


Abstract
In the present study we examined the disposition of atorvastatin, lovastatin, and simvastatin in acid and lactone forms and pravastatin in acid form in multidrug-resistant gene (mdr1a/b) knockout (KO), and wild-type (WT) mice. Each statin was administered s.c. to mdr1a/b KO and WT mice at 3.0 mg/kg (n ≥ 3 mice/time point). Blood, brain, and liver samples were harvested at 0, 0.5, 1.5, and 3 h postdose. Plasma and tissue concentrations of the acid and lactone (only the acid form was determined for pravastatin) were determined using a liquid chromatography-mass spectrometry method. Both lactone and acid were observed in plasma when lactones were administered, but only acids were detected when the acid forms were administered. The plasma and liver concentrations of acid or lactone were similar between the KO and WT mice. Two- to 23-fold higher concentrations were observed in liver than in plasma, suggesting potential uptake transporters involved. A significantly higher (p < 0.05) brain penetration in the KO compared with the WT mice was observed for lovastatin acid (but the brain/plasma ratio was low for both KO and WT mice) and lactone and simvastatin lactone but not for atorvastatin or pravastatin. The present results suggest that mouse P-glycoprotein does not affect the lactone-acid interconversion or liver-plasma distribution. Furthermore, P-glycoprotein plays a limited role in restricting the brain penetration of the acid forms of atorvastatin, pravastatin, simvastatin, lovastatin, and atorvastatin lactone but may limit the brain availability of the lactone forms of simvastatin and lovastatin.
Statins decrease intracellular cholesterol biosynthesis by reversibly inhibiting the microsomal enzyme hydroxymethyl glutaryl coenzyme A reductase. Most statins are given in the active β-hydroxy acid form except for lovastatin and simvastatin, which are administered as inactive lactone prodrugs (Shitara and Sugiyama, 2006). Both lactone and acid forms were observed in the systemic circulation after oral administration of atorvastatin (Kantola et al., 1998a), lovastatin (Neuvonen and Jalava, 1996), simvastatin (Kantola et al., 1998b; Prueksaritanont et al., 2002), and cerivastatin (Backman et al., 2002) in humans and/or animals, indicating that interconversion occurs between the lactone and acid forms of these statins. Both the acid and lactone forms of atorvastatin, simvastatin, and lovastatin have been reported to be substrates and inhibitors of CYP3A4, with the lactone forms showing stronger inhibitory effects on CYP3A4-mediated metabolism of mexazolam compared with the corresponding acid forms (Ishigami et al., 2001). Therefore, inhibition of CYP3A4-mediated metabolism has been one of the major causes of drug interaction of these statins (Shitara and Sugiyama, 2006) except pravastatin, which undergoes degradation under the acidic conditions of the stomach as well as presystemic metabolism (Hatanaka, 2000). More recently marketed statins such as rosuvastatin are minimally metabolized by P450s and cleared via hepatobiliary excretion of intact parent compound, which was shown to be mediated by drug transporters (Huang et al., 2004). Drug transporters including P-glycoprotein (P-gp, ABCB1), multidrug resistance-associated protein 2 (MRP2, ABCC2) (Yamazaki et al., 1997), breast cancer resistance proteins (BCRP/Bcrp, ABCG2) (Hirano et al., 2005b), and the hepatic uptake transporter OATP1B1 (SLCO1B1) (previously known as OATP-C or OATP2), have been suggested to contribute to the different pharmacokinetic properties of statins (Shitara et al., 2003, 2004; Chen et al., 2005).
P-gp, which is present in most tissues including liver, intestine, kidney, and brain, has been shown to limit the oral absorption and brain penetration of many drugs (Chen and Pollack, 1999; Boyd et al., 2000; Chen et al., 2003a, b; Kalvass et al., 2007). Several studies have shown that statins interact with P-gp. For example, Bogman et al. (2001) have shown that the lactone and acid forms of statins exhibit different inhibitory effects on P-gp-mediated transport of rhodamine 123 with the lactone forms of atorvastatin, simvastatin, and lovastatin exhibiting more potent P-gp inhibition than their corresponding acid forms. Consistent with the observation from Bogman's group, our recent data showed that the lactone forms of the statins were better inhibitors for P-gp-mediated transport of calcein AM (Chen et al., 2005). Furthermore, Wu et al. (2000) have shown that atorvastatin is a substrate of P-gp using Caco-2 cells. Likewise, our recent data demonstrated that both the acid and lactone forms of atorvastatin, lovastatin, and simvastatin are P-gp substrates with the acid form showing a slightly higher affinity toward P-gp compared with its lactone form (Chen et al., 2005). Overall, in vitro transport data have demonstrated differential properties toward P-gp among statins and between the acid and lactone forms of the same statin. Drug interaction studies involving statins and digoxin support a role of P-gp (Boyd et al., 2000). Many additional drugs such as verapamil (Kantola et al., 1998b) and itraconazole (Neuvonen and Jalava 1996; Kantola et al., 1998a) and dietary supplements such as grapefruit juice interact with statins and are modulators of both CYP3A and P-gp (Holtzman et al., 2006). However, data available to address the potential role of P-gp in statin disposition are scarce. So far only cerivastatin has been examined in mdr1a/b knockout mice (Kivistö et al., 2004). A 4-fold higher brain/plasma concentration ratio in the KO than in the WT mice suggests that cerivastatin brain penetration was impaired by P-gp (Kivistö et al., 2004).
In the present study we investigated the role of P-gp in the plasmabrain and plasma-liver disposition of four model statins (atorvastatin, lovastatin, and simvastatin in both acid and lactone forms and pravastatin in acid form only) in vivo using mdr1a/b KO and WT mice.
Materials and Methods
Materials. Atorvastatin, in both the acid and lactone forms were synthesized at Pfizer (Ann Arbor, MI). Lovastatin, simvastatin, and pravastatin were purchased from Sigma-Aldrich (St. Louis, MO). The acid forms of lovastatin and simvastatin were prepared by hydrolysis of the corresponding lactones under alkaline conditions. All of the other chemicals and reagents were the highest grade available from commercial sources. Structures and physical-chemical parameters of the statins used in the present study can be found in our earlier article (Chen et al., 2005).
Disposition of Atorvastatin (Lactone and Acid), Lovastatin (Lactone and Acid), Pravastatin (Acid Only), and Simvastatin (Lactone and Acid) in mdr1a/b KO and WT Mice. Male mdr1a/b KO and FVB (WT) mice (body weight ∼25 g; Taconic Farms, New York, NY) were used in the present study. Mice were housed at controlled temperature and humidity in an alternating 12-h light/dark cycle with free access to food and water before the study. Atorvastatin lactone and acid, pravastatin acid, lovastatin lactone and acid, and simvastatin lactone and acid, dissolved in 20% cyclodextrin, were injected s.c. (10 ml/kg) into the mice (n = 3-4 mice/strain/time point). Blood, brain, and liver tissues were collected at 0, 0.5, 1.5, and 3 h postdose. Plasma was obtained by centrifugation (3000 rpm for 10 min) of heparinized blood. The brain and liver tissues were rinsed in saline, blot-dried, weighed, and homogenized with saline (w/v, 1:3). All samples were stored at -20°C until analysis.
Sample Analysis. Separate standard curves were prepared for plasma, brain, and liver samples. Plasma (10 μl), brain (100 μl), and liver (50 μl) samples were transferred to a 96-well plate, followed by adding the deuterated atorvastatin acid (internal standard, 10 μl) and acetonitrile (200 μl). After vortexing, the mixture was centrifuged at 3000 rpm for 10 min, and an aliquot (10 or 20 μl) of the supernatant was analyzed using a high-performance liquid chromatography (HPLC)-mass spectrometry (MS) method.
The HPLC-MS/MS system consisted of either a Shimadzu LC-10A ternary pump (Shimadzu, Kyoto, Japan) or an Agilent quaternary pump HPLC system (Hewlett Packard, Palo Alto, CA), an autosampler, and a PE Sciex API 4000 mass spectrometer (PerkinElmer Sciex Instruments, Foster City, CA) fitted with an electrospray ionization probe operated in the positive ion mode. An Aqua C18 column (4.6 × 50 mm, 5 μm; Phenomenex, Torrance, CA) was used. The analytes were eluted with a mobile phase consisting of acetonitrile (solvent A) and ammonium acetate (10 mM, solvent B) at a flow rate of 0.5 ml/min with the following gradients: atorvastatin (acid and lactone), 50% A from 0 to 0.5 min, 50 to 90% A from 0.5 to 1.5 min, 90% A from 1.5 to 3 min, and 90 to 50% A from 3 to 3.2 min; simvastatin and lovastatin (acid and lactone), 20% A from 0 to 1 min, 20 to 90% A from 1 to 2 min, 90% A from 2 to 3 min, and 90 to 20% A from 3 to 3.2 min; and pravastatin, 20% A from 0 to 0.5 min, 20 to 90% A from 0.5 to 1.5 min, 90% A from 1.5 to 3 min, and 90 to 20% A from 3 to 3.2 min.
The compounds were monitored using the following mass transitions: atorvastatin acid, 559.3 → 440.1; atorvastatin lactone, 541.2 → 448.2; internal standard, 564.3 → 445.1; simvastatin lactone, 419.3 → 143.2; simvastatin acid, 459.2 → 343.1; lovastatin lactone, 405.3 → 199.1; lovastatin acid, 445.2 → 343.1; and pravastatin, 447.3 → 327.1. Standard curves were calculated using a weighed linear regression (1/x) of the concentration versus ratio of analyte to internal standard peak areas. The lower limits of quantitation for atorvastatin (acid/lactone), lovastatin (acid/lactone), simvastatin (acid/lactone), and pravastatin (acid form only) were 5/10, 10/10, 10/10, and 10 ng/ml in plasma, 5/5, 10/10, 10/10, and 10 ng/ml in brain homogenate, and 10/10, 40/40, 40/40, and 40 ng/ml in liver homogenate, respectively. The relative accuracy was between 80 and 120%.
Data Analysis. The mean and S.D. of concentrations and the tissue/plasma concentration ratios (wherever permitted) at 0.5, 1, and 3 h postdose were calculated for both KO and WT mice. In addition, conversion of the lactone to the acid form for each statin except pravastatin was analyzed on the basis of the acid/lactone concentration ratio after administration of the lactone form because no lactone was observed after administration of the acid form of all statins tested. Student's t test (two-tailed) was used to compare the difference between the KO and WT mice. P < 0.05 was considered statistically significant.
Results
Disposition of Atorvastatin in Plasma, Liver, and Brain.Table 1 and Fig. 1 show the plasma and tissue concentrations and tissue/plasma concentration ratios of atorvastatin. When the lactone form of atorvastatin was administered, both lactone and acid forms were observed in plasma but not in liver or brain. In contrast, when the acid form was given, only the acid form was observed, not only in plasma but also in liver and brain in both strains of mice. The brain/plasma concentration ratios at 0.5 h for the acid forms were low but similar between the KO (0.074) and WT (0.044) mice. Likewise, the liver/plasma concentration ratios at 0.5, 1.5, and 3.0 h postdose were similar between the KO and WT mice, ranging from 2.1 to 3.5 for the KO and from 1.6 to 2.4 for the WT mice.
Plasma and tissue concentrations and tissue/plasma concentration ratios of atorvastatin after a 3.0 mg/kg s.c. administration to mdr1a/b KO and WT mice
Concentration in plasma, liver, and brain at 0, 0.5, 1.5, and 3 h postdose that are not shown in the table were below the lower limit of quantitation (acid/lactone): 5/10 ng/ml in plasma, 5/5 ng/ml in brain homogenate, and 10/10 ng/ml in liver homogenate. Data are means ± S.D.
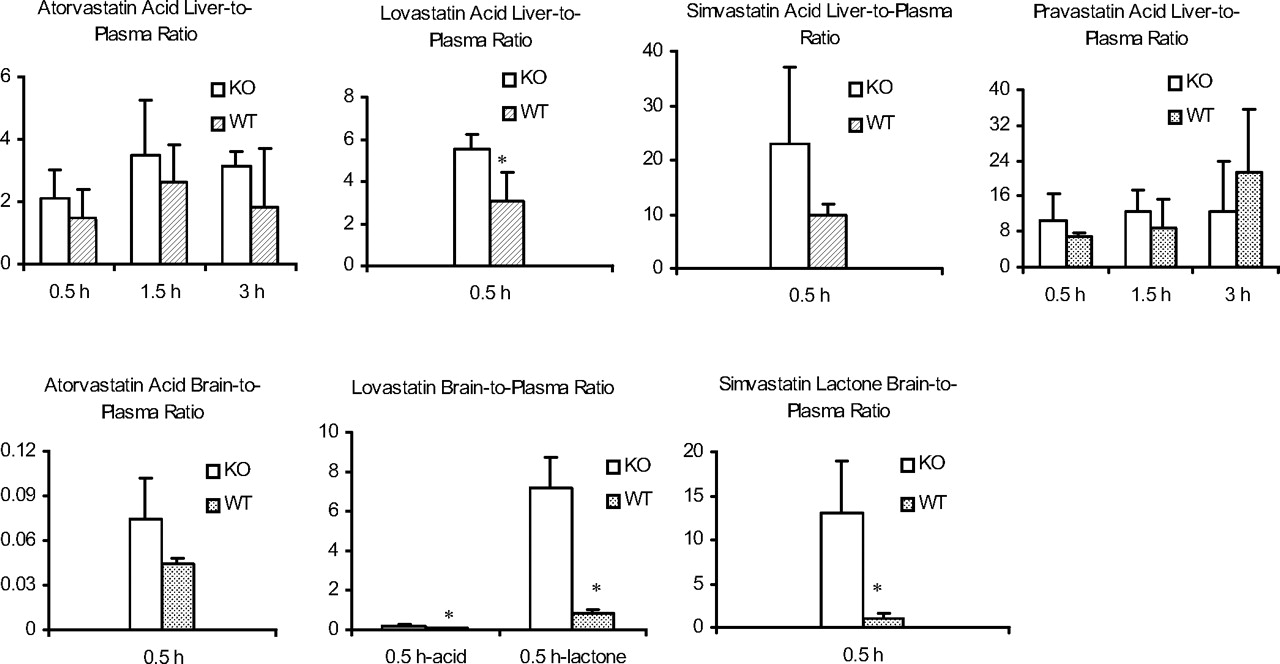
Brain-to-plasma and liver-to-plasma concentration ratios after s.c. administration of the statins (acid form only) at 3 mg/kg to mdr1a/b KO and WT mice. *, p < 0.05 between the KO and WT mice.
Disposition of Lovastatin in Plasma, Liver, and Brain. When lovastatin lactone was administered, both the acid and lactone forms were observed in plasma with no difference between the KO and WT mice (Table 2). In addition, the lactone showed a statistically higher (p < 0.05) brain penetration in the KO (7.2) compared with the WT (<0.85) mice. However, the acid form was not detected in the brain of either strain of the mice. Neither the acid nor the lactone was detected in the liver. Conversely, when the acid form was administered, no lactone form was observed in plasma, brain, and liver, but the acid form was found. Although the brain/plasma ratio of lovastatin acid after dosing of lovastatin acid was statistically higher in the KO (0.17) compared with the WT (<0.05) mice, the brain penetration for lovastatin acid was low in both strains (Fig. 1). Similar to atorvastatin, no lovastatin lactone was observed in the liver after dosing of either the lactone or the acid form. Lovastatin acid liver/plasma concentration ratios were higher than unity but similar between the two strains of mice.
Plasma and tissue concentrations and tissue/plasma concentration ratios of lovastatin after a 3.0 mg/kg s.c. administration to mdrl a/b KO and WT mice
Concentrations in plasma, liver, and brain at 0, 0.5, 1.5, and 3 h postdose that are not shown in the table were below the lower limit of quantitation (LLOQ) (acid/lactone): 10/10 ng/ml in plasma, 10/10 ng/ml in brain homogenate, and 40/40 ng/ml in liver homogenate. Data are means ± S.D.
Disposition of Simvastatin in Plasma, Liver, and Brain. The plasma, liver, and brain disposition of simvastatin is presented in Fig. 1 and Table 3. When simvastatin lactone was administered, plasma acid or lactone showed no difference between the KO and WT mice. The lactone brain/plasma concentration ratio was statistically higher in the KO (13) than the WT (0.98) mice. Neither lactone nor acid was observed in liver after dosing of the lactone form. Although after dosing of the acid form, simvastatin lactone was not observed in plasma, liver, or brain, the acid was present in plasma and liver but not detected in the brain. The liver/plasma concentration ratio at 0.5 h postdose was higher than unity and similar between the KO and WT mice.
Plasma and tissue concentrations and tissue/plasma concentration ratios of simvastatin after a 3.0 mg/kg s.c. administration to mdrl a/b KO and WT mice
Concentrations in plasma, liver, and brain at 0, 0.5, 1.5, and 3 h postdose that are not shown in the table were below the lower limit of quantitation (LLOQ) (acid/lactone): 10/10 ng/ml in plasma, 10/10 ng/ml in brain homogenate, and 40/40 ng/ml in liver homogenate. Data are means ± S.D.
Disposition of Pravastatin in Plasma, Liver, and Brain. As shown in Fig. 1 and Table 4, pravastatin acid was observed in plasma and liver but not brain after administration of pravastatin acid. The plasma concentrations and the liver/plasma concentration ratios were comparable between the KO and WT mice.
Plasma and tissue concentrations and tissue/plasma concentration ratios of pravastatin after a 3.0 mg/kg s.c. administration to mdrl a/b KO and WT mice
Concentrations in plasma, liver, and brain at 0, 0.5, 1.5, and 3 h postdose that are not shown in the table were below the lower limit of quantitation (LLOQ) (acid/lactone): 10/10 ng/ml in plasma, 10/10 ng/ml in brain homogenate, and 40/40 ng/ml in liver homogenate. Data are means ± S.D.
Discussion
Several studies have shown that both drug-metabolizing enzymes such as CYP3A and CYP2C and transporters including P-gp, Mrp/MRP2, BCRP, bile salt export pump (BSEP, ABCB11), and OATP1B1 may play a significant role in the disposition and drug interactions of multiple statins (Boyd et al., 2000; Shitara et al., 2003, 2004; Hirano et al., 2005a; Holtzman et al., 2006). Furthermore, studies have demonstrated that statins interconvert between two forms, i.e., acid and lactone in vivo (Neuvonen and Jalava, 1996; Kantola et al., 1998a,b; Backman, 2002; Prueksaritanont et al., 2002a), which exhibit differential properties toward drug-metabolizing enzymes (Ishigami et al., 2001) and transporters such as P-gp (Bogman et al., 2001; Chen et al., 2005). Understanding the mechanism of drug interactions with statins has become increasingly important because of the potential for serious adverse effects such as myopathy. However, the role of P-gp in in vivo statin disposition and potential drug interactions remains unclear. So far, only one study has examined the effect of P-gp on cerivastatin disposition using mdr1a/b KO and WT mice (Kivistö et al., 2004). The present study focuses on the examination of the role of P-gp in the in vivo disposition of atorvastatin, lovastatin, simvastatin, and pravastatin (acid form only) in both lactone and acid forms using mdr1a/b KO and WT mice. A dose of 3 mg/kg was selected after taking into consideration the clinical relevance in systemic plasma concentrations and the ability to measure by liquid chromatography/MS/MS methods.
Consistent among the four statins tested in the present study, the brain penetration of statins in the acid form, which is the pharmacologically active form, appeared to be low in both KO and WT mice, suggesting that P-gp has limited impact on the brain penetration of these statin acids. Although lovastatin acid showed a significantly higher brain/plasma ratio in the KO than the WT mice, the overall brain penetration for both strains was poor. In contrast, lovastatin lactone and simvastatin lactone showed significantly higher brain penetration in KO than in WT mice, suggesting that they are substrates of P-gp, and P-gp limits their availability to the brain.
Because both the KO and WT mice showed a similar poor brain penetration for the acid form of statins, the data imply that other efflux transporters such as MRP2 (Nies et al., 2004) and BCRP (Cooray et al., 2002) at the blood-brain barrier may have contributed to the limited brain penetration. However, few reports have shown that statin acids are substrates of these MRP or BCRP transporters, and thus further investigation is warranted.
Conversion of lactones to acids was expected and observed for atorvastatin, simvastatin, and lovastatin with different degrees (Table 5). The plasma acid/lactone concentration ratios were similar between the KO and WT mice, suggesting that enzymes/processes that mediate the conversion were unlikely to be altered due to P-gp. Interestingly, conversion from acid to lactone was not observed. No lactone was detected in plasma or brain or liver when the acid form was administered, suggesting that the conversion of acid to lactone either did not occur or occurred but the lactones were below the limit of quantitation in mice, which differs from observation in humans (Neuvonen and Jalava, 1996; Kantola et al., 1998a,b; Backman et al., 2002). The difference could be partially due to different species and/or route of administration (oral in humans versus s.c. in the present study).
Concentration ratios between acid and lactone forms at 0.5 h after a 3 mg/kg s.c. administration of atorvastatin, lovastatin, and simvastatin lactone forms to mdr la/b KO and WT mice
Data are means ± S.D.
Because only the acid form was observed in the liver after dosing of the acid form, the liver/plasma concentration ratios were calculated for the acid form only. Two interesting observations were made across the four statin acids. First, the liver/plasma concentration ratios were higher than unity, suggesting potential active uptake of these acids to the liver. Second, there was no significant difference in the liver/plasma ratio between the KO and WT mice, indicating that P-gp does not significantly affect the liver-plasma distribution of these acids.
In summary, in the present study we examined the role of P-gp in the brain-plasma and liver-plasma disposition of the acid and lactone forms of atorvastatin, lovastatin, simvastatin, and pravastatin (acid only) in mdr1a/b KO and WT mice. Our data demonstrated that P-gp does not play a significant role in limiting the access of the pharmacologically active form of these statins to the brain or their distribution to the liver.
Footnotes
-
doi:10.1124/dmd.107.015677.
-
ABBREVIATIONS: P450, cytochrome P450; P-gp, P-glycoprotein, ABCB1; MRP2 (Mrp2), multidrug resistance-associated protein 2, ABCC2; BCRP (Bcrp), breast cancer resistance protein, ABCG2; OATP, organic anion-transporting polypeptide; KO, knockout; WT, wild-type; HPLC, high performance liquid chromatography; MS, mass spectrometry.
- Received March 7, 2007.
- Accepted July 18, 2007.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}