


Abstract
The role of transporters in the disposition of (+)-2-[4-({[2-(benzo[1,3]dioxol-5-yloxy)-pyridine-3-carbonyl]-amino}-methyl)-3-fluoro-phenoxy]-propionic acid (CP-671,305), an orally active inhibitor of phosphodiesterase-4, was examined. In bile duct-exteriorized rats, a 7.4-fold decrease in the half-life of CP-671,305 was observed, implicating enterohepatic recirculation. Statistically significant differences in CP-671,305 pharmacokinetics (clearance and area under the curve) were discernible in cyclosporin A- or rifampicin-pretreated rats. Considering that cyclosporin A and rifampicin inhibit multiple uptake/efflux transporters, the interactions of CP-671,305 with major human hepatic drug transporters, multidrug resistance protein 1 (MDR1), multidrug resistance-associated protein 2 (MRP2), breast cancer resistant protein (BCRP), and organic anion-transporting polypeptide (OATPs) were evaluated in vitro. CP-671,305 was identified as a substrate of MRP2 and BCRP, but not MDR1. CP-671,305 was a substrate of human OATP2B1 with a high affinity (Km = 4 μM) but not a substrate for human OATP1B1 or OATP1B3. Consistent with these results, examination of hepatobiliary transport of CP-671,305 in hepatocytes indicated active uptake followed by efflux into bile canaliculi. Upon examination as a substrate for major rat hepatic Oatps, CP-671,305 displayed high affinity (Km = 12 μM) for Oatp1a4. The role of rat Mrp2 in the biliary excretion was also examined in Mrp2-deficient rats. The observations that CP-671,305 pharmacokinetics were largely unaltered suggested that compromised biliary clearance of CP-671,305 was compensated by increased urinary clearance. Overall, these studies suggest that hepatic transporters play an important role in the disposition and clearance of CP-671,305 in rat and human, and as such, these studies should aid in the design of clinical drug-drug interaction studies.
Toward the pursuit of novel isozyme-selective phosphodiesterase-4 (PDE4) inhibitors for the treatment of chronic obstructive pulmonary disease and asthma (Houslay et al., 2005), we have recently disclosed the de novo nicotinamide class of potent, selective, and orally active PDE4-D inhibitors, exemplified by CP-671,305 (Fig. 1), which has been selected for toxicity assessments and possible investigations in humans (Kalgutkar et al., 2004). CP-671,305 is resistant to metabolism by either phase I or phase II drug-metabolizing enzymes in liver microsomes and hepatocytes from preclinical species and human; these findings are supported by the lack of detectable metabolites in pooled plasma, urine, and/or bile from rats, dogs, and monkeys after CP-671,305 administration. In addition, CP-671,305 also does not exhibit reversible or irreversible inhibition of the activities of the major human cytochrome P450 enzymes, suggesting that the risks of clinical drug-drug interactions (DDIs) with substrates/inhibitors of cytochrome P450 enzymes are unlikely (Kalgutkar et al., 2004).
However, there exists some potential for DDIs between CP-671,305 and substrates/inhibitors of transport proteins, particularly because preliminary investigations into the clearance mechanism in rats revealed that the compound undergoes substantial biliary excretion in the unchanged form (Kalgutkar et al., 2004), suggesting that active uptake and efflux processes may be involved in the hepatobiliary excretion of CP-671,305 in the rat and therefore potentially in humans. Numerous studies have shown that intestinal and hepatic membrane transport proteins play a central role in mediating the cellular uptake and efflux of structurally diverse endogenous and exogenous xenobiotics, including drugs, and, as such, these processes can be modulated by inducers or inhibitors of transporters, resulting in clinically significant DDIs (Kim, 2006; Shitara et al., 2006). Of much interest in this context is the similarity in the elimination mechanism of CP-671,305 and the selective histamine H1-receptor antagonist and carboxylic acid derivative fexofenadine (see Fig. 1). Hepatic metabolism is also of minimal importance in the elimination of fexofenadine in the rat and human, and like CP-671,305, this lipophilic carboxylic acid is predominantly eliminated via biliary excretion in the unchanged form (Markham and Wagstaff, 1998). Biliary excretion of fexofenadine in humans is mediated by the multidrug resistance 1 gene (human MDR1, P-glycoprotein), located on the bile canalicular membrane of the liver (Cvetkovic et al., 1999). In addition, fexofenadine also functions as a substrate for hepatic uptake by human and rat organic anion transporting polypeptides (OATPs/Oatps) located on the basolateral membrane in the liver (Cvetkovic et al., 1999; Shimizu et al., 2005). In the case of fexofenadine, inhibition/stimulation of these uptake/efflux processes is known to lead to significant DDIs in humans (Hamman et al., 2001; Dresser et al., 2005) and in rat (Kamath et al., 2005).

Structures of CP-671,305 and fexofenadine.
Given some similarity in structure (the presence of a lipophilic carboxylic acid motif) of CP-671,305 with fexofenadine, we decided to systematically investigate the potential involvement of transport proteins in the hepatic uptake and biliary excretion of CP-671,305, initially using the rat as an in vivo model. The DDI potential of CP-671,305 was assessed in the rat using well characterized inhibitors of rat hepatic uptake and efflux transporters such as cyclosporin A (Kobayashi et al., 2004; Shitara et al., 2004) and rifampicin (Fattinger et al., 2000; Shitara et al., 2002). In addition, the role of multidrug resistance-associated protein 2 (ABCC2/human MRP2/rat Mrp2) in the biliary excretion of CP-671,305 was also examined in transport-deficient (TR-) rats, naturally occurring mutants of the Wistar rat that are Mrp2-deficient (Johnson et al., 2006). Additional in vitro studies were conducted in the sandwich-cultured human hepatocyte (Bi et al., 2006) to evaluate the potential of hepatobiliary transport of CP-671,305 in humans. Likewise, in vitro studies assessing the substrate properties of CP-671,305 against major rat/human hepatic uptake and efflux transport proteins were also executed. The collective findings from these studies are summarized herein.
Materials and Methods
Materials. CP-671,305 (chemical and isomeric purity >99%) was synthesized at Pfizer Global Research and Development (Groton, CT). Solvents used for analysis were of analytical or high-performance liquid chromatography grade (Fisher Scientific, Pittsburgh, PA). Cyclosporin A, verapamil, and rifampicin were purchased from Sigma-Aldrich Research (St. Louis, MO). Sodium-N-butyrate was purchased from ICN Biochemicals (Aurora, OH).
Hepatobiliary Transport Studies on CP-671,305 in Sandwich-Cultured Cryopreserved Human Hepatocytes. A detailed protocol for measuring hepatobiliary uptake of small-molecule drugs in hepatocyte sandwich culture has been disclosed by us (Bi et al., 2006). In brief, cryopreserved hepatocyte cell cultures were rinsed twice with 0.5 ml of either regular Hanks' balanced salt solution or Ca2+/Mg2+-free Hanks' balanced salt solution containing 1 mM EGTA and then equilibrated in the same buffers for 10 min at 4°C or 37°C. CP-671,305 (2 μM) in regular Hanks' balanced salt solution was then added to both sets of cultures. The cells were then incubated for an additional 10 min at 4°C or 37°C, after which time the cells were lysed with 0.5 ml of methanol, and CP-671,305 concentrations were measured by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Taurocholate (Bi et al., 2006) and midazolam were used as positive controls, and incubations were conducted in duplicate. Taurocholate concentrations were measured by LC-MS/MS as previously described (Bi et al., 2006). For midazolam, an LC-MS/MS assay similar to the one described for taurocholate was used with the following exceptions: mass spectrometry analysis was conducted in the positive-ion mode with an electrospray ionization probe voltage of 4.5 kV and collision energy of 30 eV. Midazolam was monitored using selected reaction monitoring using the transition m/z 326→291.2.
The equations used to calculate apparent uptake rate (Uptakeapp), apparent intrinsic biliary clearance (CLbile,int,app) (Liu et al., 1999a), and biliary excretion index [BEI; determined using B-CLEAR technology (Qualyst, Inc., Raleigh, NC)] (Liu et al., 1999b) over a 10- or 15-min interval are shown below. In the presence of Ca2+/Mg2+, the bile canaliculi remain intact (closed), whereas in the absence of Ca2+/Mg2+, the bile canaliculi tight junctions are disrupted (opened). Therefore, quantifying the accumulation of a test compound in the presence and absence of divalent cations allows one to determine the amount of test compound in the bile canaliculi. 


CP-671,305 Transport Studies Using Single Transporter Expression Cells. Human OATP1B1 (König et al., 2000a), OATP1B3 (König et al., 2000b), and OATP2B1 expressed in human embryonic kidney 293 (HEK 293) cells were obtained from Prof. Dietrich Keppler (DKFZ, Heidelberg, Germany), whereas rat Oatp1a1 (Eckhardt et al., 1999) and Oatp1b2 expressed in Chinese Hamster Ovary (CHO) cells were generously provided by Prof. Peter Meier (University of Zurich, Zurich, Switzerland). Rat Oatp1a4 was expressed in HEK-tetracycline inducible cells (Tet-On; Clontech, Mountain View, CA) obtained through collaboration with Xenoport, Inc. (Santa Clara, CA). On day 1 of the study, ∼2.5 × 105 cells transfected with a given OATP/Oatp isoform were seeded on a 24-well poly-d-lysine coated plate (BioCoat; BD Biosciences, San Jose, CA) and cultured overnight. To increase the expression of the OATPs/Oatps in the transfected cell lines, the medium was changed the following day, and the cells were cultured with 10 mM sodium-N-butyrate 24 h before conducting the transport assay as previously described by several laboratories (Chen et al., 1997; König et al., 2000a,b; Campbell et al., 2004; Liu et al., 2006; Treiber et al., 2007). (During validation of the transporter-transfected cell lines, we have found that induction with sodium-N-butyrate is essential to see transport activity in these cells. When sodium-N-butyrate is not used in these incubations, there is a tendency for numerous false negatives.) To induce the HEK-Tet-On cells, the medium was changed 24 h before the assay to incubation media that contained 10 mM sodium butyrate and 2 μg/ml doxycycline. On day 3, the cells were first washed with Hanks' buffered salt solution (Invitrogen, Carlsbad, CA). Uptake was measured at eight concentrations of CP-671,305 (0.5-64 μM) for 5 min at 37°C, and the assay was stopped by three washes with ice-cold uptake buffer. To verify transport activity for each cell line, parallel incubations were conducted with a positive control estrone-3-sulfate (10 μM) (Tamai et al., 2000). Uptake was shown to be linear up to 10 min (data not shown). CP-671,305 was then extracted from the cells by incubating the cells in acetonitrile for 30 min and CP-671,305 concentrations measured by LC-MS/MS. To account for the nonspecific binding of CP-671,305 to cells, uptake in untransfected HEK and CHO cells (treated with sodium-N-butyrate) was performed in parallel. In the case of rat Oatp1a4 studies, CP-671,305 uptake was also examined in HEK-tetracycline cells that lacked doxycycline and sodium-N-butyrate pretreatment to induce transporter expression. Unpublished data in-house have shown that for this inducible promoter, the level of Oatp1a4 transporter activity in cells untreated with inducers was similar to the untransfected cells. The uptake in untransfected cells was subtracted from the uptake in the transfected cells to determine the transporter activity. The data were corrected for background in untransfected cells exposed to sodium butyrate. The kinetic curves for CP-671,305 transport by rat and human OATP isoforms was generated by fitting the data to a Michaelis-Menten curve by nonlinear regression using OriginPro software (OriginLab, Northampton, MA). Represented data are an average of three separate experiments. In addition, each data point is from triplicate samples.
Analytical Methodology. Samples were concentrated on an EVX-192 Apricot Evaporex apparatus (Apricot Designs, Monrovia, CA). The residue was reconstituted to half the original volume in aqueous mobile phase before LC-MS/MS analysis. A Gilson 215 Multiprobe autosampler (Gilson Instruments, Middleton, WI) was used to deliver 25-μl injection volumes. Further sample concentration and on-line sample clean up was achieved using a single-column/valve-switching system that consisted of a Showa Denko 1.5 × 5-mm cartridge-style column (Optimize Technologies, Inc., Oregon City, OR) custom-packed with Showa Denko (Tokyo, Japan) ozone depletion potential polymeric material with a 13-μm particle size, plumbed using a 10-port two-position switching valve (Valco Instruments, Houston, TX). Two high-performance liquid chromatography pumps (model SCL-10A VP; Shimadzu Inc., Torrance, CA) delivered flow rates of 1 ml/min. The aqueous mobile phase (load step) was 98% 2 mM ammonium acetate/2% methanol. The organic mobile phase (elute step) was 10% 2 mM ammonium acetate/90% acetonitrile/methanol (50:50). Mass spectral analysis was performed on a SCIEX API 4000 triple-quadruple mass spectrometer (PE Sciex, Ontario, ON, Canada) equipped with a turbo ion spray interface. Data were acquired in negative ion mode with an electrospray ionization probe voltage of 4.5 kV. Selected reaction monitoring was used to simultaneously monitor for analyte and internal standard. The following selected reaction monitoring transitions were used for the detection of analytes: 453.5 m/z to 256.5 m/z for CP-671305 and 685 m/z to 366 m/z for internal standard. A Q2 offset voltage of 5 V was used, and collision energy was set to be 30 eV.
Human MDR 1/MDCKII-, Human ABCG2 (BCRP)/MDCKII-, and Human ABCC2 (MRP2)/MDCKII-Mediated Efflux Assay. Madin-Darby canine kidney (MDCK) II, MDR1/MDCKII, MRP2/MDCK (P. Borst, Netherlands Cancer Institute, Amsterdam, The Netherlands), and BCRP/MDCK cells (Xiao et al., 2006) were seeded at a cell density of 2.5 × 105 cells/ml in complete MEM-α media on Falcon/BD 96-well membrane inserts (Invitrogen), placed into BD feeder trays (BD Biosciences, San Jose, CA) containing 37 ml of complete MEM-α growth media (Invitrogen), and incubated (37°C, 95% humidity, 5% CO2) for 5 days. Transwell studies were conducted on day 5.
Apical-to-basolateral studies. On day 5, medium was removed from the insert, 75 μl of fresh buffer B (Hanks' balanced salt solution with added components; Invitrogen) containing CP-671,305 (final concentration, 2 μM) was added using a 96-well pipettor (Apricot Designs, Monrovia, CA), and the entire insert was placed into a 96-well collection plate (Marsh, Rochester, NY) containing 250 μl of buffer B per well, covered, and then incubated at 37°C, 95% humidity, and 5% CO2. After 5 h, 150 μl was removed from each well of the collection plate and transferred to a fresh 1.2-ml 96-well deep well plate. The remaining 100 μl of buffer B in the collection plate was removed and replaced with 100 μl of acetonitrile containing an internal standard. After mixing, 60 μl of acetonitrile was removed and added to the 150 μl of buffer B sample previously removed.
Basolateral-to-apical studies. Medium from the 96-well insert was removed and replaced with 75 μl of fresh buffer B. The insert was then placed into a fresh 96-well collection plate containing 250 μl of CP-671,305 in buffer B per well and incubated for 5 h. Afterward, 50 μl of sample from each well of the insert was removed and placed into a 1.2-ml deep well block containing 100 μl of fresh buffer B and 60 μl of acetonitrile containing an internal standard. All studies with CP-671,305 were conducted in triplicate. Parallel studies with proprietary substrates for MDR1, BCRP, and MRP2 conducted in duplicate revealed the anticipated results indicating the formation of good monolayers in the plated cells. All samples were analyzed by LC-MS/MS detection. CP-671,305 concentrations in the MDCK efflux assays were measured using our previously published high-throughput LC-MS/MS methodology (Xiao et al., 2006).
Data processing. The following equations were used to determine apparent permeability and efflux ratios. Equation 1: Papp (cm/s) = ((((receiver units - blank) · receiver cm3 · dilution factor) · donor cm3)/s)/((membrane surface area (cm2) · (T = 0 donor units - blank) · dilution factor donor cm3)); and eq. 2: Efflux ratio = basolateral-to-apical Papp/apical-to basolateral Papp.
Pharmacokinetic Studies in Rats. Animal care and in vivo procedures were conducted according to the guidelines of the Pfizer Animal Care and Use Committee. Jugular vein cannulated and bile duct-exteriorized male Sprague-Dawley rats (230-250 g), obtained from Charles River Laboratories (Wilmington, MA), were used for these studies. All animals were housed individually in metabolism cages during the course of the studies. All animals were fasted overnight before dosing, whereas access to water was provided ad libitum. Animals were fed following collection of the 2-h blood samples. CP-671,305 (3 mg/kg) in glycerol formal was administered intravenously (i.v.) via the jugular vein to three rats over 30 s, and serial plasma samples were collected before dosing and 0.033, 0.15, 0.5, 1, 2, 4, 6, 8, and 24 h after dosing. All plasma samples were kept frozen until analysis. In addition, an i.v. dose of 3 mg/kg was also given to bile duct-exteriorized rats (n = 3), where plasma, bile, and urine samples were collected on ice for up to 24 h and stored at -20°C until analysis. For the DDI studies, cyclosporin A (20 mg/kg in glycerol formal) was administered i.v. 30 min before intravenous administration of CP-671,305. Rifampicin (50 mg/kg) in saline was administered i.v. 5 min before the administration of CP-671,305. Plasma samples from the various pharmacokinetic studies were generated by centrifugation at 3000 rpm for 10 min at 4°C. Samples were stored at -20°C, pending analysis by LC-MS/MS as previously described (Kalgutkar et al., 2004).
CP-671,305 Disposition in Bile Duct-Exteriorized Normal Wistar and TR-Rats. Jugular vein and bile duct-exteriorized male Wistar rats (395-490 g) and TR- rats of the same sex (386-444 g) were purchased from Charles River Laboratories, Inc. (Wilmington, MA). CP-671,305 (1 mg/kg) in glycerol formal was administered i.v. via the jugular vein to wild-type and TR- rats (n = 3) over 30 s, and serial plasma samples were collected at 0.033, 0.15, 0.5, 1, 2, 4, 6, 8, and 24 h after dosing. All plasma samples were kept frozen until analysis. In addition, urine and bile samples were also collected on ice up to 24 h and stored at -20°C, pending analysis by LC-MS/MS as previously described (Kalgutkar et al., 2004).
Pharmacokinetic Data Analysis. Plasma concentration-time profiles were analyzed using the well established noncompartmental method in WinNonlin v2.1 (Pharsight, Mountain View, CA). Plasma clearance (CLp) of CP-671,305 was calculated as the intravenous dose divided by the area under the plasma concentration-time curve from 0 to infinity (AUC0-∞). AUC0-∞ was calculated by the linear trapezoid rule. The terminal slope of the ln(concentration) versus time plot was calculated by linear least-squares regression, and the half-life was calculated as 0.693 divided by the absolute value of the slope. The steady-state volume of distribution (Vdss) was determined using noncompartmental analysis as follows: 
where AUMC is the total area under the first moment of the drug concentration-time curve from time 0 to infinity.
Statistical Analysis. An unpaired two-tailed Student's t test was used to assess significance of differences in various pharmacokinetic parameters of CP-671,305 across groups. In instances where parameters possessed unequal variances, statistical analysis was performed with Welch's correction. One-way analysis of variance with a Bonferroni post test comparison was used to assess the difference in parameters for CP-671,305 across groups. In all cases, p < 0.05 was predetermined as the criterion for significance. All statistical analysis was performed using GraphPad Prism version 4.0 for Windows (GraphPad Software, San Diego, CA).
Results
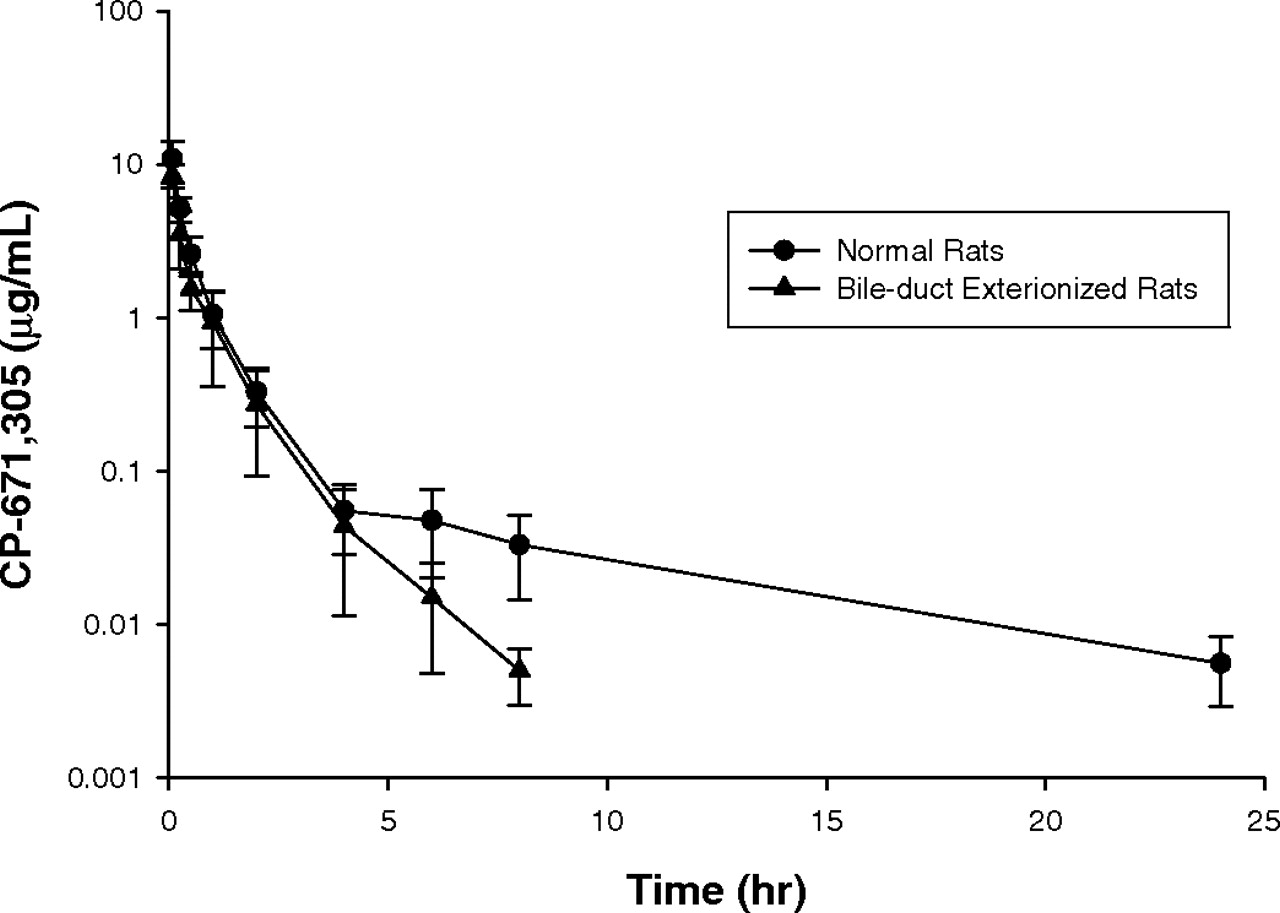
Effect of Cyclosporin A and Rifampicin on the Intravenous Pharmacokinetics of CP-671,305 in the Sprague-Dawley Rat. The pharmacokinetic parameters describing the disposition of CP-671,305 in normal and bile duct-exteriorized Sprague-Dawley rats are shown in Table 1. The mean concentration-versus-time profiles in normal and bile duct-exteriorized rats after intravenous dosing is shown in Fig. 2. After a single i.v. dose of 3 mg/kg, CP-671,305 exhibited a low CLp (8.7 ± 1.8 ml/min/kg) and a low Vdss (0.8 ± 0.3 l/kg), resulting in a mean elimination half-life of 6.5 ± 1.3 h. The corresponding AUC0-∞ was 5.96 ± 1.40 μg · h/ml. In bile duct-exteriorized rats administered a 3 mg/kg i.v. dose of CP-671,305, the half-life of CP-671,305 decreased by ∼7.4-fold (p < 0.0001). A statistically significant 1.5-fold increase in CLp (p < 0.05) was also observed after i.v. dosing to bile duct-exteriorized rats, suggestive of enterohepatic biliary recirculation of parent drug in rats. Unchanged CP-671,305 was the major component in pooled rat bile (35 ± 19%, 0-24 h) and urine (22 ± 8%, 0-24 h), respectively.
Single-dose intravenous pharmacokinetic parameters of CP-671,305 in Sprague-Dawley rats
Data are presented as mean ± S.D. from three male rats and were derived from noncompartmental analysis.

Plasma concentration-time profiles of CP-671,305 after i.v. (3 mg/kg) administration to normal (•) and bile duct-exteriorized (▴) male Sprague-Dawley rats (mean ± S.D.; n = 3).

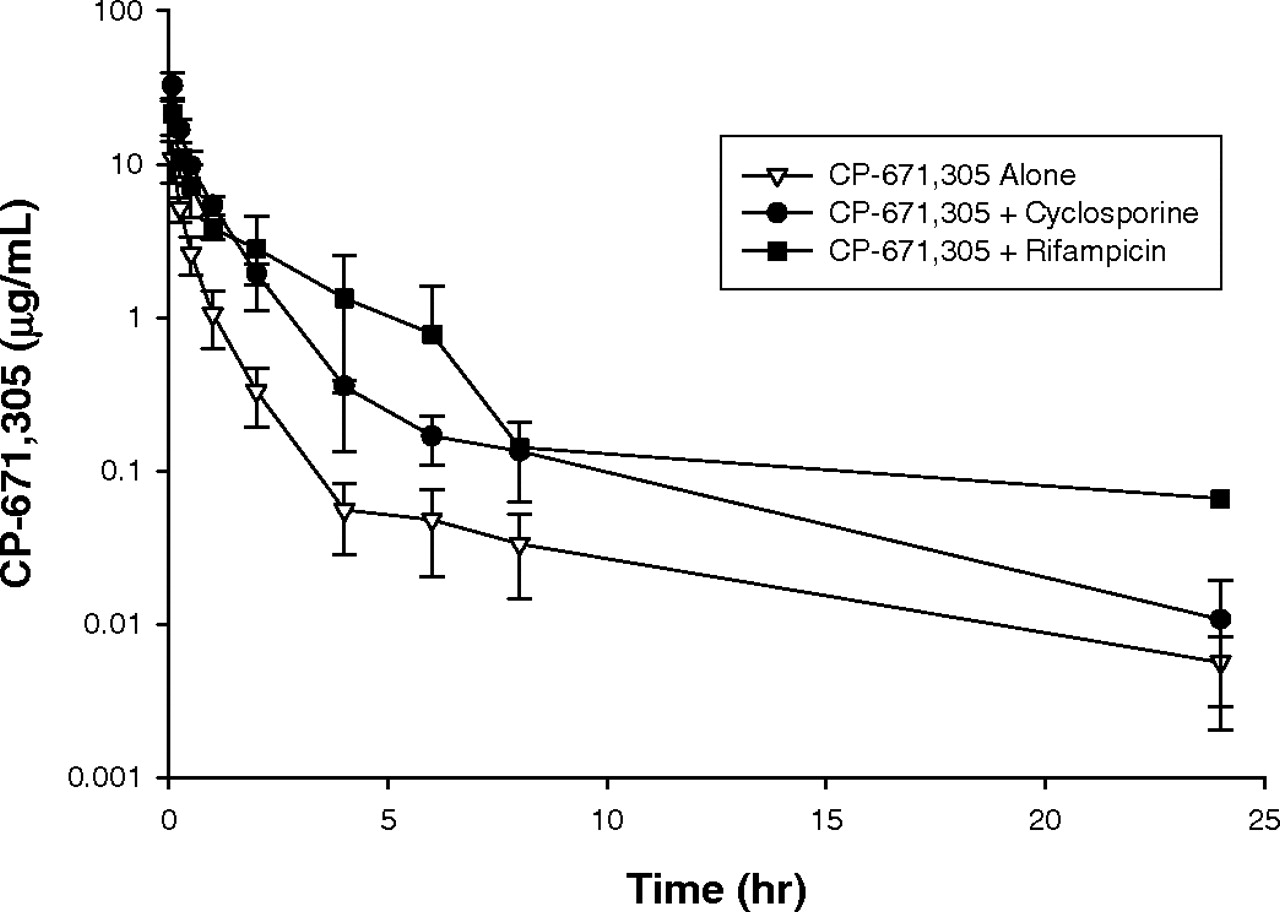
Intravenous plasma concentration-time profiles in male Sprague-Dawley rats of CP-671,305 administered alone (▿) or in combination with cyclosporin A (•) or rifampicin (▪). Rats (n = 3-4) received CP-671,305 at i.v. doses of 3 mg/kg. Cyclosporin A was given i.v. at a dose of 20 mg/kg, 30 min before CP-671,305. Rifampicin was given i.v. at a dose of 50 mg/kg, 5 min before CP-671,305.
As depicted in Table 1, i.v. administration of CP-671,305 to cyclosporin A-pretreated and rifampicin-pretreated normal Sprague-Dawley rats led to statistically significant ∼4-fold increase in CP-671,305 AUC0-∞ and a ∼4-fold decrease in CP-671,305 CLp.An approximately 2- to 4-fold decrease in the volume of distribution of CP-671,305 was also observed in cyclosporin A-pretreated and rifampicin-pretreated rats. The mean concentration-time curves in untreated, cyclosporin A-pretreated and rifampicin-pretreated rats after i.v. dosing of CP-671,305 is shown in Fig. 3.
Intravenous Pharmacokinetics of CP-671,305 in Mrp2-Deficient TR-Rats. To assess the role of rat Mrp2 in the biliary excretion of CP-671,305, the disposition of CP-671,305 was examined in wild-type and Mrp2-deficient TR- rats. As shown in Table 2 and Fig. 4, the pharmacokinetic parameters of CP-671,305 (1 mg/kg) following i.v. administration to wild-type Wistar rats and Mrp2-deficient TR- rats were comparable with no statistically significant differences in CLp, Vdss, and AUC0-∞. Interestingly, the percentage of CP-671,305 dose excreted in the bile of TR- rats decreased from 27 ± 5.4 to 9.1 ± 1.7 when compared with wild-type Wistar rats, whereas the percentage of CP-671,305 dose excreted in the urine of TR- rats increased from 24 ± 4.1 to 49 ± 5.6 in comparison with wild-type Wistar rats. Estimates of the biliary (CLbile) and renal (CLurine) clearance revealed a statistically significant (p < 0.05) ∼3.75-fold decrease and ∼1.7-fold increase in CLbile and CLurine rates, respectively, of CP-671,305 in TR- rats compared with wild-type animals.
Single-dose intravenous pharmacokinetics of CP-671,305 (1 mg/kg) in wild-type Wistar rats and Mrp2-deficient TR- rats
Data are presented as mean ± S.D. from three male rats and were derived from noncompartmental analysis.
Hepatobiliary Transport of CP-671,305 in Sandwich-Cultured Cryopreserved Human Hepatocytes. The hepatobiliary transport of CP-671,305 was examined in sandwich-cultured cryopreserved human hepatocytes using a previously described methodology (Bi et al., 2006). As seen in Table 3, relative to taurocholate, CP-671,305 exhibited moderate apparent uptake (Uptakeapp) and apparent intrinsic biliary clearance (CLbile,int,app). The corresponding mean BEI for CP-671,305 was 28. Under the present experimental conditions, taurocholate was used as a positive control and exhibited Uptakeapp, CLbile,int,app, and BEI values similar to those described previously (see Table 3) (Bi et al., 2006). The influence of temperature on the accumulation of CP-671,305 was also examined in this model. Taurocholate (Bi et al., 2006) and midazolam were used as reference compounds in this analysis. [Uptake of midazolam is not affected in the presence of 10 μM cyclosporine A, which is consistent with the passive diffusion of midazolam (data not shown).] As shown in Table 4, midazolam accumulation was not influenced by temperature suggestive of simple diffusion, whereas taurocholate accumulation decreased by ∼80% at 4°C, implicating a predominantly transport-mediated uptake process. Under these conditions, the accumulation of CP-671,305 at 4°C decreased to ∼24% of that seen at 37°C. This finding suggests that ∼76% of CP-671,305 uptake is potentially mediated via an active process involving transport proteins.
Uptake, biliary clearance and biliary excretion of CP-671,305 in sandwichcultured cryopreserved human hepatocytes
Results are presented as the mean ± S.D. for CP-671,305 (n = 3) and as the average of two separate experiments (n = 2) for taurocholate.
Effect of temperature on CP-671,305 accumulation in sandwich-cultured cryopreserved human hepatocytes
Results are presented as the mean of two replicates from each of two separate experiments.

Plasma concentration-time profiles of CP-671,305 after i.v. (1 mg/kg) administration to Mrp2-deficient TR- (▴) and wild-type (•) Wistar rats (mean ± S.D.; n = 3).
Transport of CP-671,305 by Major Hepatic Efflux Transporters. CP-671,305 (2 μM) did not reveal asymmetric permeability in the flux ratio from the basolateral-to-apical (BA) side/apical-to-basolateral (AB) side in either the parental or MDR1-MDCKII cells, suggesting that CP-671,305 is not a substrate for human MDR1 (Table 5). However, a 7- and 9.5-fold asymmetric permeability was discernible in the MDCKII cell line stably transfected with MRP2 and BCRP genes, respectively (see Table 5). These observations suggested that CP-671,305 is a substrate for potential biliary efflux mediated by MRP2 and BCRP in human.
Examining the potential of CP-671,305 as a substrate for human biliary transport proteins
Data (mean ± S.D.) are from three independent determinations.
CP-671,305 Uptake by Major Rat Hepatic Uptake Transporters. The uptake of CP-671,305 was measured in the rat Oatp-transfected cells and control cells. CP-671,305 uptake in induced (i.e., pretreated with sodium-N-butyrate and doxycycline) rat Oatp1a4 (Oatp2)-transfected HEK cells was significantly higher compared with that of the uninduced rat Oatp1a4 HEK cells (Fig. 5A). The background-subtracted CP-671,305 uptake rates were obtained as the difference in uptake rates in induced and uninduced Oatp1a4-transfected HEK cells at each of the concentrations used. Unpublished data in-house has shown that for this inducible promoter, the level of Oatp1a4 transporter activity in cells untreated with inducers is similar to untransfected cells. CP-671,305 uptake by Oatp1a4 displayed saturable transport with a Km value of 12 μM upon fitting of the data. In contrast, Km for the transport of CP-671,305 by Oatp1b2 (Oatp4) could not be determined because of weak uptake (Km > 100 μM) (Fig. 5B). Higher concentrations of CP-671,305 could not be examined due to its limited aqueous solubility. Although the comparison of CP-671,305 uptake by Oatp1a4 and Oatp1b2 suggest similar levels of uptake (Fig. 5, A and B), it is noteworthy to point out that the background uptake for Oatp1a4 and Oatp1b2 was different implicating that CP-671,305 interaction with the two transporters was different. Finally, there was no difference in CP-671,305 uptake by induced rat Oatp1a1 (Oatp1)-transfected CHO cells compared with control CHO cells (data not shown). Rat Oatp-mediated uptake in the HEK and CHO cells with a positive control estrone-3-sulfate at a concentration of 10 μM indicated transport consistent to that reported previously by Noé et al. (1997).
CP-671,305 Uptake by Major Human Hepatic Uptake Transporters. CP-671,305 uptake by human OATP2B1 (OATPB)-transfected HEK 293 cells was significantly higher compared with that of the control HEK 293 cells. The background-subtracted CP-671,305 uptake by OATP2B1 displayed saturable transport with a Km value of 4 μM upon fitting of the data (Fig. 6). (In Fig. 5A, the sigmoidal shape of the curve resulting from fitting of the data is primarily because of the 4-μM point. If the point is removed, the curve would assume the normal hyperbolic shape typical for transporter kinetics. The observation that background uptake in control cells in Figs. 5 and 6 is different is not surprising, considering that each of the transporters was transfected in different cellular backgrounds.) Human OATP2B1-mediated uptake in the HEK cells with a positive control estrone-3-sulfate at a concentration of 10 μM indicated transport consistent to that reported previously by Sai et al. (2006). In contrast, there was no significant difference in CP-671,305 uptake by HEK 293 cells that were transfected with human OATP1B1 (OATPC) or OATP1B3 (OATP8) versus control HEK 293 cells (data not shown).
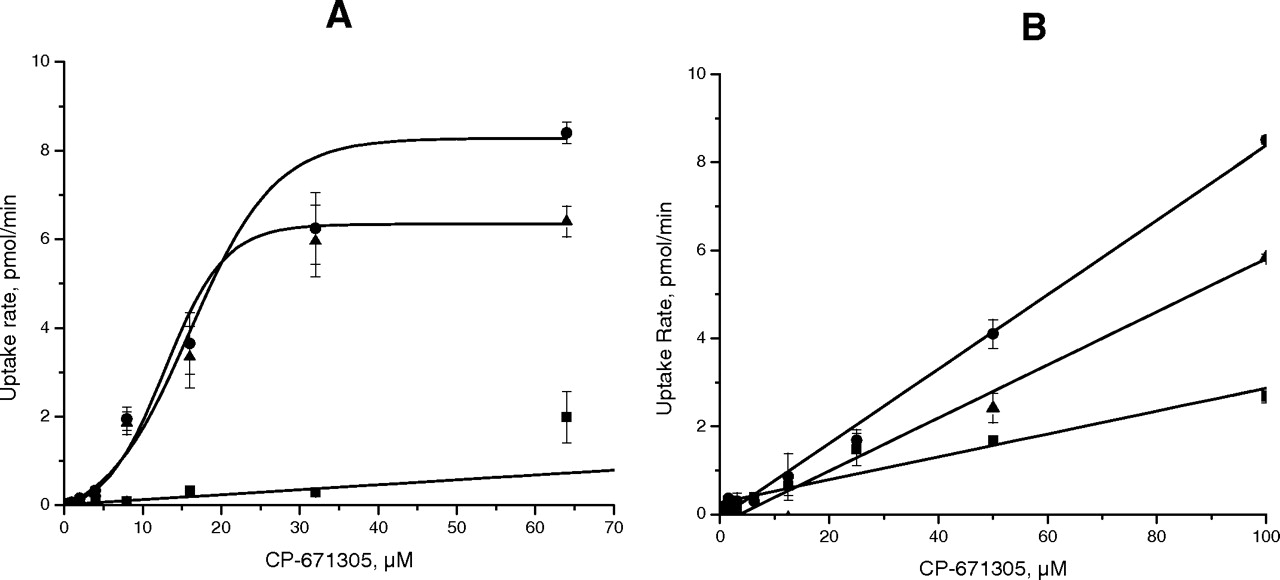
CP-671,305 uptake in rOatp1a4 (A) transfected HEK and rOatp1b2 (B) transfected CHO cells (•). Uptake in uninduced HEK cells and CHO cells was performed in parallel (▪). rOatp-specific uptake was calculated after subtracting nonspecific uptake in un-induced HEK and CHO cells (▴). Data were fitted and kinetic parameters were calculated by nonlinear regression analysis. CP-671,305 uptake rates over 5 min at concentrations from 0.5 to 64 μM in Oatp1a4-transfected HEK cells showed a single saturable transport with a Km of 12 μM.
Discussion
Over the past 15 years, a number of important human drug transporters have been identified that are expressed at the apical or basolateral side of the epithelial cells in various tissues. Most drug transport proteins which catalyze cellular uptake and efflux belong to two superfamilies, namely, the solute-linked carrier and the ABC (ATP-binding cassette) transporters, respectively (Kim, 2006; Shitara et al., 2006). The OATP and MRP combination, which represents two classes of transporters from the solute-linked carrier and ABC super-family, respectively, plays an important role in the hepatic transport of organic anions at the sinusoidal and canalicular membranes. In the human liver, OATP1B1 (also known as OATP2 or OATPC), OATP1B3 (OATP8), and OATP2B1 (OATPB) are predominant transporters responsible for the hepatic uptake of a variety of organic anionic compounds (Mikkaichi et al., 2004). Whereas Oatp1a1 (Oatp1), Oatp1a4 (Oatp2), and Oatp1b2 (Oatp4) are highly expressed in rat liver and function similarly as their human counterparts (Chandra and Brouwer, 2004). Once taken up into hepatocytes, anionic compounds and/or metabolites derived from phase II glucuronidation can undergo MRP2/Mrp2-mediated biliary excretion (Büchler et al., 1996). Besides MRP2/Mrp2, MDR1/Mdr1 and/or BCRP/Bcrp can also be involved in the active efflux of organic anions into bile (Hirano et al., 2005). Uptake via OATPs/Oatps followed by excretion via MRP/Mrp2 or other efflux transport proteins constitutes vectorial transport for the hepatobiliary excretion of drugs and modulation of either of these transport mechanisms can potentially result in a DDI as has been demonstrated in several instances (Shitara et al., 2006).
Studies on the clearance mechanism of CP-671,305 in rats revealed that a significant portion (∼35%) of CP-671,305 dose was eliminated in the unchanged form in rat bile. Consistent with this observation, assessment of the pharmacokinetic parameters of CP-671,305 in bile duct-exteriorized rats revealed ∼7.4-fold decrease in the elimination half-life of CP-671,305, suggesting that following biliary excretion, CP-671,305 effectively undergoes enterohepatic recirculation. Our finding that both cyclosporin A and rifampicin enhanced the AUC0-∞ of CP-671,305 lend further support to the hypothesis that both cyclosporin A and rifampicin exclusively inhibit active processes that modulate the hepatobiliary transport of unchanged CP-671,305 in the rat since our previous studies have demonstrated the resistance of this compound toward metabolism via phase I/II enzymes (Kalgutkar et al., 2004). More recently, using [14C]CP-671,305 as a more sensitive probe for assessments of mass balance and metabolic studies, it was shown that following oral administration at 50 mg/kg, >90% of the radioactivity was eliminated within 48 h after dose. Based on the area under the curve values, the majority of the circulating radioactivity was attributable to unchanged CP-671,305, and furthermore, ∼90-93% of the administered dose was recovered in the unchanged form in the feces and urine of the animals (Chandra Prakash, unpublished observations). Additional support finding that the resulting DDIs arise via inhibition of active hepatobiliary transport of CP-671,305 is provided from our in vitro findings in single transporter-transfected cells, which demonstrated CP-671,305 substrate properties against Oatp1a4 and Oatp1b2 isoforms that are known to be inhibited by both cyclosporin A and rifampicin (Lau et al., 2006).

CP-671,305 uptake in hOATP2B1-transfected HEK cells (•). Uptake in control HEK cells was performed in parallel (▪). hOATP2B1-specific uptake was calculated after subtracting nonspecific uptake in control HEK cells (▴). Data were fitted, and kinetic parameters were calculated by nonlinear regression analysis. CP-671,305 uptake rates over 5 min at concentrations from 0.5 to 64 μM in OATP2B1-transfected HEK cells showed a single saturable transport with a Km of 4 μM.
Use of the mutant TR- or Eisai hyperbilirubinemic rats has enhanced the facile identification of Mrp2 substrates and direct exploration of significance of Mrp2 in the hepatobiliary disposition of xenobiotics. A statistically significant ∼4-fold decrease in CLbile of CP-671,305 was observed in the Mrp2-deficient TR- rats, suggesting a potential role for Mrp2 involvement in the biliary excretion of CP-671,305. This finding provides an alternate explanation for the observed DDIs between CP-671,305 and cyclosporin A/rifampicin because in addition to inhibiting Oatp isoforms, both these compounds are well established inhibitors of rat Mrp2-mediated efflux of xenobiotics (Hesselink et al., 2005; Gupta et al., 2006). Despite a significant decrease in CLbile, the overall pharmacokinetics of CP-671,305, including CLp,Vdss, and AUC0-∞ were not dramatically different from those observed in wild-type animals. This finding was consistent with the observation that reduced biliary excretion was compensated to some extent by an increase in the renal clearance of CP-671,305 potentially due to adaptive changes in expression of other transporters in TR- rats. For example, it is known that the hepatic expression of Mrp3, which also mediates transport of organic anions across the hepatocyte basolateral membrane into sinusoidal blood, is significantly up-regulated in Mrp2-deficient rats (Hirohashi et al., 1998) and has been speculated to play a role in a compensatory shift from biliary to renal elimination for organic anions such as a acetaminophen-glucuronide (Ghanem et al., 2005) and pravastatin (Kivistö et al., 2005). Whether CP-671,305 is a substrate for human and rat MRP3/Mrp3 remains to be evaluated.
Considering the lack of discernible metabolism of CP-671,305 in human hepatic tissue, it is conceivable that CP-671,305 can undergo excretion via nonmetabolic processes in humans, which is also governed by transporters in a manner similar to the rat. Therefore, preliminary studies were initiated to examine the potential contribution of transporters in the hepatobiliary excretion of CP-671,305 in humans. Initial studies in sandwich-cultured cryopreserved human hepatocytes indicated moderate uptake from sinusoidal blood to hepatocytes and efflux of CP-67,305 into bile. Considering that a significant proportion of CP-671,305 uptake was temperature-dependent suggested an active uptake mechanism. Using single hepatic uptake transporter-transfected HEK 293 cells, we demonstrated that CP-671,305 is a substrate of the human liver OATP2B1 transporter. The potential involvement of human efflux transporters in the biliary excretion of CP-671,305 was also assessed. A lack of asymmetry in the flux ratio from BA/AB in MDR1-MDCKII cells suggested that CP-671,305 was not a substrate for MDR1. In contrast, a 7- and 9.5-fold asymmetric permeability discernible in the MDCKII cell line stably transfected with MRP2 and BCRP genes, respectively, suggested that CP-671,305 is a substrate for BCRP- and MRP2-mediated biliary efflux in human. Although the involvement of MRP2 in the biliary efflux of CP-671,305 was strengthened based on the use of the TR- rat model, the potential role of BCRP in the hepatobiliary elimination of CP-671,305 in rat and therefore in human remains less clear. Studies on the disposition of CP-671,305 in BCRP knockout mice (Zaher et al., 2006) are currently underway to better understand the contribution of this transporter in the biliary excretion of CP-671,305.
The finding that CP-671,305 undergoes uptake by Oatp1a4 in rat, whereas OATP2B1 plays a role in the uptake of CP-671,305 into hepatocytes in human is interesting. Nishio et al. (2000) have identified a rat ortholog of human OATP2B1 namely Oatp9 (Oatp2b1) or moat1 that is expressed in the liver and mediates transport of taurocholate, leukotriene C4, and prostaglandins. However, based on the transporter expression profile (mRNA levels), rat Oatp1, 2, and 4 have much higher expression than rat Oatp9 in the liver and are claimed as major rat liver Oatp transport proteins. Thus, rat Oatp9 (Oatp2b1) is not anticipated to play an important role in the hepatic uptake of CP-671,305. In the case of efflux transporters, human MRP2 and rat Mrp2 may play a crucial role in effluxing CP-671,305 into bile in human and rat, respectively. In conclusion, we have identified the uptake and efflux transport proteins that mediate the hepatobiliary excretion of the metabolically resistant PDE4-D inhibitor CP-671,305 in rat and human. Inhibition/stimulation of the OATP2B1-mediated uptake and/or MRP2/BCRP-mediated efflux processes by xenobiotics, including drugs, can lead to potential DDIs with CP-671,305 in humans as has been observed with fexofenadine. Studies are currently underway to assess the role of organic anion transporters in the renal excretion of CP-671,305 in rat and human. Overall, the collective data from these studies should aid in the proper design of clinical DDI studies involving the effects of transport inhibitors on the pharmacokinetics of CP-671,305.
Footnotes
-
doi:10.1124/dmd.107.016162.
-
ABBREVIATIONS: PDE-4, phosphodiesterase-4; CP-671,305, (+)-2-[4-({[2-(benzo[1,3]dioxol-5-yloxy)-pyridine-3-carbonyl]-amino}-methyl)-3-fluoro-phenoxy]-propionic acid; DDI, drug-drug interaction; MDR, multidrug resistance protein; MRP/Mrp, human and rat multidrug resistance-associated protein, respectively; OATP/Oatp, human and rat organic anion transporting polypeptide, respectively; TR-, transporter-deficient; LC-MS/MS, liquid chromatography-tandem mass spectrometry; Uptakeapp, apparent uptake rate; CLbile, biliary clearance; CLurine, urine clearance; CLbile,int,app, apparent intrinsic biliary clearance; BEI, biliary excretion index; HEK 293, human embryonic kidney 293; CLp, plasma clearance; Vdss, volume of distribution at steady state; BA, basolateral-to-apical; AB, apical-to-basolateral; Papp, apparent permeability; CHO, Chinese hamster ovary; BCRP/Bcrp, human and rat breast cancer resistance protein, respectively; MDCK, Madin-Darby canine kidney; AUC0-∞, area under the plasma concentration-time curve from 0 to infinity.
-
↵1 Current affiliation: Metabolism and Pharmacokinetics Department, Genentech Inc., South San Francisco, California.
- Received April 5, 2007.
- Accepted August 6, 2007.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}