculovirus-insect cell line (Supersomes) expressing CYP1A1, CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4, and CYP4A11 were purchased from BD Gentest (Woburn, MA).
Incubation Conditions.
Initial incubations to optimize metabolite formation with protein concentration (range 0.1, 0.3, 0.5, 0.7, and 1 mg/ml) and time (15, 30, 45 and 60 min) were conducted to ensure product linearity. From these studies, a typical incubation (final volume 0.2 ml) consisted of 0.3 mg/ml microsomal protein in 100 mM potassium phosphate buffer (pH 7.4). Stock solutions of (−)-OSU6162 were prepared in methanol. The drug was added to each incubation tube, and the solvent evaporated at 37°C. The residue was reconstituted with assay buffer containing microsomes. Reactions were started by the addition of NADPH (1 mM final concentration) and continued for 30 min at 37°C. For control incubations, NADPH was omitted. Reactions were terminated upon addition of 100 μl of acetonitrile (ACN), after which samples were vortex mixed and centrifuged for 15 min at 14000g. The subsequent supernatants were transferred to a HPLC autosampler vial, capped, and samples were kept refrigerated until radio-HPLC analysis.
Radio-HPLC.
Analytical separation of (−)-OSU6162 and its metabolite was achieved using a HPLC system equipped with a PerkinElmer series 200 pump and autosampler (PerkinElmer Instruments, Norwalk, CT) equipped with a chilled sample tray maintained at 8°C. The analytical column was a reverse-phase YMC-phenyl (250 × 4.6 mm, 5 μm particle size; YMC Inc., Wilmington, NC). The mobile phase consisted of water/methanol:acetonitrile/trifluoroacetic acid 85:7.5:7.5:0.1% run isocratically at a flow rate of 1 ml/min. Quantitation of (−)-OSU6162 and its metabolite was performed using a FLO-ONE Series A500 flow-through radioactivity detector (PerkinElmer Life Sciences), and peak areas were integrated with Windows-based Radio-HPLC Workstation software (FLO-ONE for Windows). UltimaFlo M liquid scintillant was introduced postcolumn at a rate of 2.5 ml/min. Rates of formation of the (−)-OSU6162 N-despropyl metabolite were determined from the fractional conversion of (−)-OSU6162 apparent from the radiochromatogram. Peaks were identified using retention time comparison with authentic (−)-OSU6162 andN-despropyl (−)-OSU6162 and structure confirmed using LC/MS.
LC/APCI/MS and Metabolite Confirmation.
The identity of the primary in vitro (−)-OSU6162 metabolite was confirmed using a Finnigan LCQ ion-trap (Thermo Finnigan MAT, San Jose, CA) operated in positive-ion atmospheric pressure chemical ionization APCI mode. The APCI vaporizer temperature was 450°C, and the discharge current and spray voltage were set at 5 μA and 4.5 kV, respectively. Nitrogen (99.9% pure; AGA Gas Inc., Maumee, OH) was employed as a drying gas at a sheath pressure of 80 psi and auxiliary flow rate of 20 ml/min, and the heated capillary was set at 250°C. Analytical separation of (−)-OSU6162 and its metabolite was accomplished using the HPLC conditions described above. Under these conditions, authentic standards of (−)-OSU6162 and the correspondingN-despropyl derivative were characterized by retention time, molecular ion, and fragmentation pattern. The collision energy used was 1.3 and 1.6 V for (−)-OSU6162 and the metabolite, respectively.
Kinetic Analysis.
Rates of formation of the N-despropyl metabolite of (−)-OSU6162 were determined in the range of 10 to 1000 μM substrate under in vitro conditions that were linear with respect to protein concentration and time of incubation. The kinetic parameters were evaluated in pooled human liver microsomes and from individual samples HL28 and HL40.
Correlation Analysis.
The rates of formation of the primary metabolite of (−)-OSU6162 (5 μM and 200 μM) were determined in a panel of liver microsomes prepared from 12 different human organ donors and compared with the catalytic activities previously characterized for specific P450 substrates (Wienkers et al., 1996). Incubations and sample work-up were carried out as described above. Coefficient of determination (r2) for enzyme activities was determined by linear regression analysis using the graphical/statistical program Prism 2.01 (GraphPad Software Inc., San Diego, CA).
Chemical Inhibition Experiments.
(−)-OSU6162 was incubated at a single concentration in pooled human liver microsomes in the presence of a panel of compounds, which interacted selectively with various cytochrome P450 enzymes. The following P450 enzyme substrates/inhibitors were examined for their ability to inhibit the microsomal metabolism of (−)-OSU6162: bropirimine/CYP1A2, BROP (200 μM); coumarin/CYP2A6, COUM (20 μM); 3-cyclopentyloxy-N-(3,5-dichloro-4-pyridyl)-4-methoxybenzamide/CYP2B6, CPMB (20 μM); sulfaphenazole/CYP2C9, SULF (5 μM); (S)-mephenytoin/CYP2C19, MEPH (200 μM); quinidine/CYP2D6, QUIN (5 μM); para-nitrophenol/CYP2E1, NTRO (50 μM); ketoconazole/CYP3A4, KETO (5 mM). The inhibitors were dissolved in ACN and were added to the incubations such that the final amount of solvent was 1%. Control incubations (minus inhibitor) also contained 1% ACN.
Incubations with Recombinant Human P450s.
The metabolism of (−)-OSU6162 was examined in microsomes prepared from a baculovirus-insect cell line (Supersomes) expressing CYP1A1, CYP1A2, CYP2A6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4, and CYP4A11. The incubations were conducted in a manner essentially as described above, with 200 mM of [14C](−)-OSU6162 and equivalent P450 protein concentrations (0.3 mg/ml) with cytochrome c reductase activities ranging between 20 to 55 nmol/(s × mg of protein) of each P450 enzyme in 100 mM potassium phosphate buffer, pH 7.4. Subsequently, the kinetic parameters were evaluated in the following selected P450 enzymes: 2D6, 3A4, 1A2, 2C19, and 1A1 using incubation conditions identical to those employed in the human microsomal kinetic studies.
P450 Inhibition Screen.
The ability of (−)-OSU6162 to inhibit select P450 enzyme activity was investigated against six different recombinant human cytochrome P450 enzyme systems (CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4). Incubations were conducted in triplicate and each incubation contained recombinant P450 microsomal protein (0.03–0.5 mg/ml), NADPH (1 mM final concentration), [14C]P450 marker substrate ([S] = Km), and (−)-OSU6162 at concentrations of 0 (minus inhibitor control), 1, 10, and 100 μM in a final volume of 0.1 ml of 100 mM, pH 7.4, potassium phosphate buffer. Incubation reactions, sample work-up, and quantitation of P450 marker metabolite formation using HPLC/radiochemical detection were conducted as previously described (Wynalda and Wienkers, 1997).
Results
Metabolism of (−)-OSU6162.
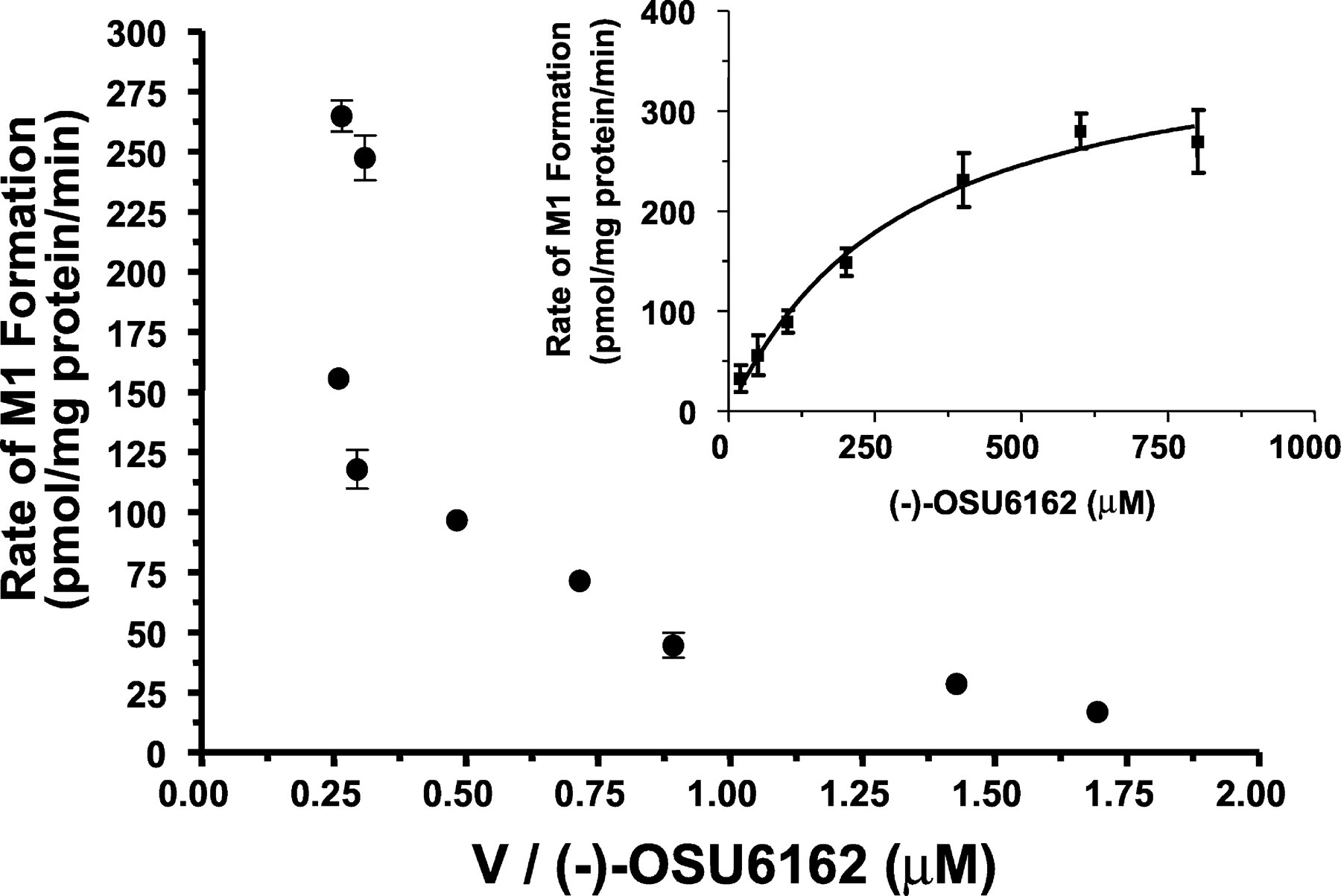
Radiochromatographic analysis of an incubation of [14C](−)-OSU6162 with human liver microsomes yielded a single metabolite peak, the retention time and MS fragmentation of which were identical with those of the authentic standard of N-despropyl(−)-OSU6162 (data not shown). Furthermore, of the (−)-OSU6162 consumed, >95% was accounted for as the N-despropyl metabolite (Fig. 1). Preliminary in vitro studies revealed that metabolite formation increased linearly with time for up to 60 min (data not shown). Unless specified, an incubation time of 30 min was used to ensure initial rate conditions for the formation of N-despropyl(−)-OSU6162. Figure2 shows a representative Eadie-Hofstee plot for (−)-OSU6162 N-depropylase activity in pooled human liver microsomes (pool of n = 10 different human livers). The plot indicated that multiple enzymes were responsible for the biotransformation of (−)-OSU6162 toN-depropyl(−)-OSU6162. Similar results were observed for incubations with HL28 and HL40 as well (data not shown).

Eadie-Hofstee plot of (−)-OSU6162 N-depropylase activity in human liver microsomes.
Inset is rate of formation of N-despropyl (−)-OSU6162 (n = 3). Incubation conditions were carried out as described under Experimental Procedures. Each data point represents the mean (± S.D.) of triplicate determinations.
Correlation Studies.
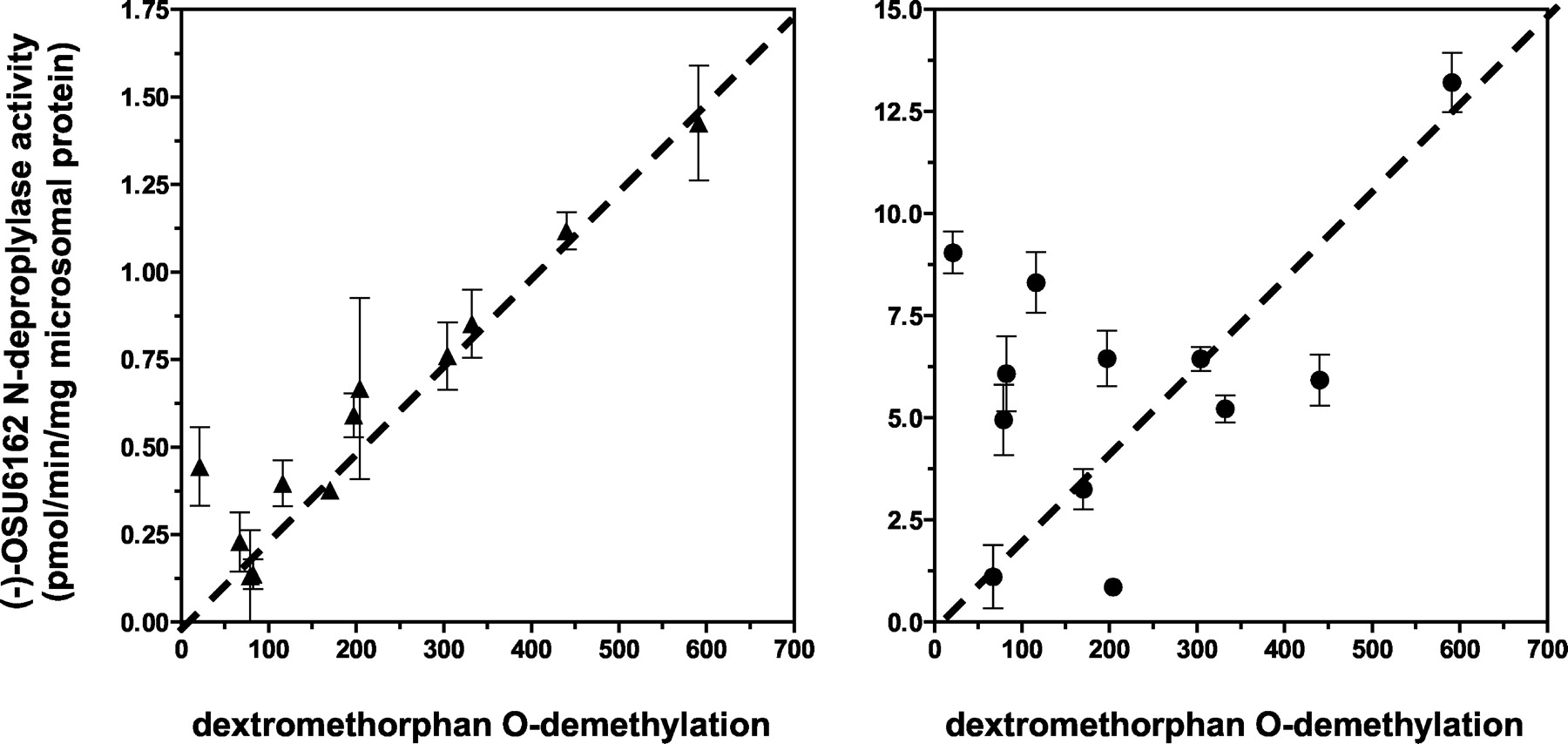
The rates of N-despropyl(−)-OSU6162 formation were determined in 12 different human liver microsomal preparations. As shown in Fig. 3A, at reduced concentrations of (−)-OSU6162 (e.g., 5 μM) the rate ofN-despropyl(−)-OSU6162 formation correlated with CYP2D6-catalyzed dextromethorphan-O-demethylase activity (r2 = 0.908). However, as depicted in Fig. 3B, at relatively high concentrations of (−)-OSU6162 (e.g., 200 μM) N-despropylase activity did not correlate with CYP2D6 activity (r2 = 0.201). Moreover, the correlation of N-despropyl (−)-OSU6162 formation tested against activities selective for other P450 forms (e.g., 7-ethoxyresorufin O-dealkylation, CYP1A2; tolbutamide hydroxylation, CYP2C9; (S)-mephenytoin 4′-hydroxylation, CYP2C19; chlorzoxazone 6-hydroxylation, CYP2E1; and testosterone 6β-hydroxylation, CYP3A4) was not significant at either concentration of (−)-OSU6162 tested (Table 1).

Correlation between dextromethorphanO-demethylation and (−)-OSU6162N-depropylase activity in a panel of human liver microsomes.
The dashed line (—-) represents the line of unity. (−)-OSU6162 (A, 5 μM or B, 200 μM) was incubated with human liver microsomes from 10 individual donors. Incubation conditions were carried out as described under Experimental Procedures. Each data point represents the mean (± S.D.) of triplicate determinations.
Correlation between rate of (−)-OSU6162 N-despropylation and standard cytochrome P450 enzyme-specific activities in a panel of human liver microsomal preparations1-a
Chemical Inhibition.
In addition to the correlation data, (−)-OSU6162 was coincubated with the following P450 enzyme-specific substrate/inhibitors: bropirimine (CYP1A2), coumarin (CYP2A6), 3-cyclopentyloxy-N-(3,5-dichloro-4-pyridyl)-4-methoxybenzamide (CYP2B6), ketoconazole (CYP3A4), (S)-mephenytoin (CYP2C19), sulfaphenazole (CYP2C9), and quinidine (CYP2D6). The agents were examined for their ability to inhibit N-despropyl metabolite formation at a substrate concentration of 20 μM. As described in Fig.4, only quinidine (a selective inhibitor for CYP2D6) strongly inhibited the formation ofN-despropyl(−)-OSU6162, whereas the other inhibitors did not show any marked effects on metabolite generation (inhibition <25%).
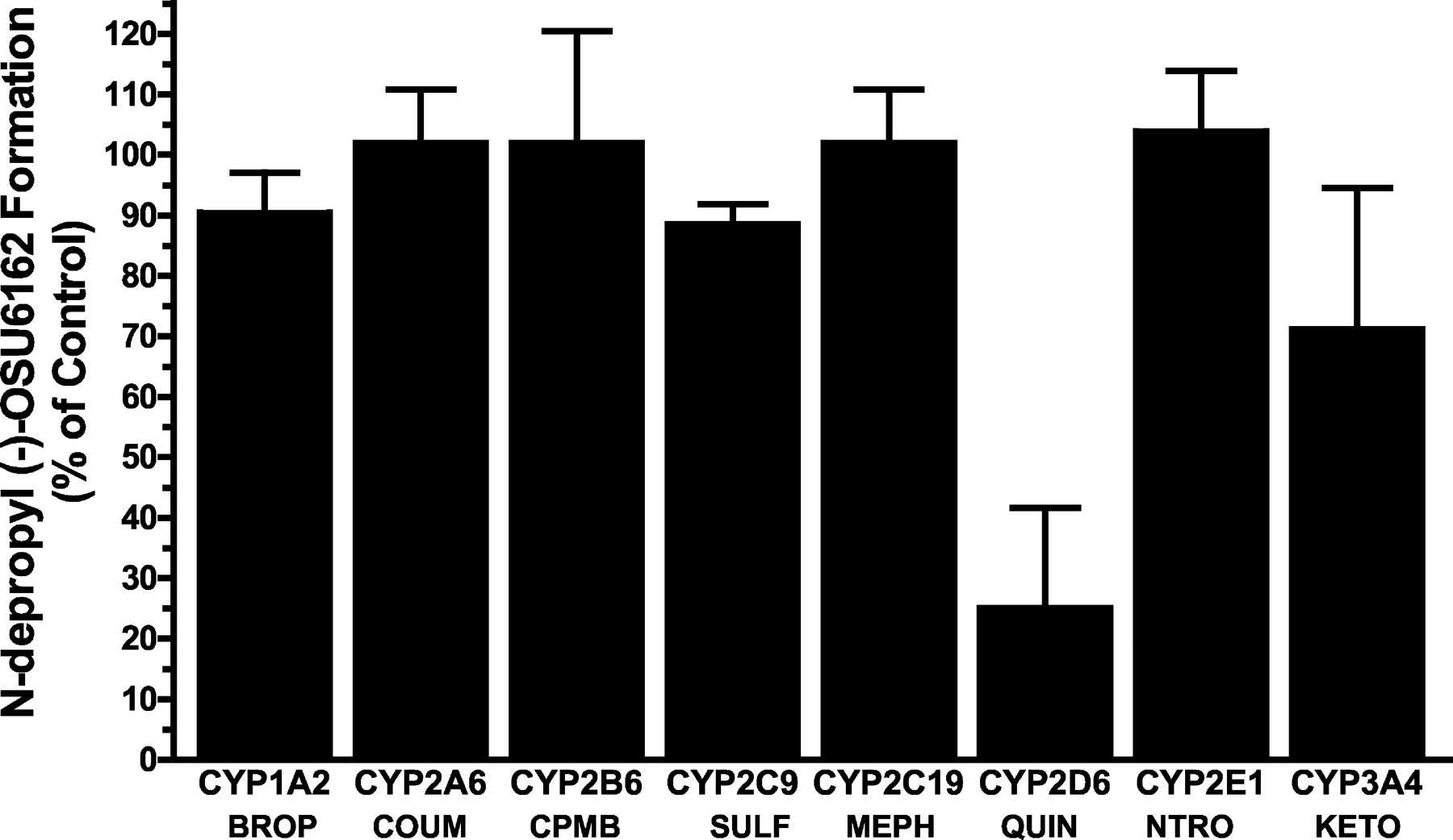
Percentage of control activities for N-despropylation of (−)-OSU6162 in the presence of various substrates and inhibitors selective for individual P450enzymes.
P450 enzyme-selective compounds were used to study their inhibitory effects on the oxidation of (−)-OSU6162 [20 μM] in human liver microsomes. The P450-selective inhibitors and/or substrates used in this experiment were 200 μM Bropirimine (CYP1A2; Wynalda et al., 1998); 50 μM coumarin (CYP2A6; Pearce et al., 1992); 50 μM 3-cyclopentyloxy-N-(3,5-dichloro-4-pyridyl)-4-methoxybenzamide (CYP2B6; Stevens et al., 1997); 10 μM sulfaphenazole (CYP2C9; Newton et al., 1995); 200 μM (S)-mephenytoin (CYP2C19;Wrighton et al., 1993); 5 μM quinidine (CYP2D6; Guengerich et al., 1986); 100 μM para-nitrophenol (CYP2E1;Tassaneeyakul et al., 1993); 5 μM ketoconazole (CYP3A4; Gibbs et al., 1999). Incubation conditions were carried out as described underExperimental Procedures. Each data point represents the mean (± S.D.) of triplicate determinations.
Incubations with Recombinant Human P450s.
Of the 10 human baculovirus-insect cell-expressed P450 isoenzymes investigated, CYP1A1, CYP1A2, CYP2C19, CYP2D6, and CYP3A4 were able to catalyze the formation of N-despropyl(−)-OSU6162. Enzyme kinetic experiments revealed that apparentKm values for (−)-OSU6162 metabolite formation by the recombinant P450s ranged from 4 to 591 μM (Table2), which was consistent with the previous multienzyme kinetic data obtained from human liver microsomes (Fig. 2). The most efficient enzyme (Vmax/Km) for total (−)-OSU6162 metabolite formation was CYP2D6, followed by CYP2C19 and then equally by CYP3A4 and CYP1A2 (Table 2). In addition to the enzymes mentioned above, CYP1A1 also exhibited (−)-OSU6162N-depropylase activity, however, since this enzyme is expressed extra-hepatically (Wrighton and Stevens, 1992), its overall contribution to (−)-OSU6162 in vivo is difficult to predict. Incubations conducted using S-9 prepared from human lung and kidney were found to posses (−)-OSU6162 N-depropylase activity albeit at a reduced rate compared with S-9 prepared from liver (data not shown).
Individual enzyme kinetic parameters for (−)-OSU6162 N-despropylation
Effect of (−)-OSU6162 on P450 Marker Substrates.
The selectivity of (−)-OSU6162 to inhibit six human P450 enzymes (CYP1A2, CYP2C9, CYP2C19, CYP2E1, CYP2D6, and CYP3A4) was evaluated using a simple in vitro inhibition screen (Wynalda and Wienkers, 1997). Of the P450 enzymes tested, only CYP2D6 catalytic activity was markedly inhibited (≈30% inhibition at 10 μM inhibitor concentration) by (−)-OSU6162 (Table 3).
The inhibitory effect of (−)-OSU6162 on specific P450 enzyme activities in recombinant P450 microsomes
Discussion
In the present study, it has been established that the hepatic biotransformation of (−)-OSU6162 involves aN-despropylation reaction, which is mediated by several P450 enzymes including CYP2D6. The involvement of CYP2D6 in the in vitro metabolism of (−)-OSU6162 is supported by several lines of evidence: 1) a good correlation between the rate ofN-despropyl-(−)-OSU6162 formation and dextromethorphanO-demethylase activity in a panel of human liver microsomes, 2) marked inhibition of (−)-OSU6162 metabolism by quinidine (a selective inhibitor of CYP2D6), and 3) a severalfold greater in vitro intrinsic clearance for (−)-OSU6162 for CYP2D6 than other P450 enzymes tested.
While inspection of the data with respect to the in vitro intrinsic clearance revealed that the majority of the N-depropylase activity on a per picomole P450 scale was largely attributed to CYP2D6, an equally important consideration (i.e., the overall expression of each P450 enzyme in human liver) is required to predict the importance of each enzyme to (−)-OSU6162 in vivo clearance. The relative abundance of the human hepatic P450s has been determined as CYP1A2 (13%), 2A6 (4%), 2B6 (<1%), 2C (20%), 2D6 (2%), 2E1 (7%), and 3A4 (30%) (Shimada et al., 1994; Rendic and Carlo, 1997). Using these values to reexamine the intrinsic clearances normalized to reflect relative abundance/activities of each form in vivo narrows the (Vmax/Km) differences between CYP2D6 and the other P450 enzymes and suggests that only about half of the (−)-OSU6162 total hepatic metabolism is predicted to be mediated by CYP2D6 and that additional human liver microsomal P450s, in particular CYP2C19 and to a lesser extent CYP1A1, CYP1A2, and CYP3A4, also contribute toward (−)-OSU6162 metabolite formation.
It is interesting that (−)-OSU6162 is a relatively good substrate for CYP2D6. Typically substrates of CYP2D6 share common structural characteristics such as the presence of at least one basic nitrogen atom, a distance of 5 or 7 Å between the basic nitrogen atom and the site of oxidation, a flat hydrophobic area near the site of oxidation, and a negative molecular electrostatic potential above the planar part of the molecule (de Groot et al., 1996; Ellis et al., 1996; Lewis et al., 1997). However, for (−)-OSU6162 the site of metabolism occurs adjacent to the lone basic nitrogen within the molecule (Fig.5). Based upon the current pharmacophore model for CYP2D6, this observation suggests that optimal catalytic binding orientation of (−)-OSU6162 (e.g., within the CYP2D6 active site) is restricted either due to steric hindrances or possibly unfavorable energy constraints for the alternative sites of aromatic hydroxylations. Therefore, the N-depropylation of (−)-OSU6162 must arise from the substrate adopting an unanticipated binding orientation within the CYP2D6 active site.
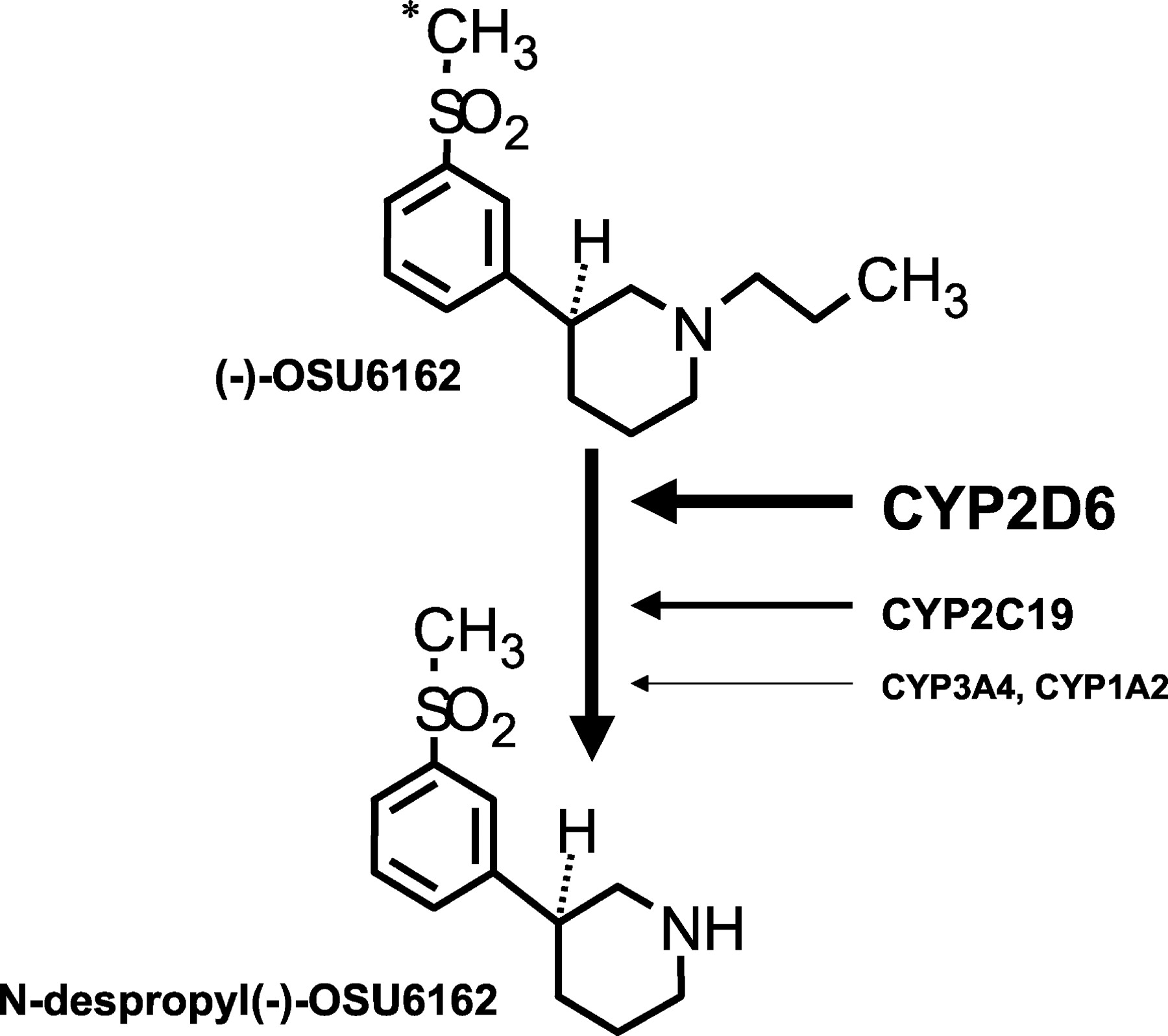
Summary of (−)-OSU6162 metabolism in human liver microsomes.
∗, denotes the site of carbon-14 label.
Although CYP2C19, CYP1A2, and CYP3A4 play a role in the metabolism of (−)-OSU6162, it is anticipated that (−)-OSU6162 will not significantly interfere with the metabolism of other drugs catalyzed by these enzymes (Table 3). Moreover, interactions with drugs metabolized by CYP2D6 are not expected due to the moderate affinity of (−)-OSU6162 (Km = 4 μM) toward the enzyme and the relatively low plasma concentrations anticipated for (−)-OSU6162 in vivo. Alternatively, because CYP2D6 may govern a large fraction of (−)-OSU6162 clearance at clinically relevant concentrations, the coadministration of other CYP2D6 inhibitors may alter (−)-OSU6162 clearance (Sindrup et al., 1996). For example, quinidine, a potent CYP2D6 inhibitor, can inhibit the in vivo metabolism of the CYP2D6 substrate, debrisoquine, to the extent that extensive metabolizer subjects receiving quinidine demonstrate debrisoquine pharmacokinetics phenotypically similar to those exhibited in CYP2D6 poor metabolizers (Brosen et al., 1987). A second example of this drastic alteration of CYP2D6 substrate pharmacokinetics is observed between the common antidepressant fluoxetine, also a potent inhibitor of CYP2D6 activity, and dextromethorphan (Otton et al., 1993). In consideration that the patient population that would benefit from the therapeutic effects of (−)-OSU6162 are likely taking other medications (Heerdirk et al., 1995; Gupta et al., 1996), the potential for drug interactions with (−)-OSU6162 exists. However, given that several P450 enzymes contribute, albeit to varying amounts, to the overall metabolic pathway of (−)-OSU6162, the risk of serious drug-drug interactions with this drug should be greatly reduced but remains untested.
In addition, the pharmacokinetics of (−)-OSU6162 will in part be dependent on CYP2D6 genotype, especially in subjects genotyped homozygous for the allelic variant forms (Fromm et al., 1997). Patients who have been phenotyped as CYP2D6 poor metabolizers have been demonstrated to achieve greater exposures of specific CYP2D6 substrates, compared with CYP2D6 extensive metabolizers (Eichelbaum and Gross, 1990). However in the case of (−)-OSU6162, given the catalytic contributions from human hepatic P450 enzymes, CYP2C19, CYP3A4, and CYP1A2, as well as the contribution from CYP1A1, the difference between poor metabolizers and extensive metabolizers may not be distinguishable given the normal variability expected within patient populations.
In conclusion, the current in vitro findings show that the dopamine D2 receptor modulator, (−)-OSU6162, appears to beN-depropylated primarily by CYP2D6. However, given that the drug is oxidized by additional hepatic P450 enzymes (e.g., CYP2C19, CYP1A2, and CYP3A4), as well as an extra-hepatic P450 (e.g., CYP1A1), extrapolation of these in vitro results to predict the magnitude to which CYP2D6 contributes to the overall clearance of (−)-OSU6162 in vivo remains to be established. Lastly, coincubation of (−)-OSU6162 did not inhibit the metabolic activity of the P450 enzymes, CYP1A2, CYP2C9, CYP2C19, CYP2E1, and CYP3A4, and only moderately inhibited CYP2D6. Therefore, in terms of predicting potential drug-drug interactions, given the predicted low dose of this agent and the poor affinity toward the human hepatic cytochrome P450 enzymes tested, clinically important interactions between (−)-OSU6162 and coadministered drugs that are metabolized by these enzymes appears unlikely.
Footnotes
- Abbreviations used are::
- (−)-OSU6162
- (S,S)-3-[3-(methylsulfonyl)phenyl]-1-propylpiperidine hydrochloride
- l-DOPA
- levodopa
- P450
- cytochrome P450
- HPLC
- high performance liquid chromatography
- BROP
- bropirimine
- CPMB
- 3-cyclopentyloxy-N-(3,5-dichloro-4-pyridyl)-4-methoxybenzamide
- COUM
- coumarin
- SULF
- sulfaphenazole
- NTRO
- para-nitrophenol
- QUIN
- quinidine
- KETO
- ketoconazole
- MEPH
- (S)-mephenytoin
- ACN
- acetonitrile
- LC/MS
- liquid chromatography mass spectrometry
- APCI
- atmospheric pressure chemical ionization
- Received June 7, 2002.
- Accepted August 30, 2002.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}