


Abstract
The flavin-containing monooxygenase (FMO) family of enzymes oxygenates nucleophilic xenobiotics and endogenous substances. Human FMO3 and FMO5 are the predominant FMO forms in adult liver. These enzymes are naturally membrane-bound, and recombinant proteins are commercially available as microsomal preparations from insect cells (i.e., Supersome FMO). As an alternative, FMO3 has previously been expressed as a soluble protein, through use of an N-terminal maltose-binding protein (MBP) fusion. In the current study, MBP fusions of both human FMO3 and FMO5 were prepared to >90% purity in the presence of detergent and characterized for biochemical and kinetic parameters, and the parameters were compared with those of Supersome FMO samples. Although MBP-FMO enzymes afforded lower rates of turnover than the corresponding Supersome FMOs, both types of FMO showed identical substrate dependencies and similar responses to changes in assay conditions. Of interest, the FMO3 enzymes showed a 2-fold activation of kcat/Km in the presence of Triton X-100. Oligomeric analysis of MBP-FMO3 also showed disassociation from a high-order oligomeric form to a monomeric status in the presence of Triton X-100. This report serves as the first direct comparison between Supersome FMOs and the corresponding MBP fusions and the first report of a detergent-based activation of kcat/Km that corresponds to changes in oligomerization.
Introduction
The family of flavin-containing monooxygenases (FMO) oxygenates a large variety of endogenous and exogenous substrates. FMO-catalyzed oxygenation generally produces polar and relatively stable, nontoxic products that are readily excreted from the body, allowing the FMOs to play an important role in metabolic detoxication (Cashman, 2008). In the adult human liver, FMO3 and FMO5 are the predominantly expressed FMO family members (Zhang and Cashman, 2006). FMO3 is known to play a significant role in human hepatic metabolism and contributes to the metabolism of many common drugs (e.g., ranitidine, amphetamine, clozapine, and tamoxifen) (for a review, see Krueger and Williams, 2005). Genetic mutations of FMO3 can result in trimethylaminuria, a metabolic disease resulting from defective trimethylamine metabolism (for a review, see Motika et al., 2007). In adults, FMO5 has hepatic mRNA levels as abundant as that of FMO3 (Zhang and Cashman, 2006). In addition, mRNA levels for FMO5 represent >50% of total FMO transcripts in human fetal liver and adult small intestine (Zhang and Cashman, 2006). However, mRNA levels do not always correspond to protein expression levels. One previous attempt to quantify human hepatic FMO (Overby et al., 1997) showed large variability in both the relative protein expression and mRNA levels, yielding poor correlation between the two. In short, the contributions of FMO5 enzyme functional activity to human chemical metabolism have not been clearly established, and efforts to pursue the issue are largely impeded because of a paucity of selective functional substrates (Zhang et al., 2007).
Initial characterization of FMOs benefited largely from early work describing the purification and kinetic evaluation of pig liver FMO1 (Ziegler and Mitchell, 1972). However, ethical and technical limitations have hindered the purification of native FMO from human tissues. Because FMOs are membrane-associated enzymes, human FMO characterization was largely conducted with liver microsomes and S9 hepatic fractions. However, any delay between time of death and tissue preparation and various practices associated with poor temperature control during tissue preparations destroy a large fraction of the FMO activity present because of its marked thermal lability (Cashman et al., 1999). In addition, once hepatic microsomes are prepared, the contributions of FMO to metabolism can be difficult to distinguish from contributions associated with cytochrome P450 (P450) enzymes that have overlapping substrate specificities with FMOs. Although P450 activity may be easily measured by taking advantage of differences in thermal lability (i.e., heat-inactivating FMO, leaving P450s intact), direct measurements of FMO activity are limited to the use of either specific P450 inhibitors or antibodies (Washio et al., 2001; Wang et al., 2008) or detergents such as Emulgen 911 or Lubrol that diminish P450 activity (Rettie et al., 1990; Venkatesh et al., 1991). However, both detergents have been shown to affect FMO activity, and the presence of these detergents may complicate subsequent analysis. Because of complications related to all of the above-mentioned issues, there have been no publications to date showing purified and functionally active native human hepatic FMO. Therefore, recombinant FMO expression systems are of great importance for FMO-relevant research.
Baculovirus-mediated recombinant expression of FMOs from insect cells was first reported in 1997 (Haining et al., 1997; Lang et al., 1998) and is arguably the most commonly used recombinant expression system for FMO. Insect cell microsomes containing various FMO forms are now commercially available (e.g., Supersome1 FMOs from BD Gentest, Woburn, MA). Although highly useful, these enzymes are not inexpensive, are provided at relatively low enzyme concentrations (i.e., 4–10 μM), and are not highly purified (typically in the range of 0.1 mg of active FMO/mg of total protein). As an alternative, recombinant expression of FMOs from Escherichia coli has also been developed using N-terminal maltose-binding protein-FMO fusions (MBP-FMO) (Brunelle et al., 1997). MBP has a well established record of increasing the solubility of proteins when used as an N-terminal tag (Kapust and Waugh, 1999; Fox and Waugh, 2003). The MBP fusion therefore not only serves as a “handle” for purification techniques but also increases the solubility of FMO, allowing for increased yield of highly purified FMO enzymes. Over the past decade, both Supersome FMOs and MBP-fused FMOs have been successfully used in a number of studies [e.g., characterization of FMO3 variants associated with trimethylaminuria using Supersome FMO3s (Yeung et al., 2007) or using MBP-FMO3s (Motika et al., 2009)], showing the applicability of both of these systems to important biological questions. However, no comparison has yet been presented for these two different systems.
In this article, new biochemical and kinetic characterizations of MBP-FMO3 and MBP-FMO5 fusion proteins are presented, and the kinetic characteristics of MBP-FMO are compared with those of the corresponding Supersome enzymes. Although differences were observed in the apparent rates of turnover, the two enzyme systems displayed identical substrate dependencies. Of interest, FMO3 also displayed a detergent-dependent activation of kcat/Km that corresponds to changes in the oligomeric state. Future studies may elucidate this as an important physiological means for regulating kinetic activity.
Materials and Methods
Reagents.
Methimazole (MMI), l-methionine, 5,5′-dithiobis(2-nitrobenzoate), glucose 6-phosphate, FAD, NADP+, NADPH, and Triton X-100 were purchased from Sigma-Aldrich (St. Louis, MO). The Native PAGE Sample Prep Kit, 4 to 16% acrylamide Bis-Tris Novex gels, and Coomassie Blue G-250 dye were purchased from Invitrogen (Carlsbad, CA). Buffers and other reagents were purchased from VWR (West Chester, PA). Synthesis of 10-[(N,N-dimethylaminooctyl)-2-(trifluoromethyl)]phenothiazine (8-DPT) was described previously (Nagata et al., 1990; Lomri et al., 1993; Zhang et al., 2003), and this product was converted to its hydrochloride salt and used as an aqueous stock solution. Supersome FMO samples (i.e., insect cell microsomal preparations of recombinant human FMO3 and FMO5) were purchased from BD Gentest. Detergents including CHAPS, n-decyl-β-d-maltoside, n-dodecyl-β-d-maltoside (DDM), n-octyl-β-d-glucoside, fos-choline-12, cymal-5, cymal-6, and cymal-7 were purchased from Anatrace Inc. (Maumee, OH).
Construction of MBP-FMO Expression Vectors.
Cloning of human FMO3 and construction of the MBP-FMO3 vector was reported previously (Brunelle et al., 1997). Human FMO5 cDNA was amplified by reverse transcriptase-polymerase chain reaction from an adult human liver cDNA library generated from total RNA isolated from a pool of human liver samples (BD Biosciences, San Jose, CA) using the following primers: gatctctagaatgactaagaaaagaattgctgtga (XbaI site italicized) and gatcctgcagccaatgaaaaacagggcagt (PstI site italicized). The FMO5 cDNA was subcloned into the expression vector pMal-c2 from New England Biolabs (Ipswich, MA) through XbaI and PstI restriction sites to create a construct encoding an N-terminal E. coli MBP fusion of human FMO5 similar to that described previously for human MBP-FMO3 (Brunelle et al., 1997). The cloned FMO5 construct used in this study matched GenBank sequence NM_001461 except for variation P351S. Although the allelic frequency for this polymorphism position is unreported, the S351 coding sequence used in the current study was identified in GenBank sequences NT_004434 and NM_001144829, indicating a prominent presence in the general population.
MBP-FMO Protein Expression and Purification.
Plasmids encoding MBP-FMO3 or MBP-FMO5 were transformed into DH1α cells, and the cells were grown in SOC media for 20 h after induction at room temperature, as described previously for MBP-FMO3 (Motika et al., 2009). Cells were then centrifuged at 6400g for 10 min at 4°C, and the resulting pellet was frozen at −20°C until further studies were done.
MBP-FMO proteins were purified via amylose affinity chromatography as described previously for MBP-FMO3 (Lattard et al., 2003) with the following modifications. The desired FMO protein was eluted with comparable results using either a linear gradient (approximately 30 column volumes) of 0 to 3 mM d-maltose or a one-step increase to 5 mM d-maltose instead of the previously reported step elution to 10 mM d-maltose. Fractions containing FMO protein were pooled and concentrated using an Amicon Ultra-15 centrifugal filter with a 50-kDa molecular mass cutoff (Millipore Corporation, Billerica, MA).
MBP-FMO3 was further purified via anion exchange chromatography using an ÄKTApurifier 10 fast-performance liquid chromatograph from GE Healthcare (Little Chalfont, Buckinghamshire, UK). Samples were loaded onto a 1-ml Q Fast-Flow (QFF) ion exchange column (GE Healthcare) equilibrated in buffer Q (50 mM Tris buffer, pH 7.4, containing either 0.5% CHAPS or 0.5% Triton X-100). The column was washed with 60 mM NaCl in buffer Q (10 ml), and protein was eluted with 250 mM NaCl in buffer Q (10 ml). The flow rate was 1 ml/min for all wash and elution steps. Fractions containing FMO3 protein, as determined by the absorbance at 450 nm, were pooled and concentrated, as described above. Protein purity was determined with SDS-PAGE followed by Coomassie Blue staining. MBP-FMO5 was purified similarly using buffer Q containing either 0.5% Triton X-100, 0.01% DDM, or no additional detergent.
MBP-FMO FAD Content Determinations.
The concentration of MBP-FMO-bound FAD was determined with absorbance at 450 nm as described previously for other flavin-containing enzymes (Wagner et al., 1999) with the following modification. To remove nonspecifically bound flavin from the samples, the MBP-FMO proteins were first precipitated using 20% PEG-8000 as described previously (Brunelle et al., 1997), and the supernatant was discarded. The pellet was resuspended in denaturation buffer (5 mM potassium phosphate, 25 mM KCl, and 3 M guanidine hydrochloride, pH 7.5). Control incubations showed that polyethylene glycol precipitation did not significantly decrease the catalytic efficiency or flavin content and recovered yields were 80 to 90%. Measurements showed that the enzyme-bound flavin absorbance coefficient for both FMO3 and FMO5 enzymes did not differ significantly from that of free FAD in the presence of guanidine hydrochloride (11,900 M−1 cm−1) (Wagner et al., 1999). This value was therefore used for determinations of the enzyme concentration under nondenaturing conditions.
In addition to the active enzyme (flavin-bound) concentration, the total MBP-FMO protein concentration for final purified protein was determined using the detergent-compatible colorimetric (DC) assay (Bio-Rad Laboratories, Hercules, CA). MBP-FMO purified after amylose chromatography was quantified for FMO concentration using quantitative SDS-PAGE analysis as described previously (Motika et al., 2009).
FMO3 and FMO5 Kinetic Assays.
FMO3-catalyzed S-oxygenation of MMI was monitored using the method described by Dixit and Roche (1984) at 37°C, as reported previously (Motika et al., 2009). QFF-purified MBP-FMO3 eluted from the Q column in buffer containing 0.5% CHAPS was used for all FMO3 kinetic assays, and assays typically contained 0.2 to 0.5 μM MBP-FMO3. Dilution of the enzyme in assay buffer yielded an assay concentration of less than 0.004% CHAPS when no additional detergent was added. This condition is referred to as the minimal detergent condition.2 In addition, assays were also run in the presence of 0.5% CHAPS or 0.5% Triton X-100. The substrate concentration dependence was determined over a range of 5 to 200 μM MMI. Supersome FMO3 was assayed using the same method. Assays contained 30 to 70 nM enzyme and were used as provided by the manufacturer with no additional manipulations. Observed rates of product formation were divided by the enzyme concentration as determined by the FAD absorbance (for MBP-FMO3) or as provided by the manufacturer (for Supersome FMO3), to afford units of minutes−1 (i.e., nanomoles of product × minute−1 × nanomoles of FMO−1). Plots of the rate of turnover versus the substrate concentration afforded kinetic parameters (i.e., kcat, kcat/Km, and Km) by fitting the data to the Michaelis-Menten equation using GraphPad Prism (version 5.01; GraphPad Software Inc., San Diego, CA). Statistical comparisons of selected data fittings were done using the extra sum of squares F-test provided in GraphPad Prism. The stability of MBP-FMO3 under the above standard assay conditions was determined by incubating 60 μg of protein in the presence of NADPH at 37°C for time intervals ranging from 5 to 30 min and then assessing the S-oxygenation activity with 80 μM MMI.
For detergent EC50 value evaluations, various concentrations of Triton X-100 or CHAPS were added to the minimal detergent assay condition described above. These assays were run in the presence of 5 μM MMI, and turnover was initiated by the addition of 0.5 μM MBP-FMO3.
In addition to MMI, oxygenation activity of MBP-FMO3 was also characterized with l-Met as a substrate. S-Oxygenation of l-Met was monitored photometrically by following NADPH consumption at 340 nm, as described previously for MBP-FMO3-catalyzed N-oxygenation of trimethylamine (Motika et al., 2009). Assays were run at 37°C, pH 8.5, under minimal detergent conditions and in the presence of 0.5% CHAPS or 0.5% Triton X-100. The concentration dependence was determined over a range of 0.25 to 50 mM l-Met, and kinetic parameters were obtained from the Michaelis-Menten equation, as described above. Assays contained 0.2 to 2 μM MBP-FMO3. Supersome FMO3 activity was also measured with this assay in the presence of 50 mM methionine and 0.1 to 0.4 μM enzyme.
FMO5-catalyzed N-oxygenation of 8-DPT HCl at 37°C, pH 8.5, was monitored with an HPLC-based assay as described previously (Zhang et al., 2007). This assay was used for both MBP-FMO5 and Supersome FMO5, and assays contained approximately 0.2 μM FMO5. The rate of 8-DPT N-oxide product formation was initially assessed at 400 μM 8-DPT and found to be a linear function of time over a range of 0 to 30 min. Subsequent assays used a 20-min incubation time. The substrate concentration dependence was determined over a range of 10 μM to 2 mM 8-DPT, and kinetic parameters were obtained by fitting the data to the Michaelis-Menten equation as described above.
Oligomeric Analysis.
The oligomeric state of the purified MBP-FMO proteins was analyzed using nondenaturing (native) PAGE and size exclusion (SEC) methods. For native PAGE analysis, purified MBP-FMO samples (approximately 6 μg) were prepared in loading buffer using the Native PAGE Sample Prep kit, according to the manufacturer's instructions, and gels were run at 150 V for 2.5 h. After destaining in 40% methanol and 10% acetic acid, the Coomassie dye yielded a limit of detection of approximately 60 ng, and no additional staining was needed. Conalbumin (75 kDa), aldolase (158 kDa), ferritin (445 kDa), and thyroglobulin (669 kDa) (GE Healthcare) were used as molecular mass standards. The electrophoretic mobilities of samples were visually compared with those of the molecular mass standards listed above.
Native PAGE analysis was also used to assess the oligomeric state of FMO after removal of the MBP moiety using factor Xa (New England Biolabs). MBP-FMO5 (4.5 mg/ml) was first incubated with 0.1 mg/ml factor Xa at 37°C for 2 h in 50 mM Tris, 150 mM KCl, pH 7.4, with 2 mM CaCl2, and 0.5 mM NADPH. Samples were then prepared for electrophoresis by diluting the enzyme into the desired detergent condition (i.e., 0 or 0.5% Triton X-100) and were analyzed as described above. We were surprised to find that FMO5 protein bands consistently showed lower staining intensity per protein mass under native conditions after factor Xa treatment, relative to MBP-FMO5. Therefore, larger protein amounts (19 μg) were used in this analysis.
Analytical SEC was done using a Superose 6 10/300 GL column from GE Healthcare. The column was equilibrated and run in buffer S (50 mM Tris, pH 7.4, 150 mM NaCl), with either 0.5% CHAPS or 0.5% Triton X-100 for MBP-FMO3 and 0.5% Triton X-100, 0.01% DDM, or no added detergent for MBP-FMO5. Protein samples (0.2 mg; 100 μl) were run with a flow rate of 0.5 ml/min, and elution volumes were compared with those of molecular mass standards (listed above) run under identical conditions.
Results
Expression and Purification of Recombinant MBP-FMO3 and MBP-FMO5.
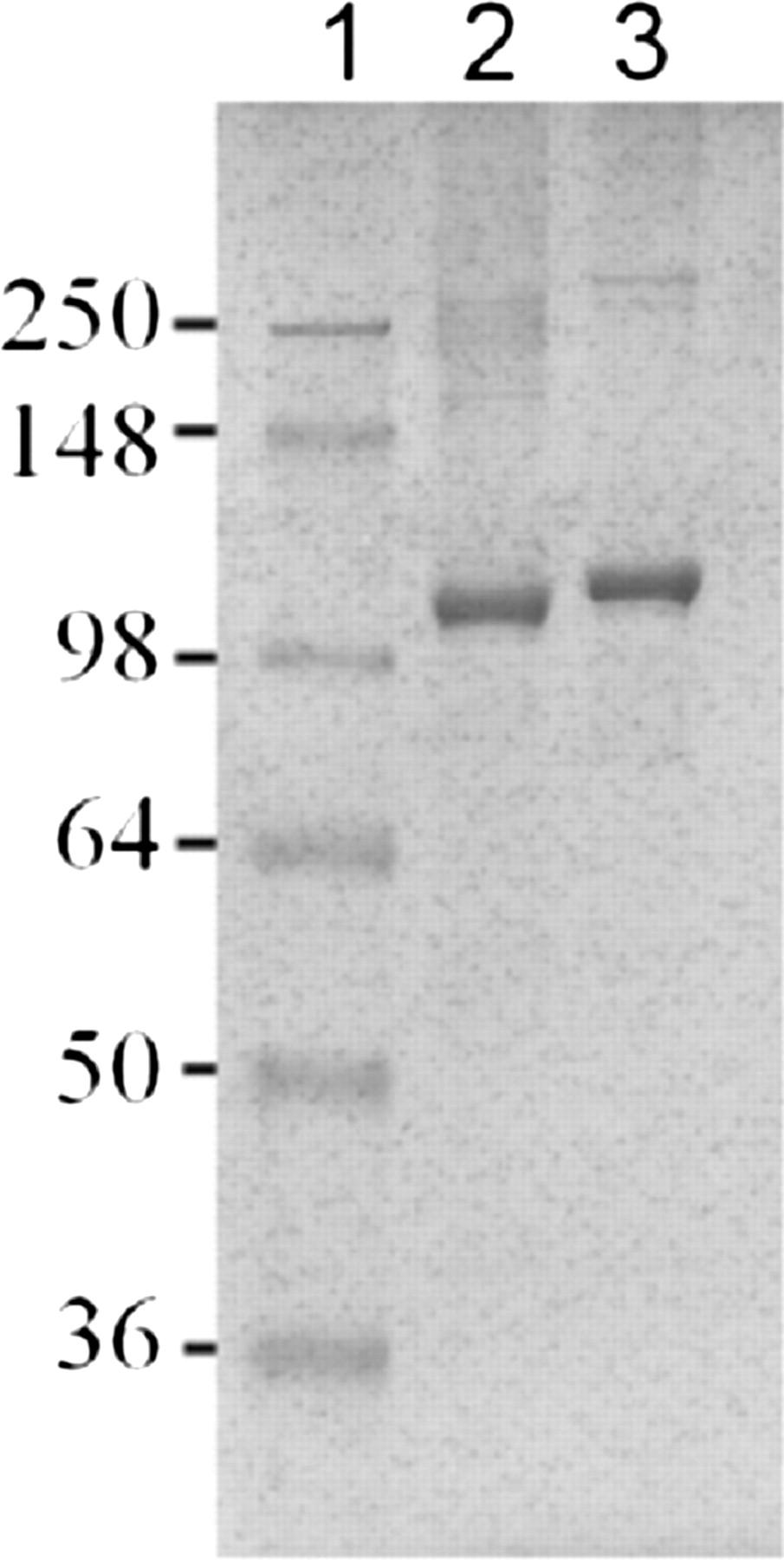
Purification of MBP-FMO3 using amylose affinity chromatography has been previously reported in the literature (Brunelle et al., 1997), and purification of MBP-FMO5 using this method yielded comparable results. Additional purification using anion exchange chromatography (i.e., QFF) was optimized for both MBP-FMO3 and MBP-FMO5. Protein yields after ion exchange chromatography, as determined by the DC assay, were approximately 2 to 4 mg/l of bacterial culture for MBP-FMO3 and 5 to 10 mg/l for MBP-FMO5. SDS-PAGE analysis of the QFF-purified MBP-FMO samples showed a major band of approximately 100 kDa (Fig. 1). This result is in agreement with the calculated molecular mass of 102 kDa for MBP-FMO3 and 104 kDa for MBP-FMO5 based on amino acid sequences. Coomassie Blue staining of highly purified MBP-FMO enzymes (i.e., 4 μg from QFF chromatography) (Fig. 1) on SDS-PAGE indicated greater than 90% purity.

SDS-PAGE analysis of MBP-FMO3 (lane 2) and MBP-FMO5 (lane 3). QFF-purified MBP-FMO samples (4 μg) were analyzed for molecular mass and purity by SDS-PAGE analysis. The numeric values indicate the molecular masses (kilodaltons) for the protein standards shown in lane 1.
MBP-FMO FAD Content Determination.
Both purified MBP-FMO3 and MBP-FMO5 exhibited a distinctive yellow color, and elution peaks monitored for FAD at 450 nm correlated with FMO selective functional activity, indicating copurification of FAD with the MBP-FMO enzymes. The FAD concentration determined by absorbance at 450 nm was compared with the total MBP-FMO protein concentrations determined with a DC assay. The molar ratio of FAD to MBP-FMO3 varied from 0.5 to 0.9 with three independent enzyme preparations. However, the enzyme functional activity of these enzyme preparations showed good reproducibility under saturating substrate concentrations [i.e., kcat (mean ± S.D.) = 8 ± 2 min−1], when the values were normalized by the active enzyme concentration as determined by the FAD content. The FAD content of MBP-FMO5 was reproducible with molar ratios of 0.6 and 0.7 from two independent enzyme preparations. Because only FMO enzymes containing bound FAD are catalytically active, experimentally determined FAD content values for MBP-FMOs and vendor-provided FAD content values for Supersome FMOs were used to define the active enzyme concentration.
MBP-FMO3 and MBP-FMO5 Thermal Stability.
Human FMO3 is a thermally labile enzyme (Cashman et al., 1999), and MBP-FMO3 will rapidly inactivate at 40°C in the absence of NADPH (Motika et al., 2009). Similar instability has been reported for mouse MBP-FMO5 in the absence of NADPH (Zhang et al., 2007), although human FMO5 has not been studied previously. Therefore, it was essential to assess the thermal stability of the MBP-FMO enzymes under the standard assay conditions. As shown in Fig. 2, MBP-FMO3 showed less than 18% loss of MMI S-oxygenation activity over a 30-min period at 37°C in the presence of NADPH. In a slightly different analysis, human MBP-FMO5 showed 8-DPT N-oxide formation with linear time dependence over a 30-min incubation period at 37°C in the presence of NADPH (Fig. 3). A time-dependent loss of activity would have caused deviation from linearity. The results clearly indicated sufficient thermal stability for both enzymes under the standard assay conditions for subsequent kinetic analysis.

Time-dependent thermal stability of MBP-FMO3. MBP-FMO3 was incubated at 37°C in the absence (●) or presence of 0.5% Triton X-100 (□) or 0.5% CHAPS (▴). After various incubation periods, catalytic turnover was determined by the addition of MMI and the resultant S-oxygenation was measured as described under Materials and Methods. The figure shows the ratio of the observed activity at the indicated incubation times (vt) relative to the activity before incubation at 37°C (v0). Data points represent the average of duplicated data points.
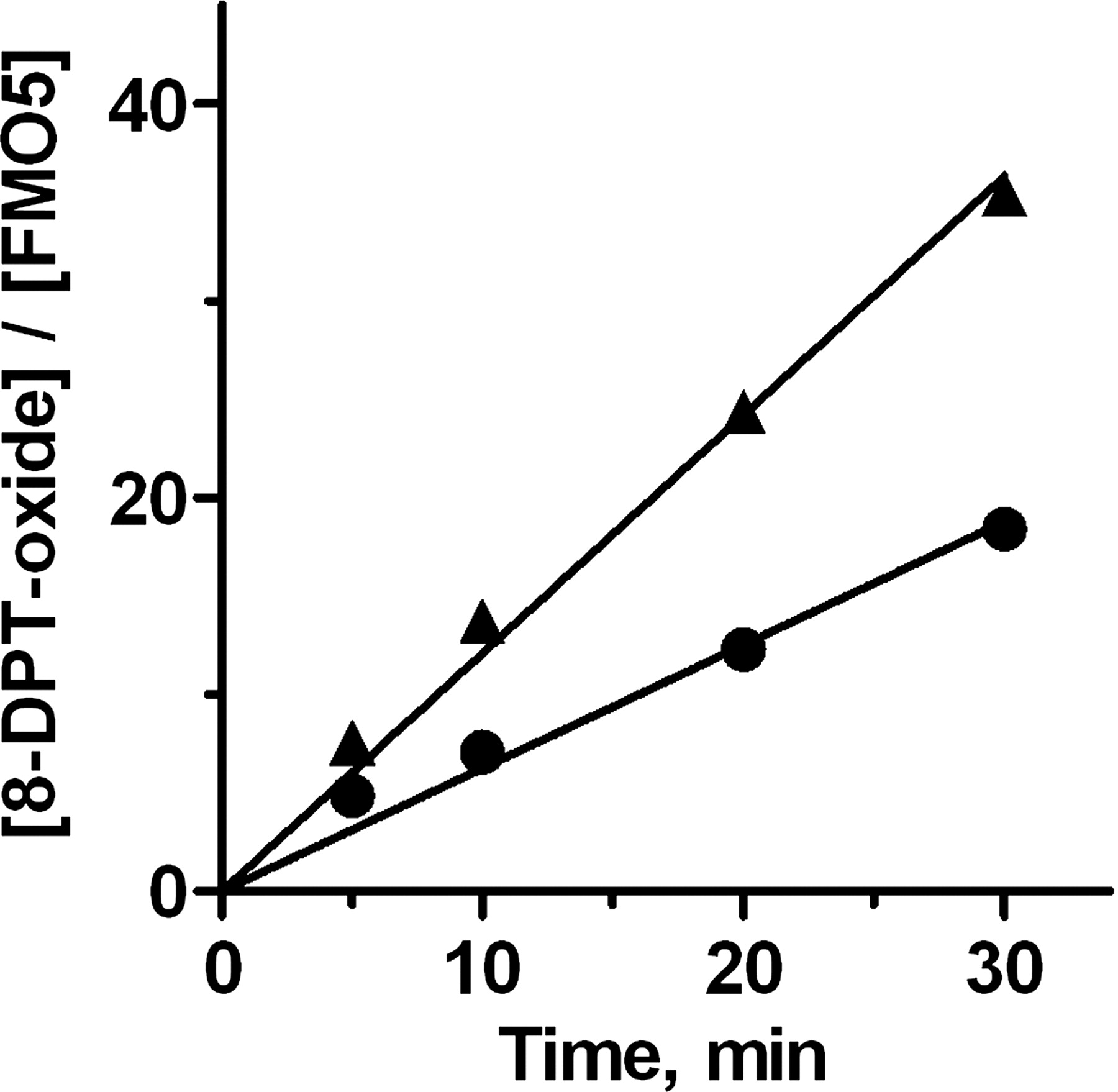
Time-dependent thermal stability of FMO5. Supersome (▴) or MBP-FMO5 (●) was incubated at 37°C in the absence of detergent, with a starting concentration of 400 μM 8-DPT. The incubations were allowed to progress for various time periods and then were assessed for 8-DPT N-oxide formation, as described under Materials and Methods. Values were normalized for the enzyme concentrations. The lines show the fit of the data to a constant rate of catalysis over the 30-min time period. Data markers represent the average of duplicated data points. The S.D.s are smaller than the data markers.
Kinetic Evaluation of Purified MBP-FMO and Supersome FMO.
Steady-state kinetic parameters (i.e., kcat, Km, and kcat/Km) were determined with highly purified MBP-FMO3 in the presence of different detergent conditions (Table 1). In the presence of minimal detergent conditions, kcat and Km were 10.8 min−1 and 32 μM, respectively, for MBP-FMO3-mediated S-oxygenation of MMI. Comparable Km and kcat values were obtained for MBP-FMO3 in the presence of 0.5% CHAPS. However, in the presence of 0.5% Triton X-100, a 2-fold increase in kcat/Km (p < 1 × 10−4) was observed. A similar detergent effect was also observed for S-oxygenation of l-Met (Table 2). The presence of Triton X-100 decreased the apparent Km value from 6.5 to 4.1 mM while modestly increasing kcat, yielding a 2-fold increase in kcat/Km (p = 1.3 × 10−3).
Steady-state kinetic parameters for FMO3-catalyzed S-oxygenation of MMI
Kinetic assays were done at 37°C, pH 8.5.
Steady-state kinetic parameters for FMO3-catalyzed oxygenation of methionine
Kinetic assays were done at 37°C, pH 8.5.
Commercially available Supersome FMO3 was assayed under the same conditions as MBP-FMO3. The apparent Km value for MMI oxygenation was in excellent agreement with the value obtained for MBP-FMO3 (Table 1). Likewise, a l-Met Km value of 6.5 mM has been reported previously for Supersome FMO3 purchased from the same manufacturer (Elfarra and Krause, 2005), in agreement with the value determined for the MBP-FMO enzyme (Table 2). In contrast to the Km values, the kcat (and kcat/Km) values obtained for Supersome FMO3-catalyzed oxygenations were approximately 3- to 5-fold greater than those observed for the MBP-FMO enzyme. Supersome FMO3 kcat values of 33 and 45 min−1 were observed in minimal detergent conditions for MMI and l-Met, respectively. These values are in good agreement with the reported kcat value of 34 min−1 with methyl p-tolyl sulfide (Haining et al., 1997).
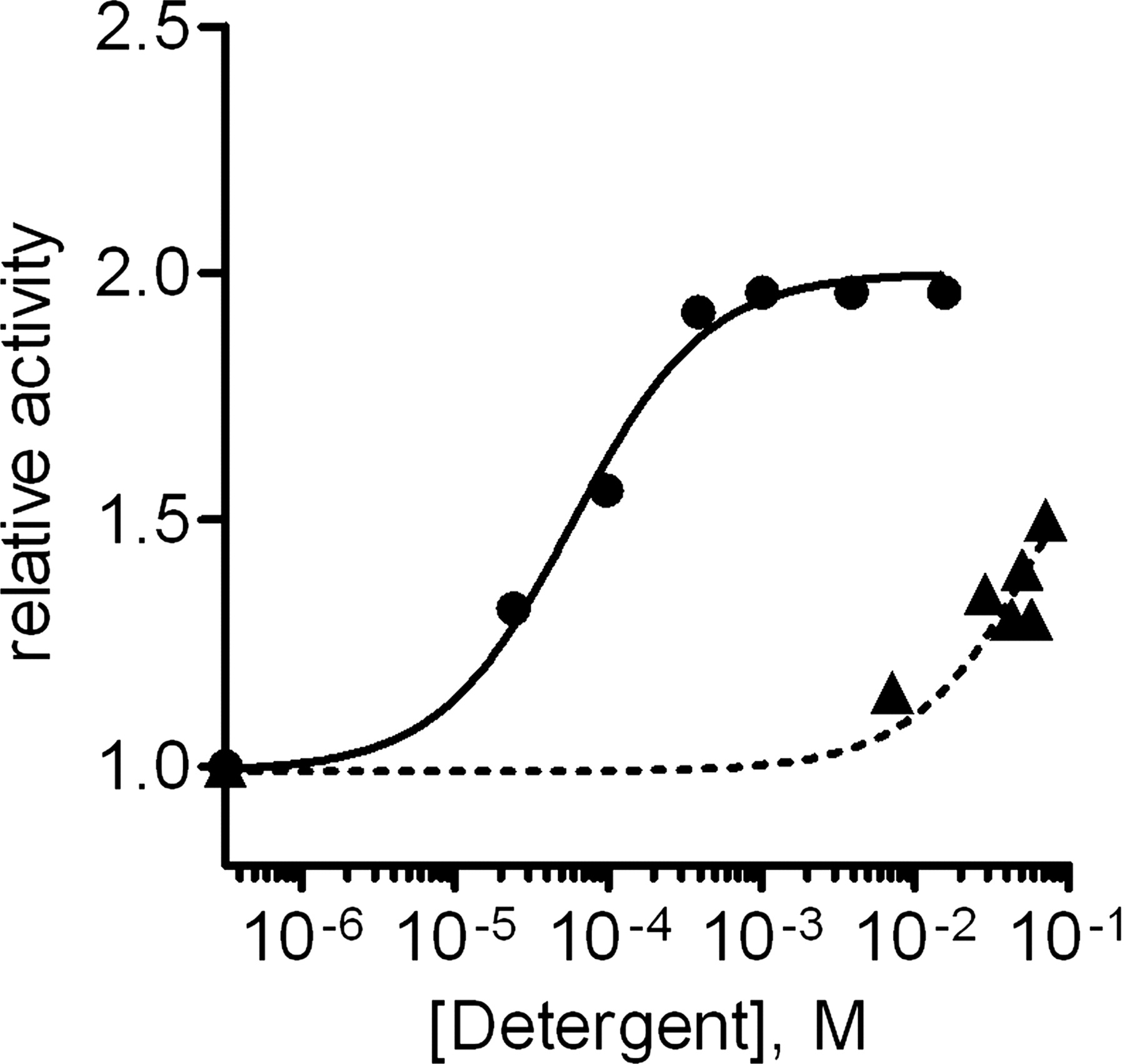
Supersome FMO3 oxygenase activity was enhanced to a degree similar to that observed for MBP-FMO3 by addition of 0.5% Triton X-100. Both enzymes showed a modest increase in kcat values (1.2- 1.3-fold) and a more significant increase in kcat/Km values (1.8-fold, p < 1 × 10−3 for Supersome FMO3 and 2.5-fold, p < 1 × 10−4 for the MBP-fusion enzyme). A similar kcat effect (1.2-fold) has also been reported for native mouse liver FMO preparations using thiourea oxygenation (Venkatesh et al., 1991). The Triton X-100-dependent increase in kcat/Km was sufficient to determine an EC50 value for activation of MBP-FMO3 in 5 μM MMI. The data fit to a sigmoidal dose response, with a 2-fold increase in activity, and an EC50 value of 57 ± 10 μM (Fig. 4) or approximately 36 ppm (using an average molecular mass value of 625 Da). This value is significantly smaller than the CMC range of 0.2 to 0.9 mM reported by the manufacturer and suggests that the activation may not require micelle formation. In contrast, CHAPS did not increase the rate of MMI oxygenation at concentrations below the reported CMC value of 7 to 8 mM but did show activation at higher concentrations, with a predicted EC50 value greater than 60 mM (Fig. 4).3

Triton X-100 (●) and CHAPS (▴) activation of MBP-FMO3 S-oxygenation of MMI. Assays were run at 37°C, pH 8.5, as described previously. Incubations contained 5 μM MMI and 0.5 μM MBP-FMO3. Observed rates of catalysis were divided by the activity observed under minimal detergent conditions (indicated by the data points on the y-axis), and the data were fit to a sigmoidal dose-response curve.
Kinetic parameters for 8-DPT N-oxygenation in the presence of MBP-FMO5 and Supersome FMO5 were also compared (Table 3). As seen for the FMO3 enzymes, MBP-FMO5 and Supersome FMO5 showed identical Km values, whereas the Supersome FMO had increased kcat and kcat/Km values (1.7-fold) (Table 3). The Supersome kcat value of 1.0 min−1 is in reasonable agreement with the kcat value of 1.4 min−1 for methyl p-tolyl sulfide supplied by the manufacturer. The difference may be attributable to the difference in assay conditions (i.e., pH 8.5 versus pH 9.5).
Steady-state kinetic parameters for FMO5-catalyzed oxygenation of 8-DPT
Kinetic assays were performed at 37°C, pH 8.5. Values are the fitting value and error from a single determination of seven substrate concentrations ranging from 10 μM to 2 mM 8-DPT.
Oligomeric Analysis of Purified MBP-FMO Proteins.
Native PAGE analysis was done with different detergent conditions in the sample loading buffer. Triton X-100-solubilized MBP-FMO enzymes indicated the presence of monomers and oligomerized proteins that were interpreted as dimers and hexamers for MBP-FMO3 and as monomers, dimers, tetramers, and hexamers for MBP-FMO5 (Figs. 5 and 6). In contrast, MBP-FMOs solubilized with other detergents tested (i.e., MBP-FMO3 solubilized in 0.5% CHAPS and MBP-FMO5 solubilized in 0.01% DDM) or minimal detergent conditions contained hexameric and larger aggregates of protein. Conceptually identical trends with regard to oligomeric organization were seen with MBP-FMO5 after cleavage of the MBP moiety via treatment with factor Xa (Fig. 6), indicating that the oligomerization was not dependent on the MBP moiety. Furthermore, the addition or removal of respective detergents changed the nature of the oligomeric state, indicating that the oligomeric states for MBP-FMOs were reversible (data not shown).
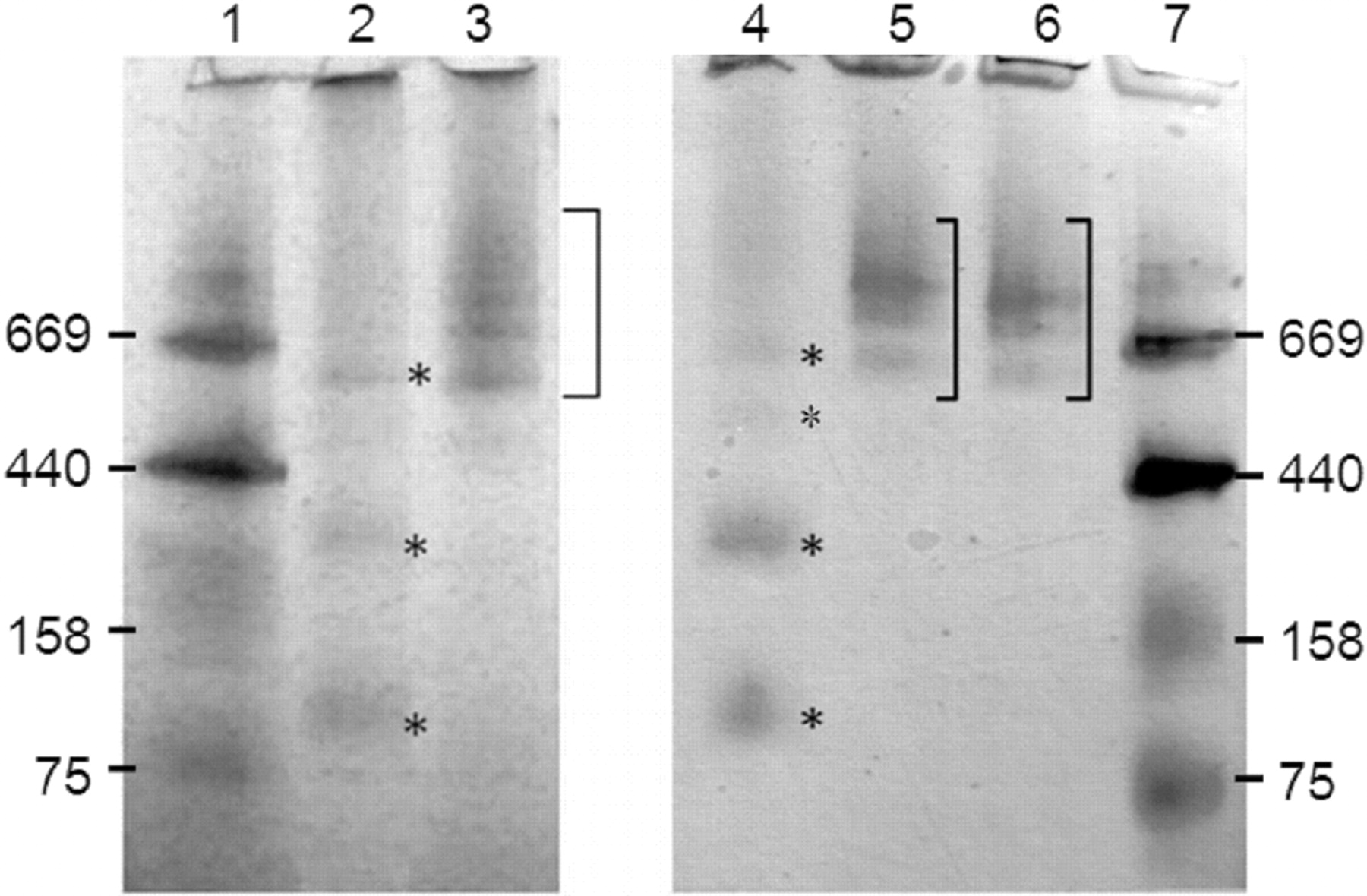
Native PAGE analysis of MBP-FMO3 and MBP-FMO5. Approximately 6 μg of MBP-FMO3 (lanes 2 and 3) or MBP-FMO5 (lanes 4–6) were solubilized with or without detergents and run on separate native PAGE gels. The mobilities of the proteins were compared with those of molecular mass standards (lanes 1 and 7) with the indicated masses (kilodaltons). The detergent conditions for MBP-FMO3 were as follows: lane 2, 0.5% Triton X-100; lane 3, 0.5% CHAPS. The detergent conditions for MBP-FMO5 were as follows: lane 4, 0.5% Triton X-100; lane 5, 0.01% DDM; lane 6, minimal detergent. Asterisks (lanes 2 and 4) or brackets (lanes 3, 5, and 6) were placed to the right of the indicated lanes to highlight FMO protein bands.

Native PAGE analysis of untreated and factor Xa-treated MBP-FMO5. Untreated (lanes 2 and 3) and factor Xa-treated MBP-FMO5 (lanes 4 and 5) were prepared in minimal detergent (lanes 2 and 4) or 0.5% Triton X-100 (lanes 3 and 5) and analyzed via native PAGE. The mobilities of the proteins were compared with those of molecular mass standards (lanes 1) with the indicated masses (kilodaltons). For lanes 4 and 5, regions showing FMO protein are indicated with brackets, and MBP released from the fusion protein is indicated with an asterisk.
In native PAGE analysis, the detergents used to solubilize proteins were included in the loading buffer but became displaced by the Coomassie Blue G-250 present in the running buffer during electrophoresis. In contrast, SEC has the benefit of maintaining a constant detergent concentration throughout the analysis. Therefore, MBP-FMO3 was further analyzed using SEC. When 0.5% Triton X-100 was present in the running buffer, a single peak of MBP-FMO3 eluted at 16.8 ml (Fig. 7A). This value was between the elution volumes for the 75- and 150-kDa protein standards run under the same conditions, consistent with a monomeric form of the enzyme. When 0.5% CHAPS was present in the running buffer for MBP-FMO3, a single peak eluted at 12.7 ml (Fig. 7B). This value is most comparable to the elution volume of the 669-kDa protein standard (12.9 ml) run under identical conditions and is consistent with a hexameric or higher order MBP-FMO3 protein. Thus, the SEC analysis supported the detergent-dependent shift in oligomeric states observed in the native PAGE analysis.

Size exclusion chromatography elution profiles of MBP-FMO3. A Superose 6 10/300 GL column was equilibrated in 50 mM Tris, pH 7.4, 150 mM NaCl, with either 0.5% Triton X-100 (A) or 0.5% CHAPS (B). Protein (200 μg) was applied to the column and eluted with the respective equilibration buffer at 0.5 ml/min flow rate. The elution of MBP-FMO3 was monitored at 450 nm based on FAD absorbance. AU, absorbance unit.
Discussion
In this study, recombinant human FMO3 and FMO5 were expressed as fusion proteins with an N-terminal MBP moiety and purified from E. coli. The steady-state kinetic parameters of the enzymes were determined and compared with those obtained from commercially available Supersome (i.e., membrane-associated) FMO enzymes. Both Supersome and MBP-fused FMOs showed kinetic activation in the presence of Triton X-100. Oligomeric analysis of MBP-FMO also showed oligomeric disassociation in the presence of Triton X-100, suggesting a possible relationship between the oligomeric state and kinetic activation.
Purification of MBP-FMO3 and MBP-FMO5 involved straightforward two-step amylose affinity and anion exchange chromatography, with levels of protein yield in good agreement with expected yields for MBP fusions expressed in E. coli (Pattenden and Thomas, 2008). The FAD contents for the final purified MBP-FMO3 and MBP-FMO5 (65–75%, molar ratio) were comparable to values reported for the Supersome FMO3 (i.e., 40–50%) (Haining et al., 1997; Yeung et al., 2007), suggesting that proper protein folding and cofactor association during expression were not grossly disrupted by the MBP fusion or subsequent purification procedures.
Kinetic studies showed that the recombinant MBP-FMO enzymes and Supersome FMOs behave very similarly. Under minimal detergent assay conditions, the MBP-FMO3 and MBP-FMO5 Km values for their respective substrates were identical to their Supersome counterparts within experimental error. However, both Supersome FMO3 and Supersome FMO5 showed greater kcat and kcat/Km values compared with their corresponding MBP-FMOs. Unlike the Km values, the apparent kcat and kcat/Km are dependent upon the apparent active enzyme concentration (i.e., as measured by FAD content). Therefore, the FAD contents for both Supersome and MBP-FMO3 were confirmed using an HPLC method (as described in Lang et al., 1998, data not shown). The observed differences in FMO activity were not attributable to differences in FAD quantification. Instead, the results suggest that some fraction of the MBP fusion FMO enzyme remains inactive, despite retaining the ability to bind FAD. Furthermore, cleavage of the MBP moiety does not yield an increase in catalytic activity, thereby ruling out a simple steric hindrance to the active site or other reversible phenomenon resulting from MBP FMO fusion.4
Of interest, during the course of kinetic characterization, an approximately 2-fold activation of kcat/Km and Km values was observed for both MBP-FMO3 and Supersome FMO3. The similar increase seen with both Supersome and MBP enzymes indicates that this phenomenon was not an artifact of the MBP fusion. The observed EC50 value determined with MBP-FMO3 was well below the CMC value, suggesting that Triton X-100 association with MBP-FMO involved a limited number of Triton X-100 molecules interacting with specific hydrophobic protein regions, rather than general micelle formation.
Although previous studies have noted detergent effects on kcat values for mammalian FMO enzymes, this is the first report of detergent-mediated activation of the bimolecular rate constant kcat/Km for these enzymes.5 Because FMO substrates do not form a Michaelis complex with the enzyme (Poulsen and Ziegler, 1995), the kcat/Km parameter reflects the rate of productive collision of substrate with 4a-hydroperoxyflavin. Therefore, the increased activity suggests greater access of the substrate to the active site in the presence of Triton X-100.
In addition to the kinetic analyses, MBP-FMOs were analyzed for their oligomeric status. Although poorly characterized in the literature, mammalian FMOs are present in higher order oligomers. Pig liver FMO1 has been described as a higher order oligomer (Ziegler and Mitchell, 1972), with one report of tetramers and octamers,6 although no experimental results have been published. Likewise, rabbit FMO2 was reported as “aggregated” under native PAGE conditions (Krueger et al., 2006), with a detergent-dependent dissociation to a dimeric state using a truncated variant. To date, there is no detailed oligomeric analysis of native human FMO enzymes. The current study found higher order oligomers for both recombinant MBP-FMO3 and MBP-FMO5 that were interpreted as hexamers. These oligomers disassociated in the presence of Triton X-100. A conceptually identical detergent-dependent oligomeric disassociation was observed after proteolytic removal of the MBP moiety from MBP-FMO5, suggesting that the oligomeric states are attributable to the FMO moieties and are not an artifact of the MBP fusions. Characterization of the Supersome enzymes using these methods was prohibited by the relatively low purity and FMO concentration of commercially available enzyme.
Although further studies will be required to fully elucidate the mechanism of Triton X-100-dependent FMO activation, it is intriguing to note that the kinetic activation correlated with the oligomeric disassociation induced by Triton X-100, suggesting that the two phenomena may be functionally related. Furthermore, a physiological mechanism of kinetic regulation based on oligomeric disassociation to improve substrate access to the enzyme active site, although purely speculative for FMOs at present, is not inconsistent with these results.
In conclusion, this study provides the only direct comparison between human MBP-FMO fusion proteins and their Supersome counterparts and is the first report of detergent-based kinetic activation of kcat/Km with a corresponding detergent-based oligomeric disassociation. MBP-fused and Supersome FMO have different apparent rates of turnover, and this difference may affect certain studies when absolute kinetic values are important. However, for many studies in which relative values are highly informative (e.g., when one is comparing the rates of one substrate with those of another or the rate of a wild-type enzyme with that of a variant), MBP-FMO has been shown to be useful (e.g., Motika et al., 2009). Furthermore, the MBP-FMO system provides more enzyme, higher levels of purity, and simpler purification schemes in comparison with Supersome FMO. These factors combine into significant advantages that show the utility of the MBP-FMO fusion proteins, especially for biochemical characterizations in which protein purity is critical. This report suggests that the MBP-FMO fusion expression system is a robust and useful alternative approach to generate high-quality recombinant FMO enzymes.
Acknowledgments.
We thank Human BioMolecular Research Institute scientist Dr. Karl Okolotowicz for chemical synthesis of 8-DPT HCl and Dr. Xueying Zheng for HPLC assays.
Footnotes
This work was supported by the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grant DK59618].
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.110.033639.
↵1 The term Supersome FMO is used throughout the manuscript to refer to any microsomal preparation of human FMO enzymes from insect cells, whether or not said preparations were purchased from a commercial source.
↵2 This calculation assumes that the CHAPS buffer passed freely through the 50-kDa filter during the enzyme concentration steps described under Materials and Methods and was not itself concentrated.
↵3 The EC50 value for CHAPS was predicted by fitting the data to a sigmoidal dose-response curve with a fixed upper limit equal to that observed in the Triton X-100-dependent trend. The reported value is the lower limit from the resulting 90% confidence interval.
↵4 MBP-FMO5 relative activity was 97 ± 17% after 3 h of incubation with factor Xa compared with the same incubation time in the absence of factor Xa. MBP-FMO3 cleavage previously yielded 112% relative activity, as reported by Brunelle et al. (1997).
↵5 In a previous study (Brunelle et al., 1997), MBP-FMO3 phenothiazine N-oxygenation decreased at Triton X-100 concentrations greater than 0.05%. This result is attributed to the hydrophobic nature of the phenothiazine substrates leading to a detergent interference with the specific assay. A similar situation was reported for decreased mouse liver FMO oxygenation of phorate in 1% Triton X-100 (Venkatesh et al., 1991).
↵6 In a recent review, Palfey and McDonald (2010) reported that pig liver FMO1 purifies as a tetramer or octamer with bound lipid. However, no primary data or literature reference was cited.
-
ABBREVIATIONS:
- FMO
- flavin-containing monooxygenases
- P450
- cytochrome P450
- MMI
- methimazole
- PAGE
- polyacrylamide gel electrophoresis
- 8-DPT
- 10-[(N,N-dimethylaminooctyl)-2-(trifluoromethyl)]phenothiazine
- CHAPS
- 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid
- DDM
- n-dodecyl-β-d-maltoside
- QFF
- Q Fast-Flow
- DC
- detergent-compatible colorimetric
- HPLC
- high-performance liquid chromatography
- SEC
- size exclusion chromatography
- CMC
- critical micelle concentration.
- Received April 2, 2010.
- Accepted August 31, 2010.
- Copyright © 2010 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}