


Abstract
Bergamottin (BG) and 6′,7′-dihydroxybergamottin (DHB) are the most abundant furanocoumarins present in grapefruit juice and have been proposed as major intestinal CYP3A4 inhibitors contributing to grapefruit juice-drug interactions. The relative contribution of BG versus DHB to the interaction potential is unclear, in part due to inconsistencies in the literature regarding inhibitory potency. To resolve these inconsistencies, the inhibitory kinetics of each furanocoumarin toward CYP3A4 catalytic activity were systematically characterized using representative probes from two distinct CYP3A4 substrate subgroups (testosterone and midazolam). With human intestinal microsomes, DHB was a substrate-independent reversible (Ki, ∼0.8 μM) and mechanism-based (KI, ∼3 μM; kinact, 0.3-0.4 min-1) inhibitor of CYP3A4. In contrast, BG was a substrate-dependent reversible inhibitor, with a Ki (13 μM) using midazolam that was 8-fold greater than that using testosterone, but a substrate-independent mechanism-based inhibitor (KI, ∼25 μM; kinact, ∼0.35 min-1). Similar trends resulted with cDNA-expressed CYP3A4, only the KI values for BG were ∼10-fold lower than with microsomes. This seemed to reflect a much greater degree of microsomal protein binding by BG compared with DHB. Differential inhibition kinetics and binding properties between BG and DHB could account in part for the apparent in vitro inconsistencies in the literature. Results also emphasize the importance of appropriate substrate selection when designing inhibition studies involving dietary constituents.
Inhibition of the prominent drug metabolizing enzyme CYP3A4 is a major mechanism underlying numerous drug-drug interactions. For some of these interactions, clinical adverse events can ensue, ranging from relatively mild and transient (e.g., excessive sedation) to life threatening (e.g., ventricular arrhythmia) (Dresser et al., 2000). In addition to drugs, some foods have been shown to inhibit CYP3A4-mediated metabolism. One such widely studied food is grapefruit juice. This juice, consumed by some people with multiple drugs at breakfast, can markedly elevate blood levels of a variety of drugs, the majority of which are substrates for CYP3A4. Grapefruit juice acts by inhibiting intestinal CYP3A4 catalytic activity during first passage of the drug from the intestinal lumen to the systemic circulation (Lown et al., 1997; Bailey et al., 1998a). Hepatic CYP3A4 activity seems to be unaffected by grapefruit juice when consumed in usual volumes. This may reflect poor absorption of the active ingredients across the intestinal wall, dilution of the active ingredients in portal blood to concentrations below their effective inhibitory concentrations, and/or to avid binding of the active ingredients to plasma proteins in portal blood.
Although grapefruit juice inhibits CYP3A4 primarily in the intestine, the potential for clinical adverse events still exists (Dahan and Altman, 2004). Accordingly, the FDA requires the labeling of several drugs (e.g., cyclosporine, sirolimus, simvastatin, lovastatin, and felodipine) to carry warnings or precautionary statements regarding grapefruit juice interaction potential (www.fda.gov/medwatch/safety.htm). Clearly, it would be desirable to identify the CYP3A4 inhibitors in grapefruit juice, because they might serve as “markers” to predict other foods that are capable of producing grapefruit juice-type interactions. In addition, it might be possible to remove these ingredients from the juice to reduce the interaction potential. It was originally believed that the flavonoids naringin and quercetin were the active ingredients, because both are present in high concentrations in the juice and have inhibitory effects toward CYP3A4 activity in vitro (Miniscalco et al., 1992; Schrag and Wienkers, 2001). However, administration of purified forms of these compounds to human subjects failed to demonstrate an effect in vivo (Bailey et al., 1993a,b; Rashid et al., 1993). Compounds termed furanocoumarins now seem to be the major substances responsible for the grapefruit juice effect. The most extensively studied furanocoumarins, largely because they are the most abundant in the juice, are bergamottin (BG) and 6′,7′-dihydroxybergamottin (DHB) (Fig. 1). Although DHB differs from BG by only two hydroxyl groups, it is 1000 times more water soluble, as evidenced by log P values of 5.6 and 2.6 for BG and DHB, respectively (SciFinder Scholar v2002; American Chemical Society, Columbus, OH).

Chemical structures of bergamottin and 6′,7′-dihydroxybergamottin.
BG and DHB have been characterized as reversible and mechanism-based inhibitors of CYP3A4 catalytic activity in vitro (Schmiedlin-Ren et al., 1997; He et al., 1998; Eagling et al., 1999; Guo et al., 2000; Ohnishi et al., 2000; Tassaneeyakul et al., 2000; Greenblatt et al., 2003) (Table 1), and studies involving human subjects support roles for both furanocoumarins as contributors to the grapefruit juice effect (Bailey et al., 1998b, 2000, 2003; Kakar et al., 2004). However, inconsistencies prevail in the literature concerning the relative inhibitory potencies of BG and DHB toward CYP3A4 activity (Table 1). For example, although DHB has always demonstrated a greater potency as a reversible inhibitor (i.e., lower IC50) compared with BG, the magnitude of the difference varied from ∼2- to >20-fold (using saquinavir and midazolam as substrate, respectively). In addition, as a mechanism-based inhibitor, DHB was 7 times more potent than BG using nifedipine as substrate, but 8 times less potent than BG using testosterone as substrate, as evidenced by the KI values (i.e., concentrations required to achieve half the maximal rate of inactivation). These differences could partly reflect variation in probe substrate used, because accumulating evidence indicates that multiple binding domains exist within the CYP3A4 active site (Hosea et al., 2000; Schrag and Wienkers, 2001; Galetin et al., 2003). Testosterone, midazolam, and nifedipine seem to bind to distinct domains and are considered representative of three separate CYP3A4 substrate subgroups (Kenworthy et al., 1999; Galetin et al., 2003). In addition, a systematic comparison of the reversible inhibitory constant (Ki) between BG and DHB has yet to be reported. Unlike an IC50, the determination of a Ki requires multiple substrate concentrations that allow the characterization of the mode of inhibition (e.g., competitive, noncompetitive, or mixed). Finally, only microsomes prepared from liver have been previously examined. Because the grapefruit juice effect occurs mainly in the intestine, microsomes prepared from this tissue should serve as a more relevant in vitro system.
Reported inhibition kinetics for BG and DHB toward CYP3A4 catalytic activity in various enzyme systems
Based on these observations, we systematically compared the reversible and mechanism-based inhibition kinetics of BG and DHB toward CYP3A4 catalytic activity using a representative probe from two major substrate subgroups (midazolam and testosterone) and human intestinal microsomes (HIM). Parallel studies were also conducted with cDNA-expressed CYP3A4. Results showed that BG and DHB differ in terms of inhibitory and binding properties, which could partly account for inconsistencies in prior studies. Results also underscore the importance of appropriate substrate selection when evaluating the interaction potential of dietary constituents.
Materials and Methods
Materials and Chemicals. Testosterone, 6β-hydroxytestosterone (6β-OH TST), cortisol, midazolam maleate, alprazolam, and NADPH were purchased from Sigma-Aldrich (St. Louis, MO). BG and psoralen were purchased from INDOFINE Chemical Co., Inc. (Hillsborough, NJ). DHB was a kind gift from the Florida Department of Citrus (Lakeland, FL). 1′-Hydroxymidazolam (1′-OH MDZ) and baculovirus insect cell-expressed CYP3A4 (Supersomes), supplemented with cytochrome b5, were purchased from BD Gentest (Woburn, MA). Intestinal microsomes were previously prepared from mucosal scrapings from the proximal (jejunal) portion of a human donor small intestine (Paine et al., 1997); these microsomes did not contain readily detectable CYP3A5 immunoreactive protein using a selective antibody to CYP3A5 (BD Gentest). Centrifree centrifugal filtration devices (YM-30) were purchased from Milli-pore Corporation (Billerica, MA). All other chemicals and reagents were of HPLC or analytical grade where appropriate.
Inhibition Kinetics of BG and DHB toward Intestinal CYP3A4 Catalytic Activity. Both the reversible and mechanism-based inhibition kinetics of BG and DHB toward CYP3A4 catalytic activity (testosterone 6β-hydroxylation or midazolam 1′-hydroxylation) were evaluated using human intestinal (CYP3A5-negative) microsomes. For comparison, similar studies were conducted with cDNA-expressed CYP3A4. Testosterone and BG were dissolved as 100-fold concentrated solutions, whereas midazolam and DHB were dissolved as 1000-fold concentrated solutions, in methanol. All incubation mixtures were equilibrated in a shaking water bath at 37°C for 5 min before initiating the reactions with NADPH. The amount of metabolite formed (6β-OH TST or 1′-OH MDZ) was linear with respect to the incubation time and amount of microsomal protein or cDNA-expressed CYP3A4. The final concentration of methanol varied from experiment to experiment (i.e., 0.2% in incubations containing midazolam ± DHB, 1.1% in incubations containing midazolam ± BG or testosterone ± DHB, and 2% in incubations containing testosterone ± BG) but was constant in all incubation mixtures within an experiment.
Reversible inhibition. Using testosterone as the CYP3A4 substrate, incubation mixtures consisted of 0.1 mg/ml of microsomal protein or 10 pmol/ml of cDNA-expressed CYP3A4, testosterone (15-400 μM), BG (0-10 μM) or DHB (0-5 μM), and potassium phosphate buffer (0.1 M, pH 7.4). Initiation of the reaction with NADPH (final concentration, 1 mM) yielded a final volume of 0.5 ml. Reactions were terminated after 15 (microsomes) or 4 (cDNA-expressed CYP3A4) min with 5 ml of ice-cold ethyl acetate. The samples were spiked with cortisol (2.5 nmol) as the internal standard, vortex-mixed, shaken for 20 min, and centrifuged (4000g × 20 min). The upper organic layer was collected and evaporated to dryness under air. The resulting residue was reconstituted in 75 μl of 50% methanol (in water), and 40 μl were analyzed for 6β-OH TST by HPLC (described below). Using midazolam as the substrate, incubation mixtures consisted of 0.1 mg/ml of microsomal protein or 4 pmol/ml of cDNA-expressed CYP3A4, midazolam (1-8 μM), BG (0-30 μM) or DHB (0-10 μM), and potassium phosphate buffer. Initiation of the reaction with NADPH (final concentration, 1 mM) yielded a final volume of 0.5 ml. Reactions were terminated after 4 (microsomes) or 2 (cDNA-expressed CYP3A4) min with 1 ml of ice-cold acetonitrile. The samples were spiked with alprazolam (30 pmol) as the internal standard, vortex-mixed, shaken, and centrifuged. The supernatant (10 μl) was analyzed for 1′-OH MDZ by liquid chromatography/mass spectometry (described below).
Mechanism-based inhibition. Primary reaction mixtures consisted of 5 mg/ml of microsomal protein or 0.4 (using testosterone) or 0.2 (using midazolam) nmol/ml of cDNA-expressed CYP3A4, BG (0-60 μM) or DHB (0-10 μM), and potassium phosphate buffer. Initiation of the reaction with NADPH (final concentration, 1 mM) yielded a final volume of 80 μl. After 0, 1, 2, 5, and 10 (or 0, 1, 2, 3, and 5) min, a 10-μl aliquot (corresponding to 50 μg of microsomal protein or 4 or 2 pmol of cDNA-expressed CYP3A4) was removed and diluted 1:50 into a secondary reaction mixture containing testosterone (250 μM) or midazolam (8 μM) and NADPH (1 mM). The secondary reactions were terminated and processed in the same manner as described for the reversible inhibition experiments.
Unbound Fraction of BG and DHB in Human Intestinal Microsomes. The degree of binding of BG and DHB to intestinal microsomes was assessed by the ultrafiltration method. BG or DHB was mixed with microsomes (5 or 0.1 mg/ml) in potassium phosphate buffer to yield a final concentration of 10 μM in a final volume of 2.5 ml. To assess the degree of nonspecific binding to the filtration device, the same concentration of BG or DHB was mixed in phosphate buffer alone. An aliquot of starting material (0.5 ml) was saved, in duplicate, before placing 0.5 ml into duplicate filtration devices. The filtration device was centrifuged at 1500g for 1 h. At least 0.08 or 0.42 ml of filtrate was collected from starting material containing 5 or 0.1 mg/ml of microsomal protein, respectively. The volume of each filtrate was adjusted to 0.5 ml with phosphate buffer, after which 2 ml of ethyl acetate was added. The samples were spiked with psoralen (10 nmol) as the internal standard, vortex-mixed, shaken, and centrifuged. The upper organic layer was collected and evaporated to dryness under air. The resulting residue was reconstituted in 60 μl of methanol, and 25 μl was analyzed for BG or DHB by HPLC (described below). The unbound fraction in the microsomal mixtures (fu,mic) was calculated as the ratio of the concentration in the filtrate to that in the starting material.
Assays for 6β-Hydroxytestosterone, 1′-Hydroxymidazolam, BG, and DHB.6β-Hydroxytestosterone. 6β-OH TST was quantified by HPLC using an Agilent 1100 liquid chromatography system (Agilent Technologies, Palo Alto, CA). Analytes were separated with a Vydac C18 column (5 μm, 4.6 × 150 mm; Grace Vydac, Columbia, MD) maintained at 22°C. 6β-OH TST and cortisol were eluted with the following gradient [time (minutes)/percentage of water/percentage of methanol] at a flow rate of 0.8 ml/min: 0:45:55, 2:45:55, 12:35:65, 20:45:55, and 30:45:55. The UV absorbance of the eluate was recorded at 248 nm. 6β-OH TST was quantified by comparing peak area ratios (6β-OH TST/cortisol) to those from a standard curve prepared with known amounts of 6β-OH TST and the appropriate biological matrix (i.e., microsomes or cDNA-expressed CYP3A4). The standard curve ranged from 0.1 nmol (limit of quantification) to 5.0 nmol, with a correlation coefficient (r2) ≥ 0.997. The intraday coefficient of variation for the quality controls (0.25 or 0.5 nmol) was ≤8.3%. The interday variation in the assay (as assessed from the slopes of the standard curves) was ≤6.1%.
1′-Hydroxymidazolam. 1′-OH MDZ was quantified by liquid chromatography/mass spectometry using an Agilent 1100 system. Analytes were separated with a Phenomenex Luna C8 column (3 μm, 2 × 30 mm; Phenomenex, Torrance, CA) maintained at 35°C. Mobile phase A consisted of 0.1% acetic acid in 95% acetonitrile/5% water; mobile phase B consisted of 0.25% acetic acid in 5% acetonitrile/95% water. 1′-OH MDZ and alprazolam were eluted with the following gradient [time (minutes)/percentage of mobile phase B] at a flow rate of 0.3 ml/min: 1.0:14, 5.0:45, 7.0:45, 7.5:14, and 12:14. Analytes were detected as their [M + H]+ ions using electrospray ionization. 1′-OH MDZ and alprazolam were detected by single ion monitoring at m/z 342.1 and 309.1, respectively. 1′-OH MDZ was quantified by comparing peak area ratios (1′-OH MDZ/alprazolam) to those from a standard curve prepared with known amounts of 1′-OH MDZ and the appropriate biological matrix. The standard curve ranged from 1.0 pmol (limit of quantification) to 150 pmol, with an r2 ≥ 0.995. The intraday coefficient of variation for the quality controls (7.5 or 15 pmol) was ≤7.8%. The interday variation in the assay was ≤7.0%.
BG and DHB. BG and DHB were quantified by HPLC using an Agilent 1100 liquid chromatography system. Analytes were separated with a YMC J'sphere ODS-M80 C18 column (4.6 × 150 mm; YMC, Inc., Wilmington, NC). BG or DHB and psoralen were eluted with the following gradient [time (minutes)/percentage of water/percentage of acetonitrile] at a flow rate of 1 ml/min: 0:90:10, 5:90:10, 30:20:80, 35:20:80, 45:90:10, and 50:90:10. The UV absorbance of the eluate was recorded at 310 nm. BG or DHB was quantified by comparing peak area ratios (BG or DHB/psoralen) to those from a standard curve prepared with known amounts of BG or DHB and phosphate buffer. The standard curve for both BG and DHB ranged from 0.25 nmol (limit of quantification) to 15 nmol, with an r2 ≥ 0.994. The intraday coefficient of variation for the quality controls (0.5 and 10 nmol) was ≤8 and 4%, respectively. The interday variation in the assay was <10%.
Data Analysis.Reversible inhibition. Estimates of the apparent Km and Vmax values were obtained from an Eadie-Hofstee plot of the data (i.e., velocity versus velocity/[substrate]) in the absence of inhibitor. An estimate of the apparent Ki was obtained from a Dixon plot (velocity-1 versus [inhibitor]). Final kinetic parameters [S50 (testosterone) or Km (midazolam), Vmax, and Ki] were obtained by nonlinear least-squares regression of the [substrate] versus velocity data using WinNonlin v4.1 (Pharsight, Mountain View, CA). Apparent intrinsic clearance, CLint, was calculated as the ratio of Vmax to S50 (testosterone) or Km (midazolam). The IC50 at each testosterone concentration was obtained from a plot of the percentage of control activity versus the natural logarithm of [inhibitor].
Mechanism-based inhibition. The natural logarithm of the percentage of remaining CYP3A4 activity was plotted against the primary reaction time. The apparent inactivation rate constant (kinact,app) at each concentration of inhibitor was determined from the slope of the initial linear decline in activity. Estimates of kinact and KI values were obtained from a Kitz-Wilson plot (i.e., kinact,app -1 versus [inhibitor]-1). Final kinetic parameters were obtained by nonlinear least-squares regression (WinNonlin) using the following equation: kinact,app = (kinact× I)/(KI + I), where I denotes inhibitor concentration. The efficiency of inactivation was calculated as the ratio of kinact to KI.
Results
Comparison of BG and DHB as Inhibitors of Intestinal CYP3A4 Catalytic Activity. The kinetics of BG and DHB as reversible and mechanism-based inhibitors of CYP3A4 catalytic activity were determined in human intestinal CYP3A5-negative microsomes and compared with those for cDNA-expressed CYP3A4.
Reversible inhibition. Within each enzyme system, regardless of CYP3A4 probe substrate, in the absence of either inhibitor, final estimates of the relevant kinetic parameters (Km or S50, Vmax, and CLint) varied less than 10% over the range of final methanol concentrations used (0.2, 1.1, or 2%). Each pair of values was therefore averaged and is presented in Table 2. With testosterone as the substrate, in the absence of inhibitor, as expected, 6β-OH TST formation in both microsomes and cDNA-expressed CYP3A4 was consistent with autoactivation, as evidenced by curved Eadie-Hofstee plots (not shown). Accordingly, the [substrate] versus velocity data were well described by a simple sigmoidal (Hill) model (Shaw et al., 1997; Galetin et al., 2003; Patki et al., 2003). The apparent S50 (which is analogous to Km in the classic Michaelis-Menten unienzyme model) determined with microsomes was roughly double that with expressed enzyme. In the presence of inhibitor, Eadie-Hofstee plots were linear, indicating no autoactivation (not shown); with an increasing concentration of either inhibitor, the slope (-Km) steepened (i.e., became less negative) while the y-intercept (Vmax) decreased, indicating that BG and DHB were mixed-type inhibitors of 6β-OH TST formation. The simple linear mixed-type inhibition model (Cornish-Bowden, 1974) best described the data. The apparent Ki for BG was approximately twice (microsomes) or similar to (cDNA-expressed CYP3A4) that for DHB (Fig. 2 and Table 2). To determine whether the apparent Ki values were reasonable values (given the sigmoidal kinetics of testosterone in the absence of inhibitor), IC50 values were also calculated at each substrate concentration (Table 3).
Reversible inhibition kinetics of BG and DHB toward CYP3A4 catalytic activity in HIM and cDNA-expressed CYP3A4.

Dixon plots showing the inhibition of testosterone 6β-hydroxylation by DHB (A) and BG (B) in human intestinal microsomes. Symbols denote the means of duplicate incubations. TST, testosterone.
IC50 values (micromolar) for BG and DHB toward testosterone 6β-hydroxylation in HIM and cDNA-expressed CYP3A4
With midazolam as the substrate, as expected, in the absence of inhibitor, 1′-OH MDZ formation in both microsomes and cDNA-expressed CYP3A4 was consistent with classic Michaelis-Menten unienzyme kinetics, as evidenced by linear Eadie-Hofstee plots (not shown). As with inhibition of 6β-OH TST formation, the simple linear mixed-type inhibition model best described the inhibition of MDZ 1′-OH formation. The apparent Km for midazolam was similar between enzyme systems (Table 2). In contrast to results with testosterone, the apparent Ki for BG with midazolam was much greater than that for DHB in both microsomes (Fig. 3) and cDNA-expressed CYP3A4, by 15- and 12-fold, respectively (Table 2).

Dixon plots showing the inhibition of midazolam 1′-hydroxylation by DHB (A) and BG (B) in human intestinal microsomes. Symbols denote the means of duplicate incubations. MDZ, midazolam.
Mechanism-based inhibition. To delineate reversible from mechanism-based inhibition, primary reaction mixtures were incubated for varying lengths of time, diluted 50-fold, and residual CYP3A4 activity was assessed at a saturating concentration of substrate. As expected, inhibition of CYP3A4 activity by BG and DHB was time- and concentration-dependent with both substrates and enzyme systems used. Using testosterone as the substrate, with microsomes, the apparent KI for BG was 11-fold greater than that for DHB (Fig. 4 and Table 4), but the maximal inactivation rate constants (kinact values) were similar between the two inhibitors, resulting in a 13-fold greater kinact/KI for DHB compared with BG (Table 4). With cDNA-expressed CYP3A4, the KI for BG was roughly double that for DHB, and the kinact values were similar, resulting in only a 2-fold greater kinact/KI for DHB compared with BG (Table 4). Using midazolam as the substrate, as observed with testosterone, the apparent KI for BG was much greater (7-fold) than that by DHB in microsomes (Fig. 5 and Table 4). Likewise, the kinact values were similar between the two inhibitors, resulting in a 6-fold greater kinact/KI for DHB compared with BG (Table 4). With cDNA-expressed CYP3A4, both the KI and kinact for BG were roughly double the corresponding values for DHB, resulting in similar kinact/KI values between BG and DHB.

Time- and concentration-dependent inhibition of testosterone 6β-hydroxylation by DHB (A) and BG (B). Double reciprocal plots of the apparent inactivation rate constant (kinact,app) as a function of inhibitor concentration (Kitz-Wilson plots) were constructed to obtain initial estimates of KI and kinact values (C and D). Symbols denote the means of duplicate incubations.
Inactivation kinetics of BG and DHB toward CYP3A4 catalytic activity in human in HIM and cDNA-expressed CYP3A4
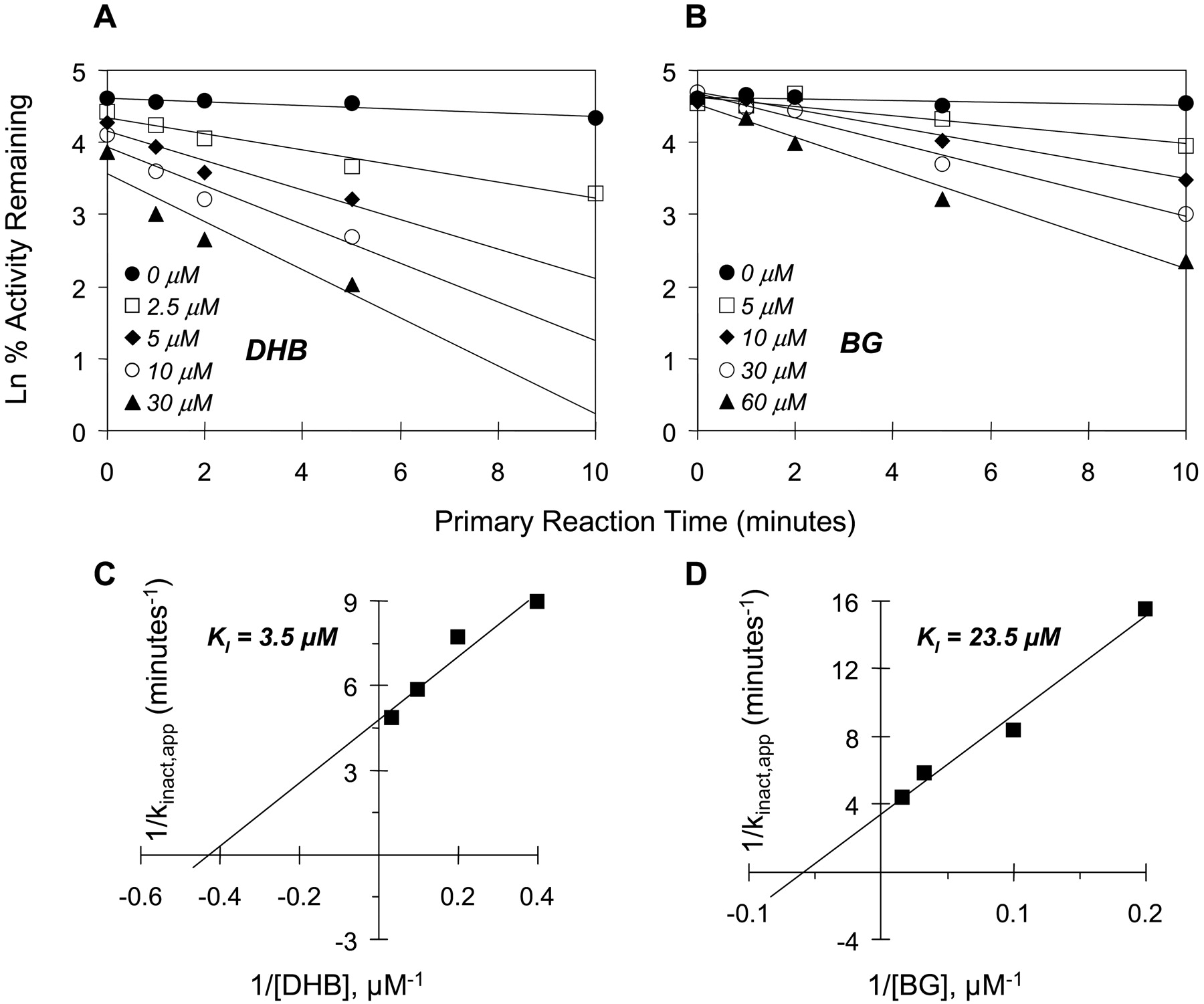
Time- and concentration-dependent inhibition of midazolam 1′-hydroxylation by DHB (A) and BG (B). Double reciprocal plots of the apparent inactivation rate constant (kinact,app) as a function of inhibitor concentration (Kitz-Wilson plots) were constructed to obtain initial estimates of KI and kinact values (C and D). Symbols denote the means of duplicate incubations.
Unbound Fraction of DHB in Human Intestinal Microsomes. To determine whether the degree of binding of BG to microsomal protein could account for the ∼10-fold difference in the KI for BG between microsomes and cDNA-expressed CYP3A4 (Table 4), the fu,mic for BG and DHB was determined (or attempted) by ultrafiltration. BG was never detected in the filtrate, even in the absence of microsomal protein; thus, the fu,mic for BG could not be determined by this method. In contrast, DHB was completely recovered in the filtrate in the absence and was easily detected in the presence of microsomal protein. At a microsomal protein concentration equal to that in the primary reaction mixture, the fu,mic of DHB was calculated as 48%. At a 50-fold lower microsomal protein concentration, which represented that in the secondary reaction mixture (and in the incubation mixtures for the reversible inhibition experiments), the fu,mic of DHB increased to essentially 100%.
Discussion
When DHB was coincubated with microsomes and the representative CYP3A4 substrate testosterone or midazolam (reversible inhibition design), CYP3A4 activity was effectively inhibited, regardless of substrate, as evidenced by similar apparent Ki values (∼0.8 μM). This is consistent with traditional theory, which predicts substrate-independent inhibition if only one site (or domain) on the enzyme is associated with inhibition. In contrast to DHB, the inhibitory potency of the more lipophilic BG was substrate-dependent, with an apparent Ki using midazolam (13 μM) that was nearly an order of magnitude greater than that using testosterone. These behaviors of BG and DHB were confirmed with cDNA-expressed CYP3A4, where the Ki for BG using midazolam (6 μM) was 12 times greater than that using testosterone, but the Ki for DHB was similar using either substrate (∼0.5 μM). The higher values obtained with microsomes compared with expressed enzyme could be due to interactions among multiple P450 isoforms in the microsomal preparations and/or to different levels of P450 reductase in microsomes versus expressed enzyme (Yamazaki et al., 1997; Backes et al., 1998).
Evidence is prevalent in the literature supporting the contention that testosterone and midazolam bind to distinct domains within the CYP3A4 active site (Hosea et al., 2000; Schrag and Wienkers, 2001; Galetin et al., 2003). Although both BG and DHB are substrates for CYP3A4 (a requirement for mechanism-based inhibition), the respective substrate binding domains are not known. The finding that BG was a more potent reversible inhibitor of 6β-OH TST formation than 1′-OH MDZ formation could be explained by preferential binding of BG to the testosterone domain, whereas DHB may bind to both the testosterone and midazolam domains. The substrate-dependent reversible inhibition of CYP3A4 by BG supports the recommended use of multiple CYP3A4 probes in the in vitro screening process for drug interaction potential (Kenworthy et al., 1999; Yuan et al., 2002). This differential inhibition by BG and other dietary constituents (flavonoids) (Schrag and Wienkers, 2001) also suggests that dietary differences could be contributing factors to the notorious lack of (or weak) inter-relationships in healthy volunteers between phenotypic trait measures of various putative in vivo CYP3A4 probes belonging to different substrate subgroups [e.g., erythromycin breath test versus midazolam clearance (Kinirons et al., 1999)].
When DHB was preincubated with the enzyme source before the addition of substrate (mechanism-based inhibition design), CYP3A4 activity was again effectively inhibited in a substrate-independent manner. Likewise, BG inhibited CYP3A4 activity in a substrate-independent manner, which contrasts with its behavior in the reversible inhibition design experiment. Mechanism-based inhibition of CYP3A4 by BG involves the formation of a reactive intermediate, recently proposed to be a furanoepoxide (Zhou et al., 2004), that seems to bind to (and modify) the apoprotein while leaving the heme intact (He et al., 1998; Zhou et al., 2004). Guo and Yamazoe (2004) recently postulated that the tricyclic furan-containing moiety is more important than the side chain for furanocoumarin-mediated CYP3A4 inactivation. Since BG and DHB differ structurally only in the side chain, it is reasonable to speculate that both inactivate CYP3A4 by the furanoepoxide binding to the apoprotein, presumably at or near their respective substrate domain(s).
Mechanism-based inhibition by both furanocoumarins occurred rapidly (within 10 min) and within the time frame of the reversible inhibition design experiments (2-15 min). As such, mechanism-based inactivation also likely occurred during the reversible inhibition experiments (although this would be attenuated in the presence of substrate). Mechanism-based inactivation could therefore account for the mixed-type reversible inhibition behavior observed with both furanocoumarins (i.e., both the Km and Vmax changed in the presence of inhibitor).
In the mechanism-based inhibition experiments, the unbound fraction for DHB in microsomes (fu,mic) was ∼50% in the primary reaction mixture (5 mg of microsomal protein/ml) and nearly 100% after the 1:50 dilution to the secondary reaction mixture (0.1 mg of microsomal protein/ml, the same concentration used in the reversible inhibition experiments). The true KI values for DHB are therefore approximately half those reported in Table 4. In contrast, the fu,mic for BG could not be determined, even in the absence of microsomal protein. This indicates that BG bound nonspecifically to the filtration device, which likely reflects the 2-fold higher log P (i.e., much greater lipophilicity) for BG compared with DHB. The unbound concentration of BG could therefore have been much lower than that for DHB in all reactions; that is, the degree of nonspecific or protein binding was much greater for BG than for DHB. In addition to interactions among multiple P450s and different reductase levels (Yamazaki et al., 1997; Backes et al., 1998), a greater degree of protein binding by BG would account for the 9- to 12-fold higher apparent KI values for BG in microsomes versus cDNA-expressed CYP3A4 (the protein concentration in the primary reaction mixtures containing microsomes was ∼5-fold greater than that in mixtures containing cDNA-expressed enzyme). Based on the “stickiness” of BG, it is possible that, on an unbound concentration basis, BG is a more potent CYP3A4 inhibitor, whether mechanism-based or reversible, than DHB.
The reversible Ki values obtained for DHB are in good agreement with the IC50 values reported in the literature (Table 1), with the exception of the high value (4.7 μM) with midazolam as the substrate (without preincubation) reported by Greenblatt et al. (2003). This disparity could reflect differences in the enzyme source (human liver microsomes versus intestinal microsomes). Alternatively, it could reflect the higher midazolam concentration used by these investigators (50 μM) compared with those used in the current work (1-8 μM), since an IC50 is substrate concentration-dependent. In addition, midazolam is a known mechanism-based inhibitor of CYP3A4 (Schrag and Wienkers, 2001; Khan et al., 2002), with reported KI values of 6 to 16 μM. Mechanism-based inhibition of the enzyme by midazolam could have thus confounded the apparent IC50 for DHB. Finally, it could be speculated that the liver microsomes used by Greenblatt et al. (2003) contained the polymorphic CYP3A5 (intestinal microsomes used in the current work did not), which shares many substrates with CYP3A4 but has often been reported to be less sensitive to inhibition (Gibbs et al., 1999; Ekins et al., 2003; Patki et al., 2003). The reversible Ki values for BG from the current study are also in good general agreement with previously reported IC50 values, with two exceptions. The high value (22 μM) reported by Guo et al. (2000) could have reflected nonspecific binding by BG, but the microsomal protein concentration in the reaction mixtures was similar to that in all other studies. Alternatively, the liver microsomes used may have contained CYP3A5. The high value (>20 μM) reported by Ohnishi et al. (2000), who used cDNA-expressed CYP3A4 (from the same source as in the current studies) and testosterone, remains unexplained.
Compared with the current work with cDNA-expressed CYP3A4, the higher mechanism-based inhibitory KI values for DHB and BG determined by Hollenberg and coworkers (Schmiedlin-Ren et al., 1997; He et al., 1998) (Table 1), who used testosterone as substrate, may reflect the use of a reconstituted, purified enzyme rather than the commercially available expressed enzyme (Shaw et al., 1997). The KI values for BG and DHB reported by Tassaneeyakul et al. (2000) (Table 1), who used human liver microsomes and nifedipine as substrate, which belongs to a third substrate subgroup (Kenworthy et al., 1999; Galetin et al., 2003), are near the values obtained in the current studies with human intestinal microsomes. Although the kinact values reported by Tassaneeyakul et al. (2000) were similar between the two furanocoumarins (∼0.07 min-1), they were much lower than those obtained in the current work, indicating that BG and DHB are less effective inactivators of CYP3A4 toward drugs belonging to the nifedipine subgroup compared with those belonging to the testosterone and midazolam subgroups.
In addition to the aforementioned in vitro inconsistencies, an apparent in vitro/in vivo inconsistency was recently reported (Bailey et al., 2003). One-quarter strength lime juice, which contained BG at a comparable concentration as grapefruit juice (but undetectable DHB), was effective as both a reversible and mechanism-based inhibitor of testosterone 6β-hydroxylase activity using recombinant CYP3A4. However, in healthy volunteers, relative to water, lime juice produced only a 20% increase in the average AUC and Cmax of felodipine (Bailey et al., 2003), the CYP3A4 substrate most commonly used in grapefruit juice-drug interaction studies. This could have reflected the high binding capacity of BG if, following lime juice ingestion, the unbound concentration of BG in enterocytes remained well below the Ki and/or KI. The minimal interaction between lime juice and felodipine may also have reflected the use of in vitro and in vivo probes belonging to different substrate subgroups. Felodipine does not belong to either the testosterone or midazolam subgroup and also seems not to bind the identical domain as nifedipine, a structural analog (Galetin et al., 2003). If lime juice reversibly inhibited intestinal CYP3A4 activity in the felodipine interaction study (which is reasonable since the juice was coadministered with the CYP3A4 probe), a greater magnitude of interaction may have resulted with a probe from the testosterone subgroup (e.g., erythromycin and cyclosporine). Utilization of in vitro and in vivo probes from the same substrate subgroup therefore seems advisable when designing in vivo interaction studies based on in vitro data.
In summary, BG, but not DHB, exhibited substrate-dependent reversible inhibition of intestinal CYP3A4 activity, with a much greater potency using testosterone compared with midazolam as substrate; however, mechanism-based inhibition by both furanocoumarins was substrate-independent and rapid. Whether substrate-dependent reversible inhibition by BG has relevance to grapefruit juice-drug interactions in vivo therefore remains unclear. BG and DHB also demonstrated a marked difference in binding properties, with the more lipophilic BG showing a much greater degree of nonspecific binding compared with DHB. These differences in inhibitory and binding properties may partly account for discrepancies reported in prior studies. These differences also emphasize the importance of proper substrate selection for the assessment of the interaction potential of not only new drug candidates, but also dietary constituents.
Acknowledgments
We thank Nicole White (School of Pharmacy, University of North Carolina) and Dr. Michael Fisher (Pfizer Central Research, Groton, CT) for help with the development of the 1′-hydroxymidazolam assay and Dr. Michael Schrag (Amgen Inc., Thousand Oaks, CA) for helpful discussions.
Footnotes
-
This work was supported by the National Institutes of Health Grants GM38149 and RR00046.
-
doi:10.1124/dmd.104.000547.
-
ABBREVIATIONS: BG, bergamottin; DHB, 6′,7′-dihydroxybergamottin; HIM, human intestinal microsomes; 6β-OH TST, 6β-hydroxytestosterone; 1′-OH MDZ, 1′-hydroxymidazolam; HPLC, high-performance liquid chromatography; P450, cytochrome P450.
- Received May 7, 2004.
- Accepted July 12, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}