


Abstract
Cytochrome P450s (P450s) metabolize a large number of diverse substrates with specific regio- and stereospecificity. A number of compounds, including nicotine, cotinine, and aflatoxin B1, are metabolites of the 94% identical CYP2A13 and CYP2A6 enzymes but at different rates. Phenacetin and 4-aminobiphenyl were identified as substrates of human cytochromes P450 1A2 and 2A13 but not of CYP2A6. The purpose of this study was to identify active site amino acids that are responsible for CYP2A substrate specificity using phenacetin as a structural probe. Ten amino acid residues that differ in the CYP2A13 and CYP2A6 active sites were exchanged between the two enzymes. Phenacetin binding revealed that the six substitution, CYP2A13 S208I, A213S, F300I, A301G, M365V, and G369S decreased phenacetin affinity. Although incorporation of individual CYP2A13 residues into CYP2A6 had little effect on this enzyme's very low levels of phenacetin metabolism, the combination of double, triple, and quadruple substitutions at positions 208, 300, 301, and 369 increasingly endowed CYP2A6 with the ability to metabolize phenacetin. Enzyme kinetics revealed that the CYP2A6 I208S/I300F/G301A/S369G mutant protein O-deethylated phenacetin with a Km of 10.3 μM and a kcat of 2.9 min-1, which compare very favorably with those of CYP2A13 (Km of 10.7 μM and kcat of 3.8 min-1). A 2.15 Å crystal structure of the mutant CYP2A6 I208S/I300F/G301A/S369G protein with phenacetin in the active site provided a structural rationale for the differences in phenacetin metabolism between CYP2A6 and CYP2A13.
The functional human cytochrome P450 2A enzymes include CYP2A13, which is expressed primarily within the respiratory tract with the highest level of mRNA in nasal mucosa, and CYP2A6, which contributes to 1 to 10% of the total P450 in the liver (Su et al., 2000; Zhang et al., 2006). These enzymes metabolize a variety of small cyclic compounds including coumarin, nicotine, aflatoxin B1 (AFB1), naphthalene, styrene, toluene, 4-aminobiphenyl, and phenacetin (Su et al., 2000; Bao et al., 2005; Nakajima et al., 2006; Fukami et al., 2007, 2008). Despite their close identity, a difference of only 32 amino acids, CYP2A13 and CYP2A6 can metabolize the same substrate at different rates or with different metabolites. For example, although both CYP2A13 and CYP2A6 metabolize AFB1, only CYP2A13 metabolizes AFB1 to carcinogenic/toxic epoxide metabolites (He et al., 2006). In the case of 4-aminobiphenyl, a major carcinogen found in tobacco smoke, and phenacetin, an analgesic and antipyretic withdrawn from the market after its implication in human nephropathy, only CYP2A13 is efficient in metabolism, whereas CYP2A6 either metabolizes these substrates at barely detectable rates or not at all (Flower et al., 1985; Nakajima et al., 2006; Fukami et al., 2007).
The overall structure of CYP2A13 is very similar to that of CYP2A6, but there are differences in the identity of 10 amino acids surrounding the active site (Smith et al., 2007). These differences account for an active site in CYP2A13, which is 15 to 20% larger than the active site of CYP2A6 (Smith et al., 2007). The active site volume for CYP2A13 is reported at 307 Å3, a volume intermediate to that of CYP2A6 (250 Å3) and CYP1A2 (375 Å3) (Sansen et al., 2007b; Smith et al., 2007). CYP2A13, CYP2A6, and CYP1A2 each have active sites that are planar and highly hydrophobic and are lined with a number of aromatic phenylalanine residues. In these respects, the active sites are complementary to their generally compact, aromatic substrates (Sansen et al., 2007b; Smith et al., 2007). Thus, the observed differences in substrate metabolism are probably due to a few amino acid side chains that differ between the CYP2A13 and CYP2A6 active sites rather than to global structural differences.
We selected phenacetin as a substrate probe to determine which amino acids within the active site might be responsible for CYP2A substrate specificity because it has been extensively studied throughout the last 20 years (Fischbach and Lenk, 1985a,b). To determine the role that individual amino acids play in CYP2A phenacetin metabolic capability, a series of single and multiple mutants of CYP2A13 and CYP2A6 were examined in this study. Each resulting protein was examined for phenacetin binding (KD) and the ability to catalyze phenacetin O-deethylation. Overall, these results identify four amino acid residues that play key roles in determining the ability of CYP2A enzymes to metabolize phenacetin. To relate these functional findings to structural differences of the two active sites, we crystallized a CYP2A6 quadruple mutant containing all four key amino acids as a complex with phenacetin. This structure, combined with the known structures of CYP2A13 and CYP2A6, provides a structural rationale for the roles of the amino acids at positions 208, 300, 301, and 369 in controlling phenacetin metabolism by human CYP2A enzymes.
Materials and Methods
Chemicals and Reagents. Phenacetin and acetaminophen were purchased from Sigma-Aldrich (St. Louis, MO) at the highest purity available.
Protein Design, Expression, and Purification. Rat NADPH cytochrome P450 oxidoreductase and cytochrome b5 were expressed in Escherichia coli and purified as described previously (Smith et al., 2007). The CYP2A13 and CYP2A6 proteins were engineered by truncating the N-terminal transmembrane sequence (Δ2–23), modifying the N terminus and adding four histidine residues to the C terminus. These modifications were made to increase protein expression and facilitate purification as described previously (Smith et al., 2007). Mutations were produced using either pKK2A13dH or pKK2A6dH as a template and synthetic oligonucleotides (Genosys, Woodlands, TX) containing the desired nucleotide substitutions and selected silent mutations (Table 1) using the QuikChange method (Stratagene, La Jolla, CA). For construction of multiple mutants, the desired nucleotide changes were either incorporated simultaneously using one oligo if the changes were in close proximity (e.g., 300/301 and 365/366) or iteratively by adding an additional mutation to a template that already contained one or more mutations. All genes were completely sequenced to verify the desired mutation(s) and the absence of unintended mutations. The cytochrome P450 2A proteins were all expressed in E. coli TOPP-3 cells (Stratagene) and purified as described previously (Smith et al., 2007).
Sequences are shown below for one of two complementary oligonucleotides used in the construction of the indicated mutation(s) Bold indicates changes from the wild-type sequence. Underline indicates the location of the desired mutation. Italics indicate additional synonymous changes that alter a restriction site and were used to facilitate identification of plasmids containing the desired nonsynonymous mutation. Multiple mutations were made by sequential addition of changes using these same oligonucleotides.
The CYP2A6 I208S/I300F/G301A/G369S protein used for crystallography was purified as described, except that 1% ANAPOE-35 (Anatrace, Maumee, OH) was used instead of 4.8 mM Cymal-5 (Anatrace). This protein was additionally purified on a Superdex 200 16/60 gel filtration column (GE Healthcare, Uppsala, Sweden) in buffer A (50 mM KPi, pH 7.4, 20% glycerol, 0.5 M NaCl, and 1 mM EDTA). The eluting CYP2A6 I208S/I300F/G301A/G369S fractions were pooled and concentrated by centrifugal ultrafiltration with 5 mM phenacetin in buffer A. Detergent, 0.2% ANAPOE-X-405 (Anatrace), was added to the concentrated protein before crystallization.
Reconstitution of Enzymes. CYP2A13, CYP2A6, and mutants thereof were reconstituted with reductase and cytochrome b5 for 20 min at room temperature before use. A 1:6:2 ratio of CYP450 enzymes to reductase to cytochrome b5 was used for all metabolism experiments as this ratio gave maximal metabolism (data not shown).
Phenacetin O-Deethylation. The phenacetin O-deethylation metabolism assay procedure was modified from the method reported by Fukami et al. (2007) for use with purified CYP2A protein. Reconstituted protein (30 pmol of cytochrome P450, 180 pmol of NADPH reductase, and 60 pmol of cytochrome b5) was added to 100 mM potassium phosphate buffer (pH 7.4) with phenacetin to a total volume of 300 μl. Single activity measurements were determined using a phenacetin concentration of 250 μM and a NADPH concentration of 500 μM. Kinetics were determined using phenacetin concentrations of 0 to 250 μM and an NADPH concentration of 1 mM. Reactions were initiated with the addition of NADPH and proceeded for 30 min during incubation in a water bath at 37°C. Reactions were terminated with 15 μl of 60% perchloric acid. Samples were centrifuged for 5 min at 5000 rpm, and the supernatant was removed for analysis by high-performance liquid chromatography. High-performance liquid chromatography analysis was accomplished using a 4.6 × 250 mm Luna C18 column (Phenomenex, Torrance, CA) with a flow rate of 1 ml/min and an isocratic mobile phase consisting of 8% acetonitrile with 50 mM potassium phosphate solution (pH 4.9). The injected sample was monitored with UV absorption at 245 nm. Acetaminophen standards were prepared in the same manner as the samples except for the absence of the reconstituted protein system. Preliminary experiments showed that the absence of protein did not affect the standard curve (data not shown).
Spectral Difference Assays. Spectral difference assays were completed and analyzed as described by Schlicht et al. (2007). Phenacetin concentrations ranged from 0.5 to 275 μM with measurements at ≥10 intervening concentrations.
Protein Crystallization, Data Collection, and Structure Determination. Crystals of 100 μM CYP2A6 I208S/I300F/G301A/G369S with 5 mM phenacetin were grown by hanging drop vapor diffusion. The protein was equilibrated against a 750-μl solution of 30% polyethylene glycol 3350, 0.10 M Tris, pH 8.5, and 0.2 M ammonium sulfate. For data collection, crystals were flash cooled in liquid nitrogen after being immersed in a cryoprotectant consisting of 350 μl of synthetic mother liquor and 300 μl of 100% ethylene glycol. A native data set was collected at beamline 9-2 at the Stanford Synchrotron Radiation Laboratory (Stanford, CA) using a 0.98 Å wavelength and temperature of 100 K. Data processing was completed using Mosflm (Leslie, 2006) and Scala (Evans, 2006). The CYP2A6 I208S/I300F/G301A/G369S mutant structure was solved by molecular replacement with the program PHASER (McCoy et al., 2007). The search model was CYP2A6 complexed with coumarin [Protein Data Bank (PDB) 1Z10, molecule B]. Model building and refinement were done iteratively using COOT (Emsley and Cowtan, 2004) and REFMAC5 (Murshudov et al., 1997) in the CCP4 suite (Collaborative Computer Project, Number 4, 1994). The final model exhibited an R value of 0.21 and Rfree value of 0.27 with four molecules in the asymmetric unit. Well defined electron density was present above the heme prosthetic group in molecules A, B, and D of the asymmetric unit but not in molecule C. Data collection and processing statistics are provided in Table 2.
Data collection and refinement statistics
Structure Validation and Analysis. The CYP2A6 I208S/I300F/G301A/G369S structure was validated using WHAT-IF (Vriend, 1990) and PROCHECK (Laskowski et al., 1993). The Ramachandran plot showed 90.7% of residues in the most favored regions, 8.6% in the additional allowed regions, 0.4% in the generously allowed regions, and 0.2% in the disallowed regions. Consistent with previous structures of CYP2A6, F42 was in the disallowed region for all four molecules, but was clearly defined in the 2|Fo| - |Fc| electron density map (Yano et al., 2005; Sansen et al., 2007a). Coordinates have been deposited in the Protein Data Bank (PDB code 3EBS). All voids used in structural analysis were calculated using VOIDOO (Kleywegt and Jones, 1994) with a probe radius of 1.4 Å and a grid mesh of 0.3 Å.
Results
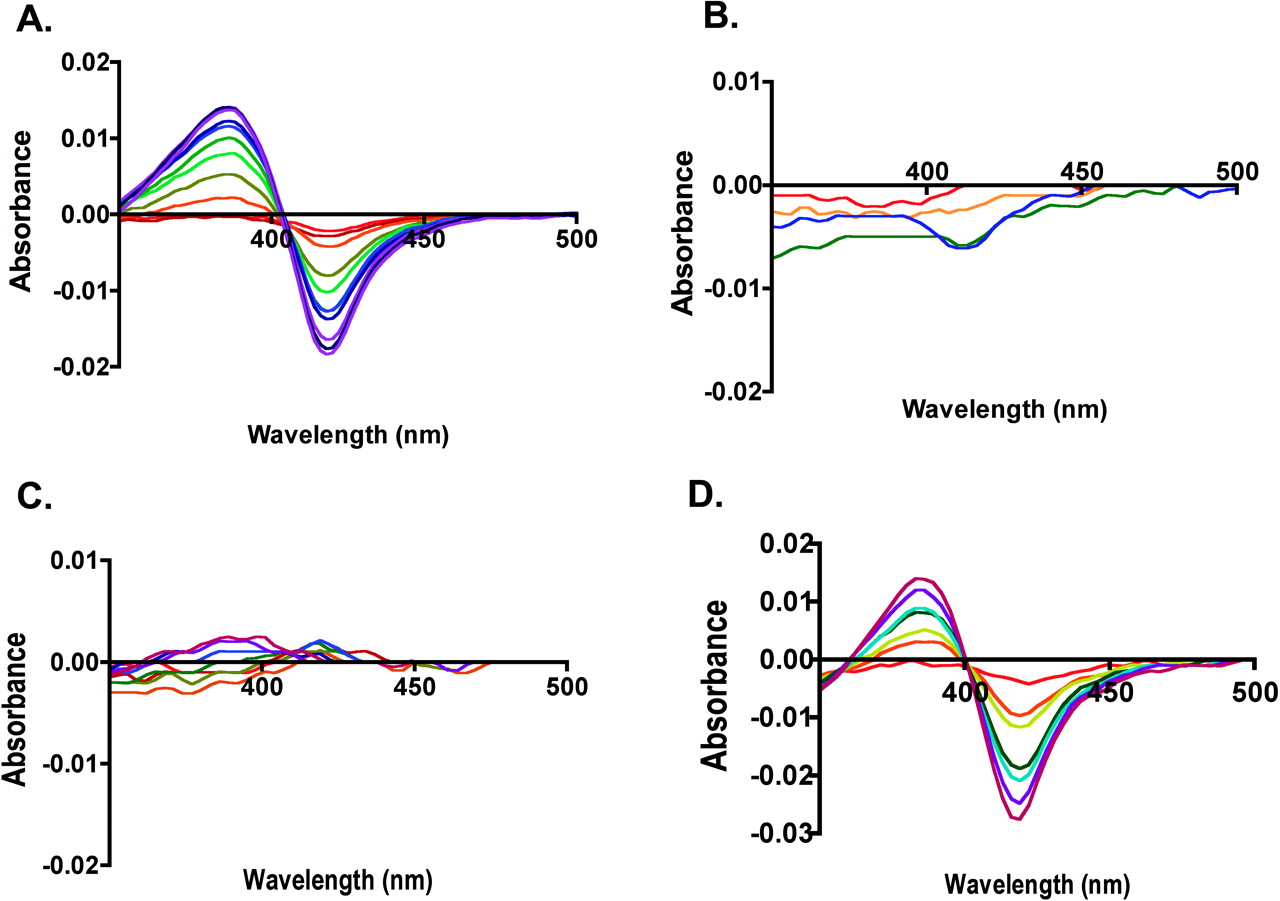
Phenacetin Dissociation Constants for CYP2A13, CYP2A6, and CYP2A13→2A6 Single-Site Mutant Enzymes. CYP2A13 and CYP2A6 had very different spectral responses upon addition of phenacetin. Titration of CYP2A13 with phenacetin yielded a classic type I spectral shift with a strong peak at 384 nm and trough at 419 nm, reflecting the iron transition from hexacoordinate low spin to penta-coordinate high spin (Fig. 1A). The KD value for this binding was 34 μM. In contrast, at phenacetin concentrations up to 250 μM, CYP2A6 demonstrated very little spectral shift above the baseline noise level (Fig. 1B), suggesting that either phenacetin did not bind or did not bind close enough in the active site to displace the water molecule present on the heme iron in the enzyme resting state.
Of the 10 single substitutions made in CYP2A13, several types of alterations were observed in phenacetin binding affinity (Table 3). One mutant, the Leu to Ile substitution at position 366 (L366I), significantly increased binding affinity by a 15-fold decrease in KD to 2.2 μM. A second set of mutated proteins including L110V, A117V, and H372R had smaller effects on phenacetin binding, causing a <2-fold decreases in the KD values. A third set of mutations including A213S, G369S, S208I, and M365V still yielded CYP2A13-like spectral shifts but with 2 to 4-fold decreases in phenacetin affinity. In contrast, a fourth set of enzyme mutations at positions 300 and 301 demonstrated substantially reduced spectral responses to phenacetin titration. Titration of F300I with phenacetin revealed only a small trough at 420 nm, whereas titrations of CYP2A13 A301G had essentially no spectral shift (Fig. 1C).
Phenacetin dissociation constants (KD) Details of titration given in text and Fig. 1.
Phenacetin O-Deethylation Activity. Phenacetin O-deethylation activity was initially determined for all CYP2A enzymes at 250 μM phenacetin. This concentration was selected because it was ∼25 times the KD for CYP2A13 in preliminary experiments (data not shown). At this concentration, CYP2A13 demonstrated an activity of ∼3.4 pmol/min/pmol, whereas CYP2A6 had activity that was only 0.08 pmol/min/pmol, only barely above our lower limit of detection (Tables 4 and 5, respectively).
Phenacetin O-deethylation rates and comparisons for CYP2A13 and mutations thereof All assays used 250 μM phenacetin.
Phenacetin O-deethylation rates and comparisons for CYP2A6 and mutations thereof. All assays used 250 μM phenacetin.
Of the 10 CYP2A13 singly substituted proteins, 5- and 3-fold increases in phenacetin O-deethylation were observed for A117V and L366I, respectively, whereas all other CYP2A13 mutations resulted in markedly decreased phenacetin metabolism (Fig. 2). The individual CYP2A13 mutations L110V, S208I, F300I, A301G, M365V, and G369S each reduced phenacetin metabolism to less than 15% of the wild-type activity, with residues at positions 300, 301, 208, and 369 having the largest effects. The A213S and H372R mutations had much less dramatic effects on activity with rates ∼1 pmol/min/pmol.
Most of the 10 CYP2A6 single mutants were similar to CYP2A6 in that they had little or no phenacetin O-deethylation activity (Fig. 3). However, V110L, I300F, and S369G had low but significant levels of activity (0.2–0.3 pmol/min/mol) (Table 5). Because these results suggested that the ability of CYP2A13 to metabolize phenacetin must be the result of several additive differences in the human 2A active sites, multiple mutants at key positions identified by the binding and metabolism studies were generated and characterized. The doubly mutated 2A6 protein V365M/I366L had almost no activity. However, the double mutant CYP2A6 I300F/G301A had a moderate increase in activity to 0.6 pmol/min/pmol over either of the single mutants. Addition of the I208S or S369G substitutions to the 300/301 double mutant yielded further increases in phenacetin activity to 0.8 and 1.0 pmol/min/pmol, respectively (Table 5). The quadruple mutant CYP2A6 I300F/G301A/S369G/I208S had activity of 3 pmol/min/pmol, nearly equivalent to that of CYP2A13 wild type (Fig. 3).
Phenacetin Dissociation Constants for CYP2A6→2A13 Multiple Mutant Enzymes. Phenacetin dissociation constants were then determined for those CYP2A6 multiple mutant proteins that demonstrated significant phenacetin metabolism (Table 3). As for CYP2A6, the CYP2A6 G301A mutant protein gave essentially no spectral shift with phenacetin concentrations up to 250 μM. However, the single CYP2A6 I300F substitution did have a slight spectral shift, but a reliable KD value could not be ascertained because of the very low absorption change. Combination of these mutations (I300F/G301A) yielded phenacetin binding with a high KD of 103 μM. Addition of I208S (I208S/I300F/G301A) decreased the KD to 63 μM. By comparison, addition of G369S to the I300F/G301A double mutant had a larger impact, reducing the KD to 13 μM, which is a greater affinity than that of wild-type CYP2A13 (KD 35 μM). Combination of all four substitutions (I208S/I300F/G301A/G369S) yielded an intermediate KD of 21 μM (Fig. 1D).
Phenacetin O-Deethylation Kinetic Parameters. Kinetic parameters were subsequently determined for all proteins with phenacetin O-deethylation activity >0.7 pmol/min/pmol in the single-concentration studies above. In addition, because phenacetin has been used as a marker substrate for CYP1A2, the full-length version of this enzyme was also characterized alongside the CYP2A enzymes. CYP2A13 and CYP1A2 both had catalytic efficiencies of ∼0.36 μM-1 min-1 (Table 6). Despite their similar catalytic efficiencies, there was a marked difference in their kinetic parameters. CYP1A2 had a kcat of 9.68, 3 times greater than that of CYP2A13, but the Km of CYP1A2 was also approximately 3 times higher than the Km of CYP2A13 (Fig. 4A).
Kinetic parameters for CYP1A2, CYP2A13, and mutants of CYP2A13 and CYP2A6 Data represent the average of two independent experiments each with all concentrations conducted in triplicate.
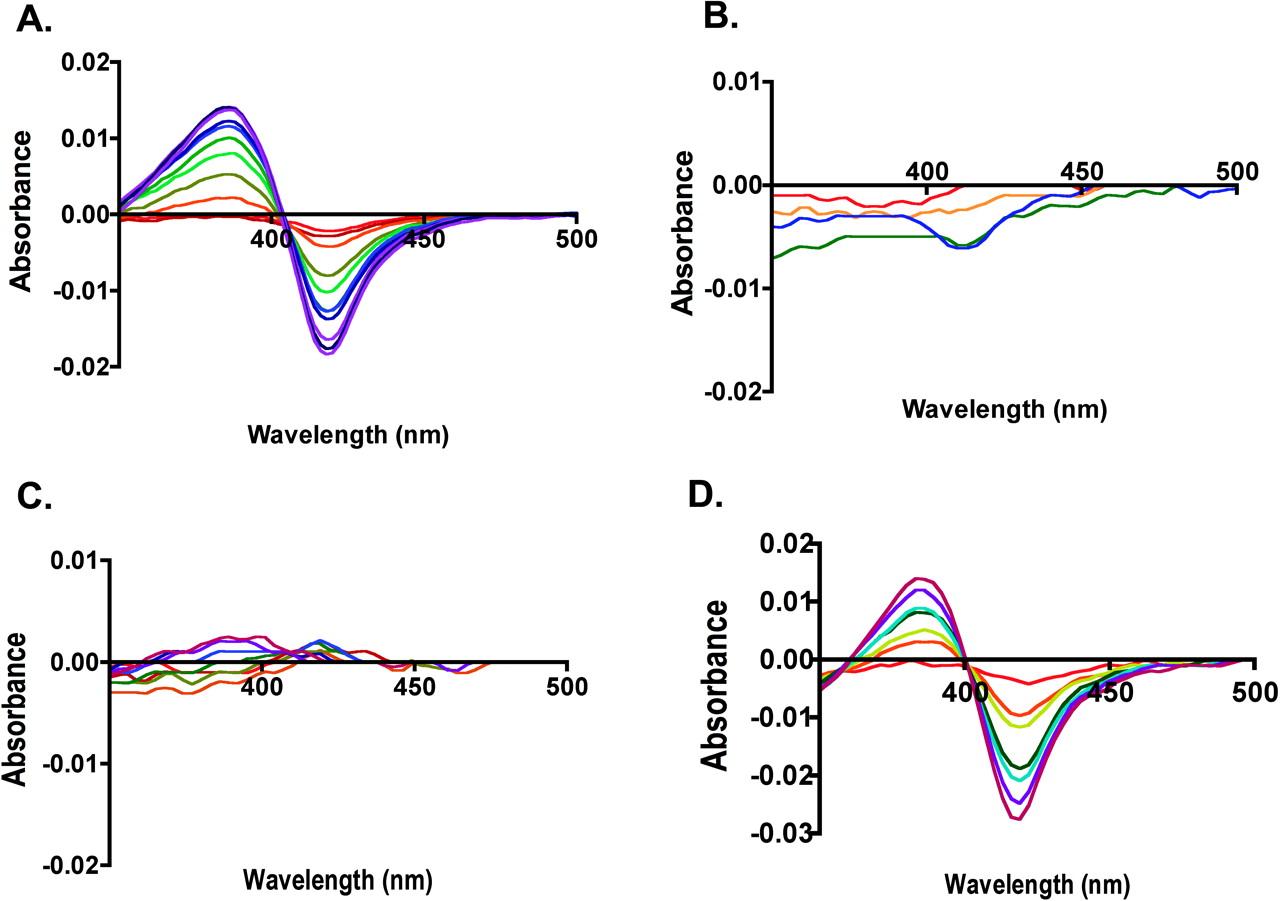
Spectral changes upon phenacetin binding to (A) CYP2A13, (B) CYP2A6, (C) CYP2A13 A301G, and (D) CYP2A6 I208S/I300F/G301A/S369G. Increasing concentrations of phenacetin during the titrations are indicated by spectra scans colored from red (low concentration 0.5 μM) to indigo (high concentration 250–300 μM).
Phenacetin metabolism by the CYP2A13 mutant enzymes varied considerably (Table 6). Two CYP2A13 mutations caused phenacetin metabolism at rates significantly higher than that of either CYP2A13 or CYP1A2. A117V has a 7.7-fold increase in kcat compared with CYP2A13 (Fig. 4A). A concomitant 5-fold increase in Km resulted in a catalytic efficiency for the mutant that was 1.7-fold higher than that for CYP2A13. L366I had a kcat 3.6-fold higher than that for CYP2A13 (Fig. 4A), but similar increases in Km yielded a catalytic efficiency similar to that of CYP2A13 (Table 6). A second set of CYP2A13 single mutants consisting of A213S, M365V, and H372R had decreases in kcat (Fig. 4B) and catalytic efficiencies at or below that observed for CYP2A13 (Table 6).
Although the phenacetin O-deethylation activity for all of the 2A6 single mutants were too low to determine reliable kinetic parameters, several of the multiple mutants had significant activity, thus permitting kinetic analysis (Table 6). The CYP2A6 triple mutants I300F/G301A/I208S and I300F/G301A/S369G both had kcat values of ∼0.9 pmol/min/pmol, which was significantly increased from that of CYP2A6 and approximately one third of that observed for CYP2A13 (Fig. 4C). However, the Km for the triple-mutant CYP2A6 I300F/G301A/S369G was 4.9 μM or approximately half that for CYP2A13, whereas the triple-mutant CYP2A6 I300F/G301A/I208S had a Km of 38.9 μM or 3.6 times higher than that for CYP2A13 (Table 6). As a result, the catalytic efficiency of I208S/I300F/G301A was the lowest observed, whereas the catalytic efficiency of I300F/G301A/S369G was almost half that of CYP2A13. Finally, the quadruple mutant CYP2A6 I208S/I300F/G301A/S369G had catalytic efficiency very similar to that of CYP2A13 as a result of an almost identical Km and a kcat only slightly lower than that for CYP2A13 (Fig. 4C).

Effects of mutation on phenacetin O-deethylation rates by CYP2A13 mutant proteins.
Structure of 2A6 I208S/I300F/G301A/S369G with Phenacetin. Although analysis of the wild-type enzyme structures and docking studies suggested that the ability of these mutations to interconvert phenacetin activity were primarily due to a relief of steric clashes in the active site near Phe300, the phenacetin-complexed structure of the CYP2A6 I208S/I300F/G301A/G369S mutant was determined to further evaluate this idea. The overall tertiary structure of the CYP2A6 quadruple mutant was very similar to those of both wild-type CYP2A6 and CYP2A13 with root-mean-squared deviations of the Cα atoms of 0.39 and 0.47 Å, respectively. These values were similar to the root-mean-squared deviations among the four molecules of the CYP2A6 quadruple mutant asymmetric unit (0.29–0.48 Å). The greatest variation in tertiary structure was observed in the solvent exposed loops.
All four mutations were clearly evident in the active site. Furthermore, electron density that corresponded to phenacetin was present in three of the four protein molecules. This density indicated that phenacetin oriented with the acetamide directed toward residue 300 and the ethoxy group oriented toward the heme iron, consistent with the experimentally observed O-deethylation (Fig. 5A). Phenacetin seems to have some flexibility in orientation as the molecule varied somewhat between all three molecules, in terms of the plane of the phenyl ring and the flexible ethoxy tail (Fig. 5B). However, in each molecule there is a conserved hydrogen bond (2.9–3.14 Å) between Asn297 and the amide oxygen of phenacetin, and the distance from the subterminal carbon and the iron was 4 to 4.5 Å.
The orientation of the Leu370 side chain also varied between molecules. Molecules A and C had the leucine side chain oriented so that it is rotated away from the phenacetin (Fig. 5B). In molecule A, the terminal carbon of phenacetin ethoxy chain projects toward this side chain, and the repositioning of Leu370 makes additional space available to the ligand. This side chain had previously been noted to have an alternate conformation in molecule A of the CYP2A6 structure complexed with coumarin (PDB 1Z10) (Yano et al., 2005). Because of the rotation of Leu370 away from the active site, the active site volumes for molecules A and C are 300 Å3 and 297 Å3, respectively. In contrast, the active site volume is reduced in molecules B (270 Å3) and D (272 Å3) (Fig. 5C). Replacement of the adjacent Ser369 by the glycine present in CYP2A13 may result in additional flexibility for Leu370.
The mutated side chains Phe300 and Ala301 were located in the same orientation in all four molecules. Phe300 forms the top portion of the active site, whereas Ala301 forms part of the side (Fig. 5B). The Ser208 side chain is rotated toward the active site in molecule A, whereas in molecules B, C, and D it is angled away from the active site (Fig. 5B). The rotation of this side chain does not seem to affect the active site volume.
The active site of the quadruple mutant is primarily hydrophobic, with residue Asn297 as the most orientating feature. Despite the small active site volume, phenacetin has some degree of flexibility within the active site, as indicated by its different orientation in the three molecules.
Discussion
Experimental analysis of phenacetin binding and metabolism by human CYP2A enzymes together with the structure of the CYP2A6 I208S/I300F/G301A/G369S mutant provides insights into the substrate specificity of the human cytochrome P450 2A family. The quadruple mutant is able to metabolize phenacetin with a Km nearly identical to that for CYP2A13 and a kcat ∼75% of that for CYP2A13. The ability to create phenacetin metabolism in CYP2A6 where essentially none existed previously demonstrates that these four residues largely control the ability to bind and metabolize phenacetin in human 2A enzymes. To further understand the contributions of these four residues, the structure of the quadruple CYP2A6 mutant was determined in complex with phenacetin.
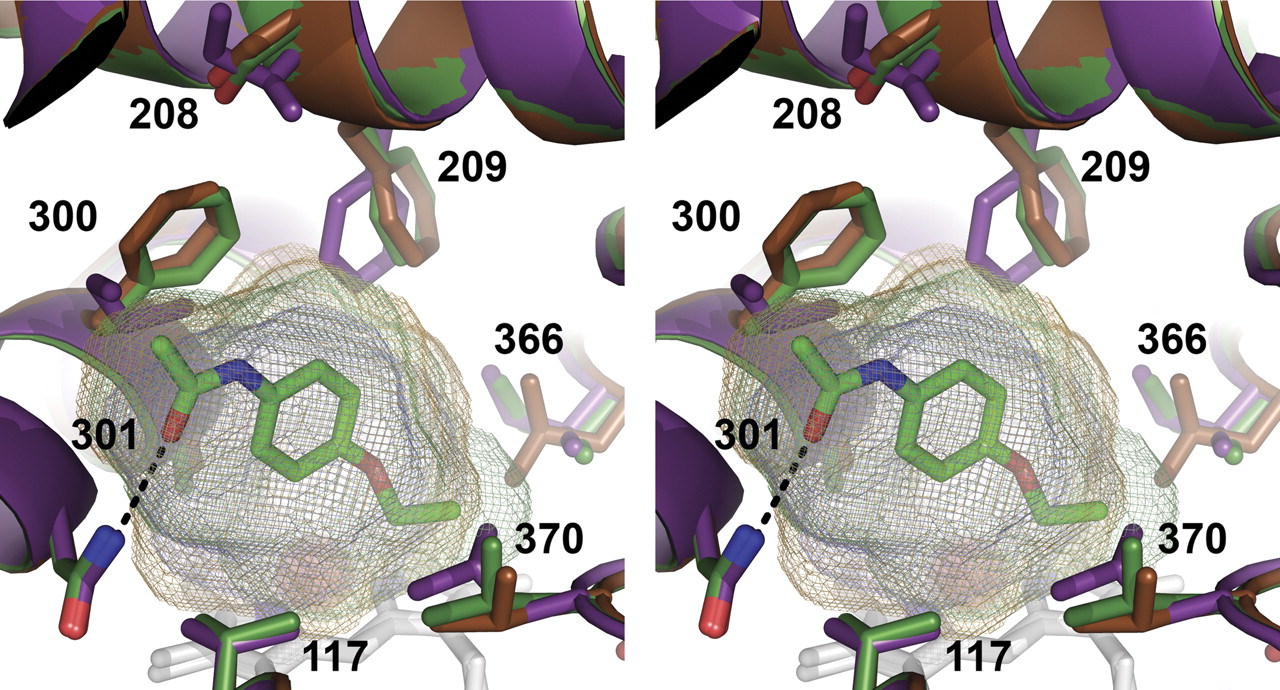
Comparisons of the CYP2A13, CYP2A6, and CYP2A6 I208S/I300F/G301A/G369S active sites give insight into how these four mutations significantly alter CYP2A functionality (Fig. 6). CYP2A6 has one of the smallest human P450 active sites, with a volume of 214 to 230 Å3 for the four CYP2A6 molecules (PDB 1Z10). CYP2A13 has a larger active site, with a volume of ∼304 Å3 (PDB 2P85). All of the quadruple mutant molecules had intermediate volumes from 270 to 300 Å3, depending primarily on the Leu370 conformation.

Effects of mutation on phenacetin O-deethylation rates by CYP2A6 mutant proteins.
The residues 300 and 301 seem to play the most critical roles in phenacetin binding and metabolism, consistent with their close interaction with phenacetin in the crystal structure. The phenylalanine at position 300 is torsioned away from ligand in CYP2A13 and the quadruple mutant, resulting in additional active site space in this region to accommodate the phenacetin acetamide. Substitution of the phenylalanine to the smaller isoleucine in CYP2A6 actually causes a decrease in active site space available to the ligand because the isoleucine side chain projects directly into the active site. Of particular note is a steric clash between CYP2A6 I300 and the terminal carbon of the amide end of phenacetin, which is particularly evident when the active site cavities are viewed (Fig. 7). Functional effects have also been observed for a CYP2A6 I300V mutation in a random mutagen-esis screen for increased indole metabolism (Wu et al., 2005). At position 301, alanine seems to be essential for CYP2A phenacetin binding and metabolism. The methyl side chain of alanine is directed toward the face of phenacetin just proximal to the ring. Substitution to glycine may remove the packing interactions needed to stabilize the phenacetin in the active site. This residue has not previously been identified as altering CYP2A function.
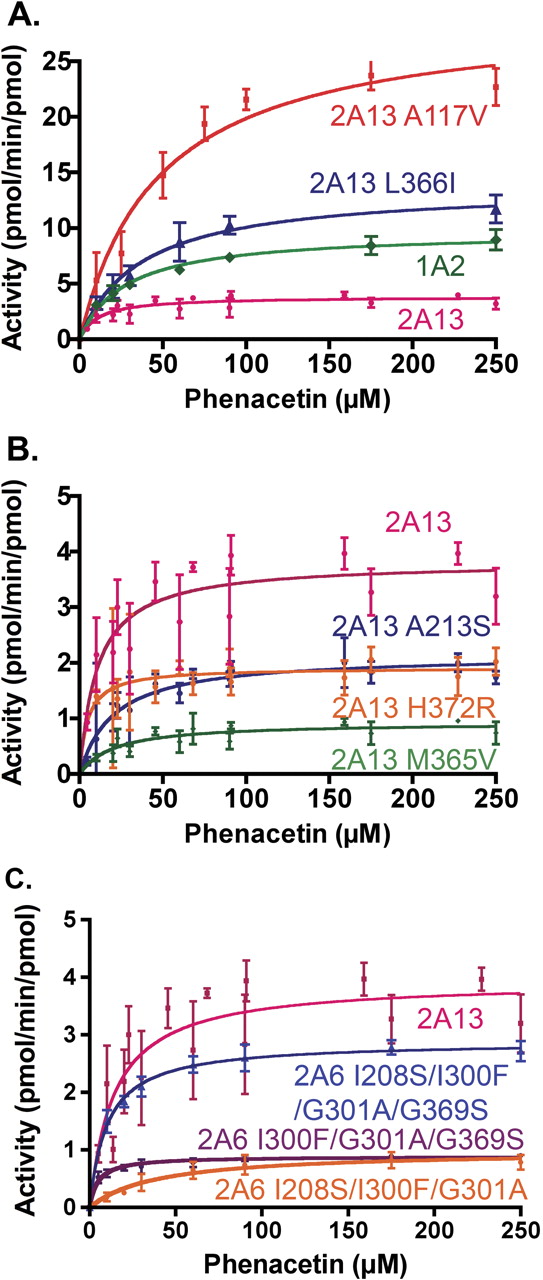
A, contains phenacetin metabolism by CYP1A2, CYP2A13, and CYP2A13 mutant proteins. B, contains phenacetin metabolism by CYP2A13 and CYP2A13 mutants. C, contains phenacetin metabolism plots by CYP2A13 and CYP2A6 mutants. Data represent a global fit to two independent experiments with each point performed in triplicate.
The third mutated residue essential for phenacetin metabolism is residue 208. In CYP2A13 and the quadruple mutant, this residue is a serine and is located in helix F directly above residue 300. In addition to the reduction in side chain size compared with the isoleucine found in CYP2A6, Ser208 is torsioned away from the active site in the majority of the quadruple mutant molecules. This creates space that probably facilitates torsion of Phe300 away from the active site, indirectly increasing space for phenacetin binding as described above. In addition, the residue at position 208 affects the orientation of the adjacent conserved Phe209 (Fig. 6). The orientation of Phe209 away from phenacetin in the quadruple mutant is identical to the position of Phe209 in CYP2A13 and also creates active site volume that would better accommodate phenacetin (Fig. 7). The CYP2A13 mutation S208I was previously shown to alter metabolism of the nicotine-derived procarcinogen NNK (He et al., 2004a). Mutation at position 209 has also been shown to alter the specificity of mouse CYP2A enzymes (Negishi et al., 1996).
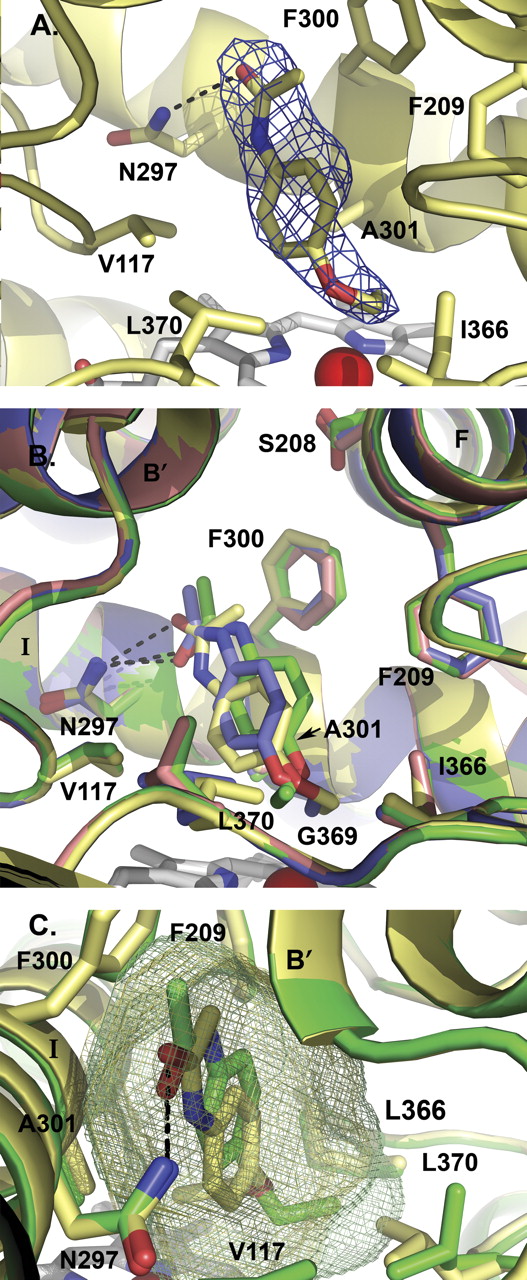
The CYP2A6 I208S/I300F/G301A/G369S structure. A, electron density surrounding the phenacetin ligand found in molecule B of the CYP2A6 I208S/I300F/G301A/G369S structure. B, structural overlay of the four nonidentical molecules found in the CYP2A6 I208S/I300F/G301A/G369S asymmetric unit, high-lighting the differences in phenacetin and Leu370 positions. Molecules A (green), B (yellow), and D (blue) contain one molecule of phenacetin, whereas molecule C (pink) did not have electron density for phenacetin. C, active site volume comparison between molecule A (green mesh, 300 Å3) and molecule B (yellow mesh, 270 Å3). The difference in active site volume among the four CYP2A6 quadruply mutated protein molecules seems to depend largely on the position of Leu370. All structure figures were made with PyMOL (DeLano, 2002). Hydrogen bonding to Asn297 is indicated by black dashed lines.
The fourth residue, 369, is located on the opposite side of the active site cavity and somewhat more distant from the phenacetin molecule. The serine found at this position in CYP2A6 is oriented toward the proximal side of the heme, where it interacts with the heme propionate (Fig. 6). In CYP2A13 and the quadruple mutant, a glycine is found at this position and the contact with the heme propionate is instead mediated by a water molecule stabilized via interaction with the protein backbone. Instead, the residue at position 369 may play an indirect role in phenacetin binding by modulating the position of the adjacent Leu370 residue. In all six molecules of CYP2A13, Leu370 is positioned with its side chain is oriented alongside the active site cavity. In the 1Z10 structure of CYP2A6, three of the four of the molecules have Leu370 directed into the active site cavity, reducing the volume available to ligands (Fig. 6). However, one of the four CYP2A6 molecules was modeled with two alternate conformations: 50% occupancy extending into the active site like the remaining CYP2A6 molecules and 50% occupancy oriented alongside the active site as in CYP2A13. In the CYP2A6 quadruple mutant structure, two of the four molecules have the Leu370 side chain in the CYP2A13 position and in the other two molecules Leu370 is in the CYP2A6-dominant orientation. Thus, additional flexibility imparted when glycine is at 369 may facilitate movement of Leu370 so that it is more likely to be positioned alongside the cavity as seen in CYP2A13. The added flexibility of Leu370 to adopt this second conformation would result in more active site space available to ligands and probably contributes to increased phenacetin binding and metabolism.

Comparison of the active sites of CYP2A6 (purple, PDB 1Z10, molecule B), CYP2A6 I208S/I300F/G301A/G369S with phenacetin (green, molecule A), and CYP2A13 (orange, PDB 2P85, molecule A). The four residues mutated in the CYP2A6 quadruple mutant, identical to the residues found in CYP2A13, are labeled in bold. The hydrogen bond from phenacetin to Asn297 is shown as a dashed black line.

Stereo view comparison of the active site cavities of CYP2A6 (purple protein and mesh, PDB 1Z10), CYP2A13 (orange protein and mesh, PDB 2P85), and CYP2A6 I208S/I300F/G301A/G369S (green protein and mesh). Steric clashes between the phenacetin and CYP2A6 residues 300, 209, and 370 are evident.
The CYP2A6 I208S/I300F/G301A/G369S crystal structure also gives structural insight into the two CYP2A13 mutations that resulted in increases in phenacetin kcat substantially above that of the CYP2A13 wild-type enzyme. The first, CYP2A13 A117V, had a 40% decrease in KD, a 4.5-fold increase in Km, and an overall 2-fold increase in catalytic efficiency, suggesting multiple effects in binding and catalysis. The CYP2A13 A117V mutation has been shown previously to be important for coumarin and NNK metabolism (He et al., 2004b). Although the valine side chain of the quadruple mutant projects further into the active site toward the phenacetin than the CYP2A13 alanine, the reduction in volume in this portion of the active site complements and may be beneficial in orientating the phenacetin within the active site (Fig. 7). The L366I mutation had an overall catalytic efficiency similar to that of wild-type CYP2A13, with ∼3.5-fold increases in both Km and Vmax. However, this mutation also dramatically increased binding affinity. The CYP2A13 L366I mutation is located close to residues 365 and 369 near the alkyl chain of phenacetin. Mutation from the leucine found in CYP2A13 to the isoleucine in CYP2A6 would also probably increase the size of the active site cavity.
CYP2A13 L110V and M365V also reduced phenacetin O-deethylation activity at 250 μM to less than 15% of the wild-type CYP2A13 enzyme. These mutations were not incorporated into the multiple mutant series because the quadruple mutant substitutions were sufficient to closely approximate the kinetic parameters observed for the CYP2A13 enzyme. In CYP2A13, the M365 side chain extends into the active site next to Leu366, whereas in CYP2A6 the shorter valine side chain at this position does not actually form part of the active site as the side chain of Ile366 extends further and shields Val365 from the active site (Fig. 6). It is possible that the CYP2A13 M365V mutant exhibits a loss in activity because an increase in active site volume in this region is not conducive to phenacetin stabilization in the active site. In contrast, the L366I substitution results in a beneficial effect on binding and metabolism. It seems likely that the structural results of mutations at adjacent residues 365 and 366 may be related as their mutation in CYP2A13 resulted in opposing effects. The effect of the CYP2A13 L110V mutation is more difficult to explain. This residue is not located in the active site; instead it is located in the B′ helix across from residue 208. Furthermore, although the CYP2A13 L110V mutant has a significant reduction in phenacetin metabolism activity, it causes a small increase (less than 2-fold) in phenacetin binding affinity. Further investigation will be needed to determine why this mutation causes such a striking reduction in phenacetin O-deethylation.
The crystal structure of CYP2A6 I208S/I300F/G301A/G369S together with the phenacetin binding and metabolism data clearly indicates that the ability of CYP2A13 to metabolize phenacetin is the aggregate result of several differences from the CYP2A6 active site. This is why incorporation of individual 2A13 residues into CYP2A6 at each of the 10 active site positions where the parent enzymes differ yielded no difference or only small increases in phenacetin metabolism. Changes in phenacetin metabolism were observed with the multiple mutants, which is consistent with the fact that multiple residue alterations are necessary for phenacetin to bind in CYP2A6. The two 2A6 triple-mutant proteins (2A6 I208S/I300F/G301A and 2A6 I300F/G301A/S369G) had significant phenacetin metabolic activity (kcat of ∼1 min-1), indicating that increasing the active site volume around residue 300 and either 370 or 209 permits enough volume for phenacetin to bind, albeit not as well as in CYP2A13. The quadruple mutant (2A6 I208S/I300F/G301A/S369G) has phenacetin metabolism parameters nearly identical to those of CYP2A13 as well as an active site volume substantially larger than that of CYP2A6. The active site of this quadruple mutant closely resembles that of CYP2A13. Some of the CYP2A6 residues (Val117 and Ile366) are actually beneficial to phenacetin metabolism and may be reflected in the slightly increased binding affinity of the quadruple mutant (KD of 21 μM compared with a KD of 34 μM in CYP2A13). The ability to create phenacetin metabolism in CYP2A6 where essentially none existed previously demonstrates that these four residues largely control the ability to bind and metabolize phenacetin in human 2A enzymes.
In conclusion, this study identifies four amino acid residues, 208, 300, 301, and 369, that are responsible for controlling the metabolism of phenacetin in the human cytochrome 2A family. The crystal structure of CYP2A6 I208S/I300F/G301A/S369G and comparison with the active sites of CYP2A13 and CYP2A6 suggests that the ability of CYP2A enzymes to metabolize phenacetin is largely the culmination of steric effects of the substituted side chains themselves and their effects on the positions of adjacent side chains Phe209 and Leu370. Although phenacetin is no longer used clinically as an analgesic, it was used in these studies as a model substrate because of the difference in affinity between CYP2A6 and CYP2A13. Further investigations will determine whether these same residues are responsible for differences in selectivity for other CYP2A ligands such as the procarcinogen NNK.
Acknowledgments
We thank Jenny Morrison, Chad Schroeder, and Melanie Blevins for construction of the CYP2A13 mutants and Linda Blake, Naseem Nikaeen, Matthew Axtman, Agnes Walsh, and Kyle Bailey for construction of the CYP2A6 mutants. We also thank Christopher Wood for assistance in expressing and purifying some of the CYP2A13 mutants. Full-length human CYP1A2 was a generous gift from Dr. Fred Guengerich (Vanderbilt University School of Medicine, Nashville, TN). We thank Linda Blake for critical reading of the manuscript. Crystals were grown and initially screened using the facilities of the Protein Structure Laboratory core facility at The University of Kansas. We acknowledge access to the facilities and excellent support from the staff at Stanford Synchrotron Radiation Laboratory (SSRL). The SSRL is operated by the Department of Energy, Office of Basic Energy Sciences. The SSRL Biotechnology Program is supported by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program and by the Department of Energy, Office of Biological and Environmental Research.
Footnotes
-
This work was supported by National Institutes of Health Grant GM076343 (to E.E.S.).
-
Portions of this work were previously presented as follows: DeVore NM, Smith BD, Urban MJ, Nikaeen N, Blake LC, and Scott EE (2008) Identification of active site residues essential for the metabolism of phenacetin to acetaminophen in human 2A cytochromes P450. Proceedings of Experimental Biology 2008; 2008 April 5–8; San Diego, CA. Federation of American Societies for Experimental Biology, Bethesda, MD.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.108.023770.
-
ABBREVIATIONS: P450, cytochrome P450; AFB1, aflatoxin B1; PDB, Protein Data Bank; NNK, nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone.
- Received August 6, 2008.
- Accepted September 5, 2008.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}