


Abstract
Thioacetamide (TAA) is a potent hepatotoxicant and has been widely used to develop experimental liver fibrosis/cirrhosis models. Although the liver toxicity of TAA has been extensively studied, little is known about its potential influence on UDP-glucuronosyltransferases (UGTs) associated with the development of liver fibrosis. The study presented here aimed to uncover the regulation patterns of UGTs in TAA-induced liver fibrosis of rats. Potential counteracting effects of hepatoprotective agents were also determined. TAA treatment for 8 weeks induced a significant transcriptional up-regulation of the major UGT isoforms, including UGT1A1, UGT1A6, and UGT2B1, accompanied with the dramatic elevations of most typical serum biomarkers of liver function and fibrosis scores. Upon TAA intoxication, the mRNA and protein levels of the major UGT isoforms were increased to 1.5- to 2.5-fold and 2.5- to 3.3-fold of that of the normal control, respectively. The hepatoprotective agents Schisandra spp. lignans extract and dimethyl diphenyl bicarboxylate could largely abolish TAA-induced up-regulation of all three UGT isoforms. However, enzyme activities of UGTs remained unchanged after TAA treatment. The dissociation of protein expression and enzyme activity could possibly be attributed to the inactivating effects of TAA, upon a NADPH-dependent bioactivation, on UGTs. This study suggests that the transcriptional up-regulation of UGTs may be an alternative mechanism of their preserved activities in liver fibrosis/cirrhosis.
Introduction
UDP-glucuronosyltransferases (UGTs), a group of membrane-bound enzymes that reside in the endoplasmic reticulum, catalyze the conjugation of UDP-glucuronic acid to the hydroxyl, carboxyl, amine, or thiol group of numerous structurally diverse endogenous substances and xenobiotics (Radominska-Pandya et al., 2005). Because the addition of glucuronic acids generally increases the water solubility of substances and thus facilitates elimination, UGTs play an important role in detoxifying or inactivating endogenous and xenobiotic substances (Wang et al., 2010). Alternatively, UGT-catalyzed glucuronidation could contribute to the bioactivation and/or toxification of some substances, as in the case of acyl glucuronidation of carboxylic drugs such as nonsteroidal anti-inflammatory drugs (Southwood et al., 2007; Koga et al., 2011). Nevertheless, the dysregulations of UGTs caused by various pathological factors and exogenous toxicants could trigger significant changes of pharmacological, toxicological, and/or pathological consequences of various substances.
The dysregulations of cytochromes P450 in hepatic fibrosis/cirrhosis have been extensively studied (Gomez-Lechon et al., 2009; Morgan, 2009). In contrast, the current understanding of UGT dysregulation in hepatic injury remains very limited, and the previous results are largely controversial. UGT activities were found largely preserved in liver diseases, possibly because of the activation of latent UGT enzymes (Desmond et al., 1994), the increased expression in remaining viable cells (Debinski et al., 1995), and the increased contribution of extrahepatic UGTs (Omar et al., 1996). However, more recent studies focusing on the mRNA levels of individual UGT isoforms from clinical patients with liver diseases (Congiu et al., 2002) and mice with inflammation (Richardson et al., 2006) indicated down-regulations of most UGT isoforms. The major limitation of previous studies concerning UGT regulation in liver injury is that the expression and enzyme activity of individual isoforms have not been concomitantly evaluated, resulting in inconsistencies across various studies with different measures. In addition, results obtained from clinical patients may be complicated by other confounding factors, including gene polymorphisms and concomitant drugs, tobaccos, and dietary components. Therefore, it is important to simultaneously determine the expression and enzyme activity of individual UGT isoforms in the experimental models of liver injury for better understanding of liver injury per se on regulating UGTs.
Thioacetamide (TAA), originally used as a fungicide, is a potent hepatotoxicant and has been widely used in developing experimental liver fibrosis/cirrhosis models mimicking the human liver fibrosis/cirrhosis (Nozu et al., 1992; Kang et al., 2005). As the largest metabolic organ in biological systems, the liver, endowed with many types of drug-metabolizing enzymes, plays a critical role in the metabolic elimination of many endogenous substances and exogenous toxicants. Therefore, extensive understanding of the dysregulation of drug metabolizing enzymes in liver injury should be an essential step for exploring pathological processes and consequences of liver diseases. It had been previously reported that most cytochrome P450 isoforms were downregulated in a TAA-induced rat cirrhosis model (Nakajima et al., 1998). In contrast, the profile of UGT regulation in TAA-induced liver injury models remains largely unknown.
Considering that some recent reports suggested a dysregulation of most individual UGT isoforms in liver injury, we hypothesized herein that TAA-induced fibrosis/cirrhosis would also result in a dysregulation of UGT isoforms. The major purpose of this study was to test the hypothesis by concomitant evaluation of the mRNA, protein, and enzyme activity of major rat UGT isoforms including UGT1A1, UGT 1A6, and UGT 2B1 in a TAA-induced liver injury model. In addition, it was of great interest to determine whether hepatoprotective agents would be effective in counteracting TAA-induced dysregulation of UGTs. For this purpose, Schisandra spp. lignan extract (SLE) and dimethyl diphenyl bicarboxylate (DDB), both of which had been well proven to possess powerful hepatoprotective effects against liver injury induced by various pathological factors(Gao et al., 2005; Abdel-Hameid, 2007; Xie et al., 2010), were included in this study to further verify our hypothesis.
Materials and Methods
Chemicals and Reagents.
TAA was obtained from Jiahui Medicine Chemical Co. Ltd. (Anhui, China). DDB and SLE were obtained from Qingze Science and Technology Co. Ltd. (Nanjing, China). Bicinchoninic acid protein assay kit, SDS-polyacrylamide gel electrophoresis, and sample loading buffer were purchased from Beyotime Institute of Biotechnology (Jiangsu, China). UGT1A1, UGT1A6, and UGT2B antibodies (goat anti-rat IgG) were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Bilirubin was purchased from the Laboratory for the Control of Drugs of Jiangsu Province (Nanjing, China). Testosterone (TES), 4-methylumbelliferone (4-MU), 4-MU-O-glucuronide, glucose 6-phosphate, NADP+, glucose-6-phosphate dehydrogenase, UDP-glucuronic acid (UDPGA), alamethicin, and d-saccharic acid 1,4-lactone monohydrate were all purchased from Sigma-Aldrich (Shanghai, China). The purity of all of the chemicals was proven to exceed 99%. High-performance liquid chromatography (HPLC)-grade acetonitrile and methanol were obtained from Merck (Darmstadt, Germany). Deionized water was purified using a Milli-Q system (Millipore Corporation, Billerica, MA).
Animals and Treatment.
Male Sprague-Dawley rats (200–250 g) were caged and fed at 20–25°C, with a 12-h light/dark cycle and free access to food and water. The animals were acclimatized to the environment for 1 week and fasted with free access to water for 12 h before each experiment. All animal experimental procedures were approved by the Animal Care and Use Committee of the China Pharmaceutical University and were in accordance with the guiding principles for the use of animals in toxicology adopted by the Chinese Society of Toxicology. Animals were randomly divided into five groups with six individuals in each group. Four groups of rats were injected with TAA (200 mg/kg i.p., 3% in physiological saline) twice a week for 8 consecutive weeks to induce hepatic fibrosis (Barash et al., 2008). Another group of rats serving as a normal control was treated with normal saline. After an initial TAA treatment for 4 weeks, the rats were intragastrically administered with 1% sodium carboxymethyl cellulose (CMC-Na; TAA control), SLE low dose (100 mg/kg, suspended in 1% CMC-Na), SLE high dose (400 mg/kg, in 1% CMC-Na), or DDB (200 mg/kg, in 1% CMC-Na) once a day, respectively, for another 4 weeks. All of the rats were weighed before treatment and at appropriate time points throughout the whole experimental duration and were observed daily for signs of illness. Rats were sacrificed by cervical dislocation under ether anesthesia at the end of the 8th week. Blood samples were collected for biochemical determinations; the liver tissues were quickly removed, weighed, and frozen immediately in liquid nitrogen until analysis.
To determine the acute effects of TAA on regulating UGTs, rats were injected with a single dose of TAA at 200 mg/kg i.p., whereas the normal control group of rats was injected with normal saline. Six hours after treatment, the rats were sacrificed under ether anesthesia, and the livers were quickly removed and immediately frozen in liquid nitrogen. The liver samples were used for the determination of mRNA levels and enzyme activities of UGTs.
Hepatic Function Biomarkers and Fibrosis Determination.
Hepatic function biomarkers, including alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (AP), albumin (ALB), γ-glutamyl transpeptidase (γ-GT), and total bilirubin (T-Bil), were determined by an automatic blood biochemical analyzer (Beckman counter LX20; Beckman Coulter, Inc., Fullerton, CA).
Slices of the same part of each rat liver were cut off and fixed in phosphate-buffered 10% formaldehyde solution and then embedded in paraffin wax. Sections of liver tissues with 5-μm thickness were cut and stained with hematoxylin-eosin according to standard procedures and then examined for histopathological changes under the microscope (Olympus BH2; Olympus, Tokyo, Japan). The extent of fibrosis was graded, according to the semiquantitative METAVIR scoring system, as no fibrosis (score 0), portal fibrosis without septa (score 1), portal fibrosis with few septa (score 2), numerous septa without cirrhosis (score 3), and established cirrhosis (score 4). The assessment of the extent of fibrosis was performed in blind by an experienced pathologist.
RT-PCR Analysis.
Frozen liver samples were crushed with a glass mortar in liquid nitrogen. RNA was extracted using a RNA extraction kit (Keygene, Shanghai, China) according to the manufacturer's instructions. The RNA pellet was dried and dissolved in RNase-free water and stored at −80°C until use. RNA concentrations were determined with a spectrophotometer at 260 nm. For cDNA synthesis, a reaction media containing 5 μg of total RNA, 1 μl of oligo dT (10 μM), 1 μl of dNTPs (10 mM), and RNase-free water to a final volume of 15 μl was heated at 65°C for 5 min and then immediately placed in an ice-water bath for 5 min. To each sample, 0.5 μl of RNase inhibitor (40 U), 2 μl of 10× avian myeloblastosis virus reaction buffer, 1 μl of dithiothreitol (1 M), and 1 μl of reverse transcriptase (avian myeloblastosis virus) was added and kept at 37°C for 60 min and then at 70°C for 15 min. The cDNA samples were kept at −80°C until use.
Primers for rat UGT1A1, UGT1A6, UGT2B1, and glyceraldehyde phosphate dehydrogenase (GADPH) were designed using the primer select software (Primer Premier 5.0; PREMIER Biosoft International, Palo Alto, CA) on the basis of previous reports (Nozu et al., 1992; Kang et al., 2005). Sequences of the polymerase chain reaction (PCR) primers are as follows (sense and antisense for each isoform, 5′–3′, respectively): UGT1A1 (554 bp), ACG AAG TGG TGG TCA TAG CA, CTG TAA GAT TTC AGT GGC AAG; UGT1A6 (270 bp), TGG TGC TAG TGC CAG AAG TCA A, GAG CAT CAA ACT GGT TCT CCC T; UGT2B1 (439 bp), GTC ACG GTT CTT GTA TCT TCG G, GAA CAA CAG GCA CAT AGG AAG G; and GAPDH (352 bp), CGG GAA GCT TGT CAT CAA TGG, GGC AGT GAT GGC ATG GAC TG. The PCR mixture containing 5 μl of 10× Taq buffer, 1 μl of dNTPs (10 mM), 2 μl of each primer (10 μM), 2 μl of cDNA template, 0.5 μl of Taq DNA polymerase, and 4 μl of MgCl2 (2.5 mM) was initially denatured at 95°C for 90 s, followed by 32 cycles of amplification for UGT1A1, 30 cycles for UGT1A6, 28 cycles for UGT2B1, or 30 cycles for GAPDH. Each cycle was performed as the following steps: 90 s at 94°C (denaturation); 45 s at 59.5°C for UGT1A1, 59°C for UGT1A6, 56°C for UGT2B1, or 55°C for GAPDH (annealing); and 45 s at 72°C (extension). The final extension was performed at 72°C for 10 min. Then, 8 μl of the PCR-amplified mixture was subjected to electrophoresis on 1.5% agarose gel and visualized by ethidium bromide staining. The gel was then photographed by UV gel image formation (Gel DocTM XR; Bio-Rad Laboratories, Hercules, CA), and the gray scale was determined by a digitalized software (Quantity one; Bio-Rad Laboratories).
Western Immunoblotting.
Liver tissue proteins (70 μg) were resolved by SDS-10% polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane at 180 mA for 60 min using a wet transfer method (Bio-Rad Laboratories, wet blotter). After transferring, the membranes were blocked with 5% nonfat dry milk in Tris-buffered saline/Tween 20 buffer (TBST) at 37°C for 90 min. The membranes were then incubated overnight at 4°C with each diluted antibody (1:200 for UGT1A1, 1:150 for UGT1A6, and 1:200 for UGT2B) in 5% nonfat dry milk-TBST and washed 3 times with TBST for 15 min each time. For references, β-actin was detected using a polyclonal antibody (1:200). A peroxidase-conjugated rabbit anti-goat IgG (BioWorld Products, Inc., Visalia, CA) (1:5000) was used as second antibody and incubated with the membranes for 60 min at 37°C. The membranes were washed extensively again (three times, 15 min/time) with TBST before detection using enhanced chemiluminescence detection reagents (BioWorld). After exposure in the darkroom, the film was then photographed and the signal intensity was detected by a digitalized software (Quantity one, Bio-Rad). The protein levels of UGTs were normalized to that of the reference band β-actin.
Preparation of Rat Liver Microsomes.
For enzyme activities analysis of UGTs, each group of rats' livers were pooled together and the hepatic microsomal suspensions were prepared by differential centrifugation as described previously (Hao et al., 2007) and resuspended in 0.25 M sucrose/1 mM EDTA solution, pH 7.4. Protein concentrations were determined with a bicinchoninic acid protein assay kit according to the manufacturer's instructions. Final protein concentration was adjusted to approximately 10 mg/ml with the same buffer. The suspensions were frozen and maintained at −80°C until use.
Enzyme Activities Assay of UGTs.
Enzyme activities of UGTs in rat liver microsomes were determined using the typical substrate bilirubin for UGT1A1, 4-MU for UGT1A6, and TES for UGT2B1. For the UGT1A1 test, the pooled and alamethicin-permeabilized microsomes (2 mg/ml) from each group were incubated for 30 min at 37°C in the presence of 10 mM MgCl2 and 320 μM bilirubin in a final volume of 0.2 ml. The reactions were initiated by the addition of 20 mM UDPGA and terminated by the addition of 200 μl of ice-cold glycine/HCl (2 M, pH 2.7). The bilirubin glucuronidation was assayed by the electrophotometrical determination of metabolites as described elsewhere with minor modifications (Gordon et al., 1983). Bilirubin glucuronides were determined by measuring the absorbance at 550 nm using a detection kit on the basis of the conversion of mono- and di-bilirubin glucuronides to dipyrrolic azo derivatives. Because bilirubin is a light-sensitive compound, all handling procedures for bilirubin were performed under dim light.
For the UGT1A6 and UGT2B1 tests, the pooled and alamethicin-permeabilized microsomes (2 mg/ml) of each group were incubated at 37°C with 4-MU (500 μM) for 5 min or TES (75 μM) for 18 min in the media containing 10 mM MgCl2 in a final volume of 0.2 ml in phosphate-buffered saline, pH 7.4. The reactions were initiated by the addition of 20 mM UDPGA and terminated by the addition of 200 μl of ice-cold acetonitrile. After centrifugation, 10 μl of supernatant was injected into a HPLC system (Shimadzu, Kyoto, Japan) for the detection of 4-MU, TES, and their glucuronides. The UV detector was set at 220 nm for the detection of 4-MU and its glucuronide and at 250 nm for TES and its glucuronide, respectively. Separation was achieved on a Diamonsil C18 column (150 × 4.6 mm, 5 μm; Dikma Technologies, Inc., Lake Forest, CA). A mobile phase consisting acetonitrile and 50 mM ammonium phosphate buffer [30:70 (v/v)] was used for TES and its glucuronide detection. For 4-MU and its glucuronide detection, the mobile phase was composed of acetonitrile and 20 mM ammonium phosphate buffer [10:90 (v/v)]. The flow rate was set at 1.0 ml/min in both cases.
To determine the potential inactivating effects of TAA on UGTs, TAA (5, 50, and 200 μM) was preincubated with rat liver microsomes for 0.5 h with or without the addition of NADPH before the addition of UDPGA and probe substrates to initiate the UGT-catalyzed reactions. Effects of hydrogen peroxide (0.2, 1, and 5 mM) preincubation were also determined likewise. In another set of incubations, DDB (10, 50, and 200 μM) or GSH (1 and 5 mM) was added concomitantly with TAA in the UGT1A6 activity test.
TAA Disposition in Liver Microsomes.
To investigate whether the hepatoprotective agents were influential on the TAA disposition, an assay of NADPH-dependent metabolic depletion of TAA was performed in the in vitro liver microsome incubation system. The incubation mixture contained 1 mg/ml microsome protein, TAA (0.1 mM), NADPH-regenerating system, and the hepatoprotective agent SLE (0–500 μg/ml) or its lignan component schizandrol A (0–200 μM) and deoxyschizandrin (0–100 μM), or DDB (0–500 μM), in a final volume of 200 μl. NADPH-regeneration system was finally added to initiate the reaction. All of the incubations were conducted at 37°C for 0.5 h and terminated by adding ice-cold acetonitrile. The concentration of remaining TAA was determined by HPLC-UV (Chilakapati et al., 2005). Five replicates were performed for each of the incubations.
Statistical Analysis.
Data are expressed as mean ± S.D., and statistically significant differences were assessed with the one-way analysis of variance followed by post hoc analysis (Dunnett's test) in most cases, except for the liver fibrosis scores, which were analyzed by a χ2 test. All statistical analyses were performed with SPSS software version 16.0 (SPSS Inc., Chicago, IL); the difference was considered statistically significant when the probability value was less than 0.05 (P < 0.05).
Results
Body and Liver Weight.
TAA treatment for 8 weeks caused a significant growth retardation of rats, as evidenced by the significant decrease of final body weight and body weight gains. Although the absolute liver weights were not changed significantly, TAA treatment for 8 weeks induced a significant increase of liver weight index. SLE or DDB treatment exerted a slight ameliorating effect on the body weight loss, but little impact on the increase of liver weight index, induced by TAA (Supplemental Table 1).
Histopathology.
Induction of hepatic fibrosis was notable in rats exposed to TAA for 8 weeks, characterized with a fibrosis score of 2.83 ± 0.41. SLE and DDB exerted a significant attenuating effect on TAA-induced hepatic fibrosis, as evidenced from histopathological examinations (Supplemental Fig. 1) and fibrosis scores (Table 1).
Liver fibrosis scores of rats with TAA intoxication for 8 weeks
The extent of fibrosis was graded according to the semiquantitative METAVIR scoring system and scored as 0, 1, 2, 3, and 4, respectively. The liver fibrosis for rats of normal control was graded as zero.
Serum ALT, AST, AP, Albumin, γ-GT, and Total Bilirubin.
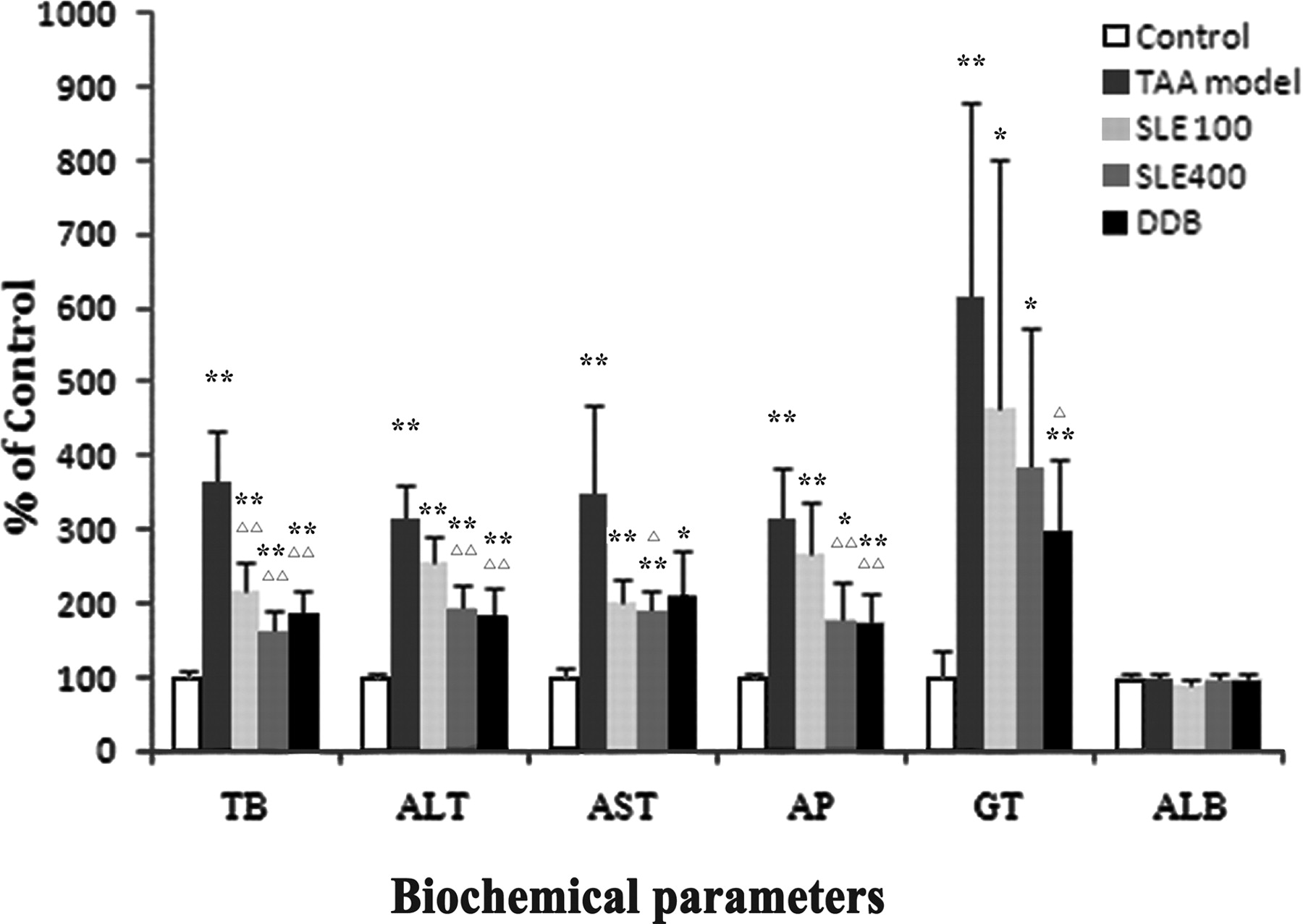
Typical serum biomarkers of hepatic functions including ALT, AST, AP, ALB, γ-GT, and T-Bil were determined. The results are shown in Fig. 1. Compared with the normal control, the levels of all serum biomarkers except ALB were significantly increased after TAA treatment for 8 weeks, indicating a serious hepatic injury caused by TAA. Rats treated with SLE and DDB showed remarkably lower levels of serum biomarkers compared with those in the TAA group.

Biochemical parameters of liver function after 8 weeks of TAA intoxications. The serum level of T-Bil, ALT, AST, AP, γ-GT, and ALB for the control group is 2.96 ± 0.25 μM, 44.40 ± 3.21 IU/l, 149.25 ± 18.01 IU/l, 191.80 ± 10.66 U/l, 1.20 ± 0.45 U/l, and 19.80 ± 0.84 g/l, respectively. Data are expressed as percentage (%) of control (mean ± S.D., n = 6). *, P < 0.05 and **, P < 0.01 compared with the control group; ▵, P < 0.05 and ▵▵, P < 0.01 compared with TAA group.
mRNA Levels of UGTs.
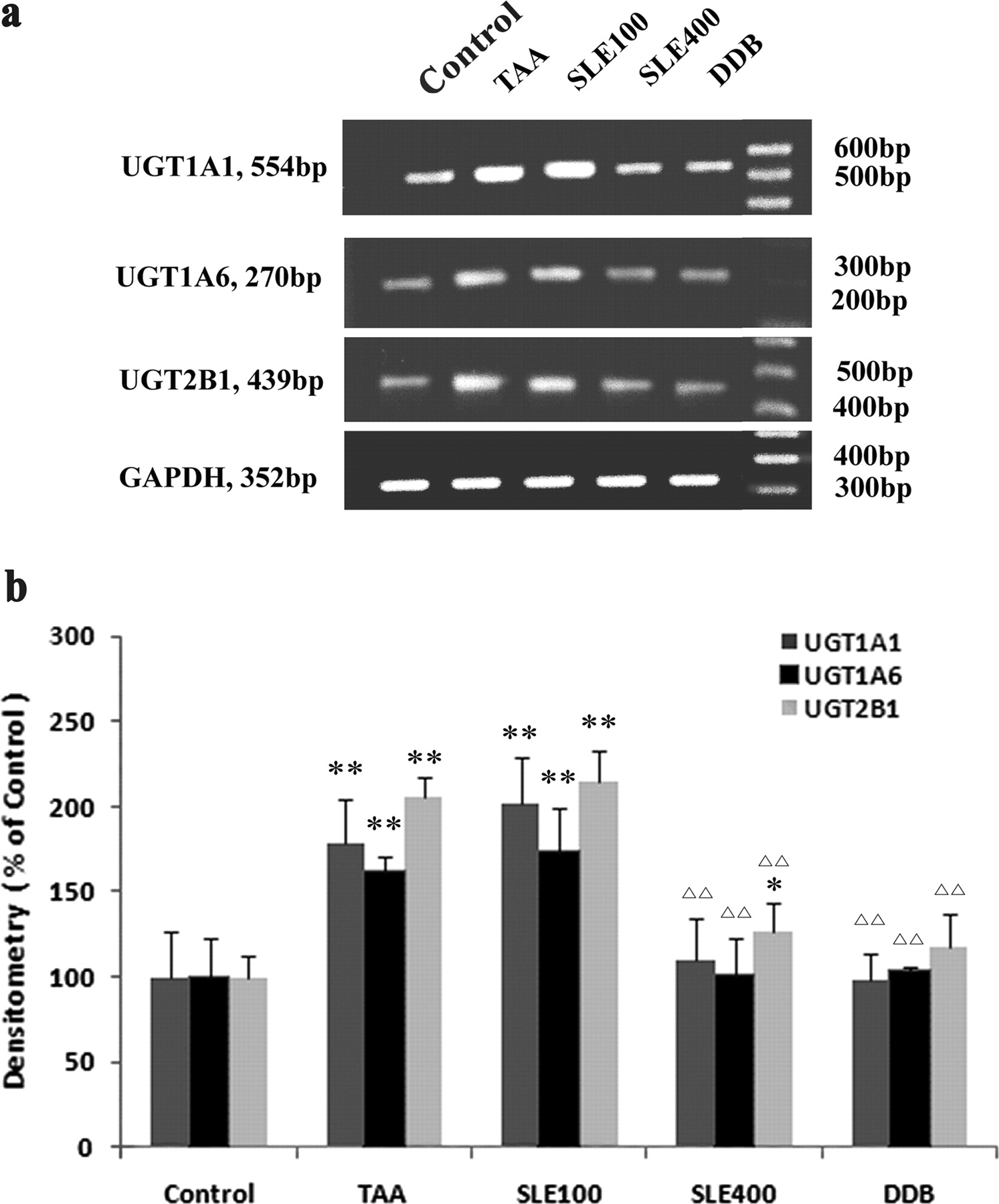
The mRNA levels for the major rat liver UGTs including UGT1A1, UGT1A6, and UGT2B1 were determined by RT-PCR. TAA treatment significantly increased the mRNA levels of all three UGT isoforms in rat livers, with UGT1A1 increased to 182%, UGT1A6 to 157%, and UGT2B1 to 206% of those in the control group (Fig. 2). It was of interest to find that treatment with the hepatoprotective agents SLE and DDB exhibited a significant counteracting effect on the TAA-induced adaptive mRNA up-regulation of UGTs. The mRNA levels of all three UGT isoforms in the SLE (400 mg/kg) and DDB treatment groups were even restored to the normal levels (97–132% of the normal control).
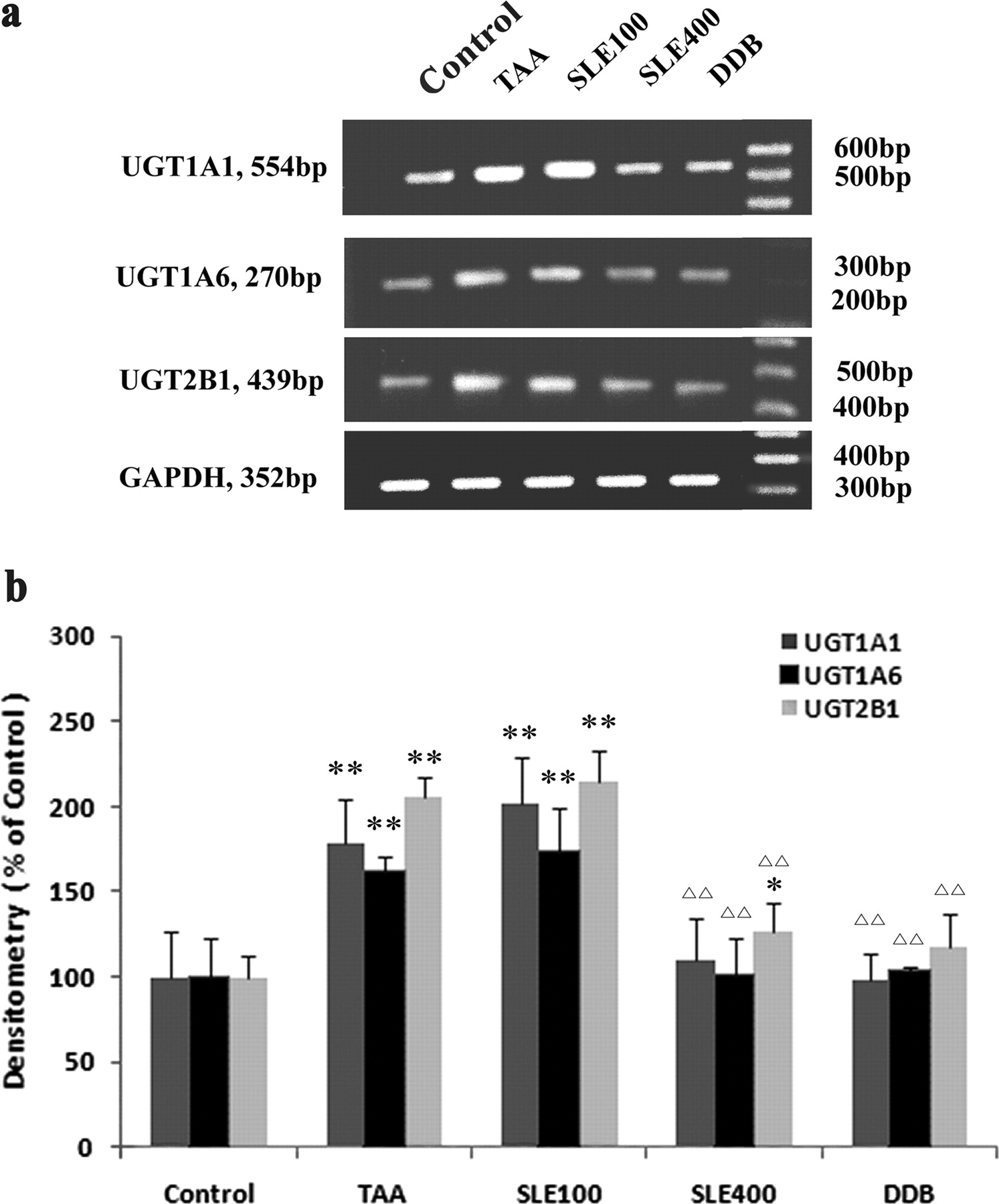
RT-PCR analysis of the mRNA levels of UGT1A1, UGT1A6, and UGT2B1. a, image of RT-PCR results; b, semiquantitative data analysis of RT-PCR results. The mRNA levels of UGTs were normalized to that of GADPH in the same preparation and quantitated by densitometry. Data are expressed as percentage (%) of control, and bars represent mean ± S.D. of six individual liver samples. **, P < 0.01 compared with the control group; ▵▵, P < 0.01 compared with TAA group.
Protein Expression of UGTs.
For assessing the protein expression levels of the major rat liver UGTs, a Western blotting analysis using the polyclonal antibody of UGT1A1, UGT1A6, and UGT2B was applied in this study. In accordance with the mRNA levels, TAA treatment resulted in a nearly 3-fold increase of the protein levels of all three UGT isoforms compared with the normal control (Fig. 3). SLE and DDB treatment significantly counteracted the TAA-induced up-regulation of UGT protein levels. High-dose SLE (400 mg/kg) and DDB treatment restored the UGT protein levels back to normal levels (97–135% of normal control).
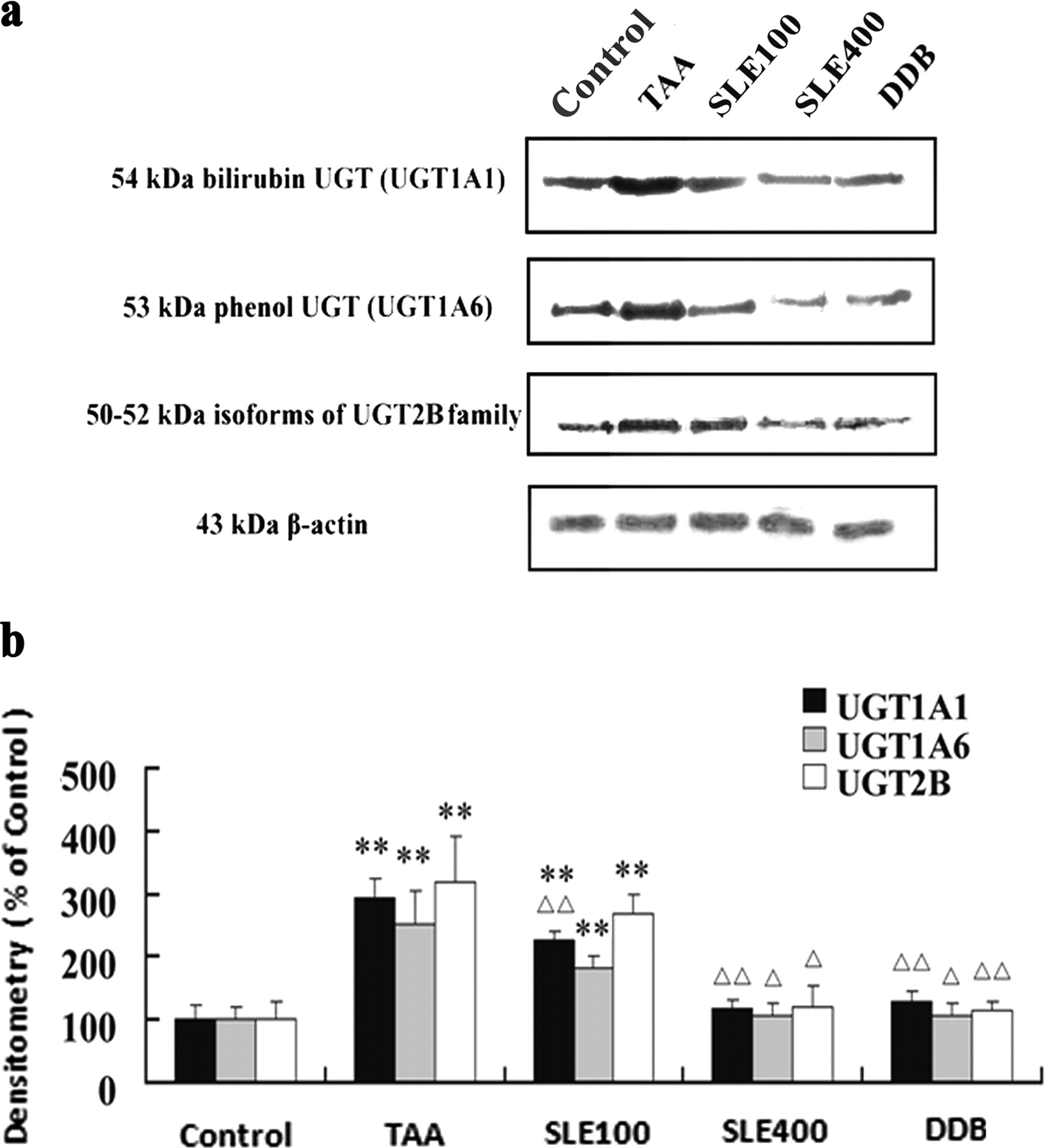
Immunoblot analysis of the protein levels of UGT1A1, UGT1A6, and UGT2B1. a, image of Western blot results; b, semiquantitative data analysis of Western blot results. The protein levels of UGTs were normalized to that of β-actin in the same preparation and quantitated by densitometry. Each of the UGTs protein blots is a representative of six liver samples. Densitometry data are expressed as percentage (%) of the control, and bars represent mean ± S.D. of six liver samples. **, P < 0.01 compared with the control group; ▵, P < 0.05 and ▵▵, P < 0.01 compared with TAA group.
To evaluate the regulating effects of SLE and DDB per se on UGTs, SLE (400 mg/kg) or DDB (200 mg/kg) was administered intragastrically to healthy rats once a day for 2 weeks, and the protein levels of hepatic UGTs were determined by western blotting. As a result, no significant differences were found between the hepatoprotective-agent-treated groups and the control group, suggesting that SLE and DDB themselves have little direct effect on regulating UGTs (Supplemental Fig. 2).
Enzyme Activities of UGTs.
Enzyme activities for the major rat UGT isoforms UGT1A1, UGT1A6, and UGT2B1 were determined using the typical substrate bilirubin, 4-MU, and TES, respectively, in pooled liver microsome incubation systems. The incubation conditions including microsomal protein levels and incubation time were optimized to ensure a linear production of glucuronidation metabolites of each substrate.
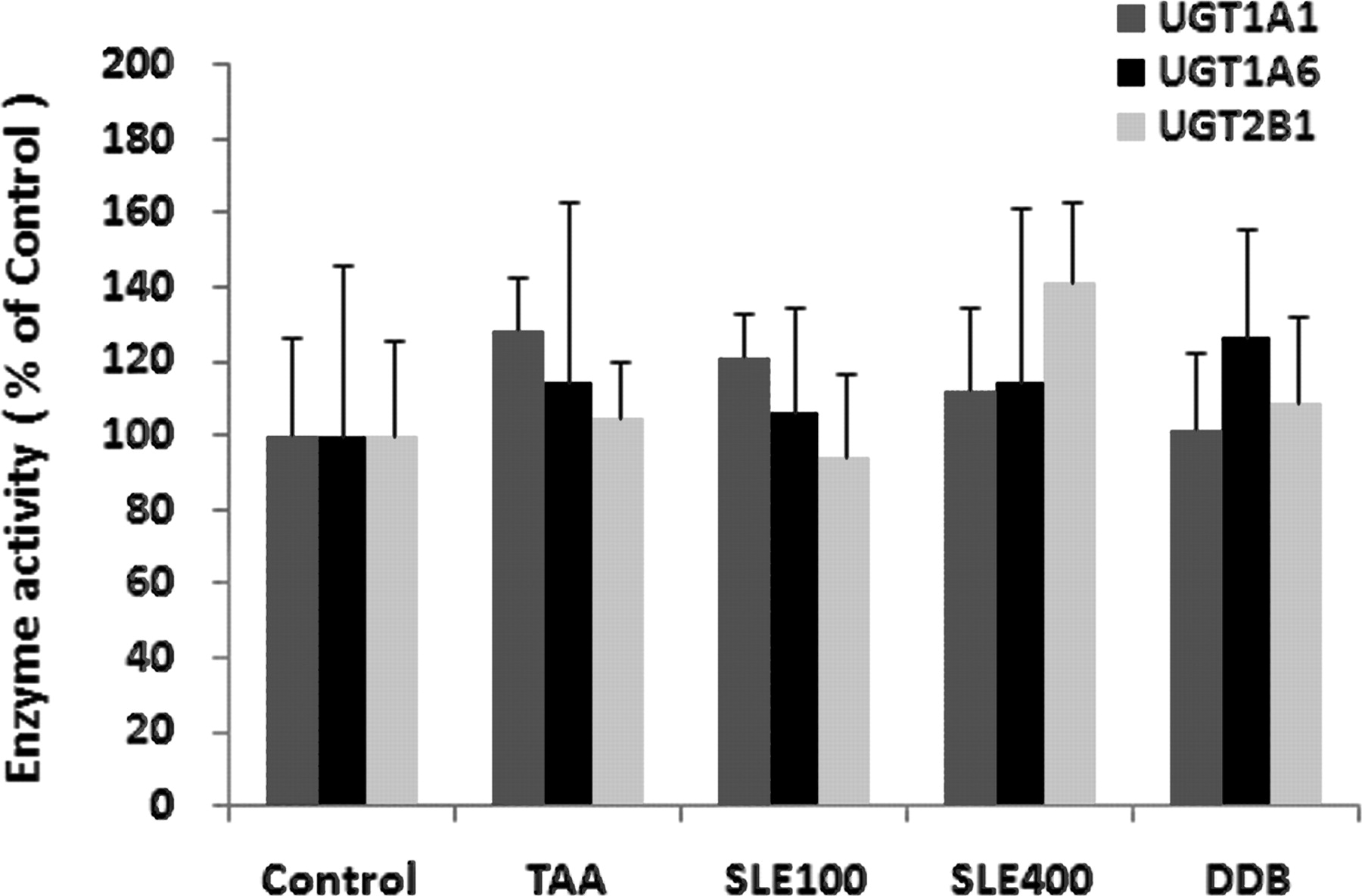
In contrast to the UGT mRNA and protein expressions, it was surprising to find that TAA treatment exhibited little effect on the enzyme activities of all three UGT isoforms tested in the microsomal incubation systems. In addition, neither SLE nor DDB treatment showed significant influence on the enzyme activities of UGTs compared with either the normal control or TAA group (Fig. 4).

Enzyme activities of UGT1A1, UGT1A6, and UGT2B1. Bilirubin (320 μM), 4-MU (500 μM), or TES (75 μM) was used as the typical substrate for UGT1A1, UGT1A6, and UGT2B1, respectively. Data are expressed as percentage (%) of control; bars represent mean ± S.D. of five replicates.
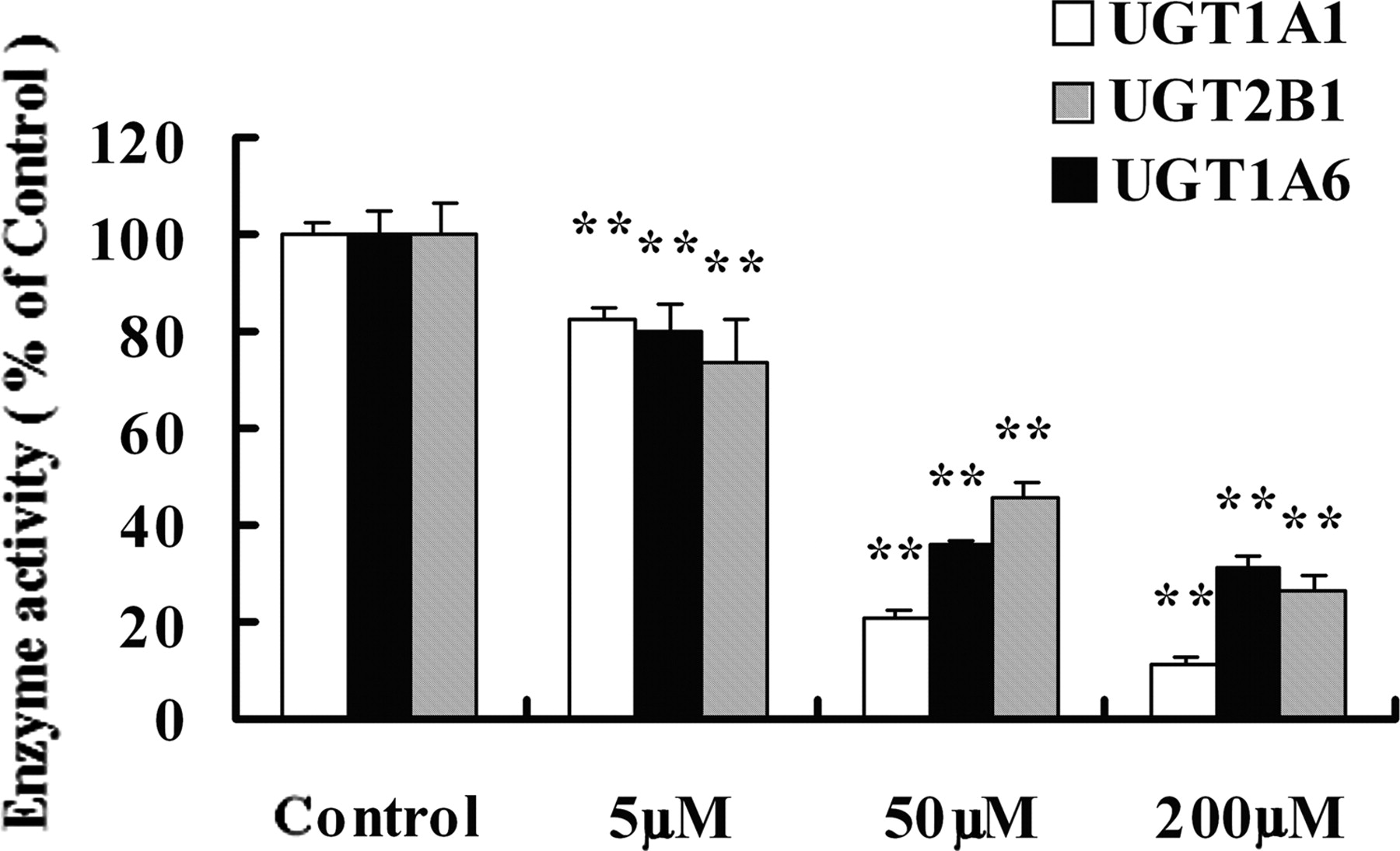
The lack of correlation between protein expression and enzyme activity upon TAA intoxication prompted us to hypothesize that TAA may directly inactivate UGTs. To test this hypothesis, TAA (0, 5, 50, and 200 μM) was preincubated in liver microsomes with the addition of NADPH for 0.5 h, and then the remaining enzyme activities of UGTs were determined by adding the respective probe substrates as described under Enzyme Activities Assay of UGTs. The preincubation of TAA without the addition of NADPH showed little effect on UGT activities. In contrast, TAA preincubation with NADPH dramatically and concentration-dependently inactivated the enzyme activities of all three UGT isoforms (Fig. 5). Likewise, hydrogen peroxide pretreatment significantly inactivated the enzyme activities of UGTs (Fig. 6). Taking the UGT1A6 test as an example, the addition of DDB and GSH partially abolished TAA-induced inactivation of enzyme activity (Fig. 7).

The inactivating effects of TAA on the enzyme activities of UGT1A1, UGT1A6, and UGT2B1. TAA was preincubated in liver microsomes with the addition of NADPH regenerating system for 0.5 h before the enzyme activity assay of UGTs. The preincubation of TAA without the addition of NADPH had little effect on UGT activities. Data are expressed as percentage (%) of control, and bars represent mean ± S.D. of five incubations. **, P < 0.01 compared with the control group.

The inactivating effects of hydrogen peroxide on UGT1A1, UGT1A6, and UGT2B1. Hydrogen peroxide (200 μM, 1 mM, and 5 mM) was preincubated with microsomes for 0.5 h before the enzyme activity test of UGTs. Data are expressed as percentage (%) of control; bars represent mean ± S.D. of five incubations. **, P < 0.01 compared with the control group.

The protective effects of DDB and GSH against the inactivating effect of TAA on UGT1A6. NADPH regenerating system was included in all groups of samples except the TAA 200 μM (−) group. Data are expressed as mean ± S.D. of five incubations. **, P < 0.01 compared with the TAA 200 μM (−) group; ▵▵, P < 0.01 compared with the TAA 200 μM (+) group.
Acute Effects of TAA on Regulating UGTs in Rats.
To observe the acute effects of TAA on regulating UGTs, rats were intraperitoneally injected with a single dose of TAA (200 mg/kg). The mRNA levels and enzyme activities of liver microsomal UGTs after a 6-h TAA treatment were determined. As shown in Fig. 8, a single dose of TAA treatment exerted little influence on the mRNA levels and enzyme activities of most UGTs, except for a slight reduction of UGT2B1 activity.

Acute effects of TAA on regulating mRNA and enzyme activities of UGTs in rats. a, mRNA levels of UGT1A1, UGT1A6, and UGT2B1. The mRNA levels of UGTs were normalized to that of GADPH in the same preparation and quantitated by densitometry. b, enzyme activities of UGTs. Data are expressed as percentage (%) of control; bars represent mean ± S.D. of triplicate samples. *, P < 0.05 compared with the control group.
Effect of DDB and SLE on TAA Disposition In Vitro.
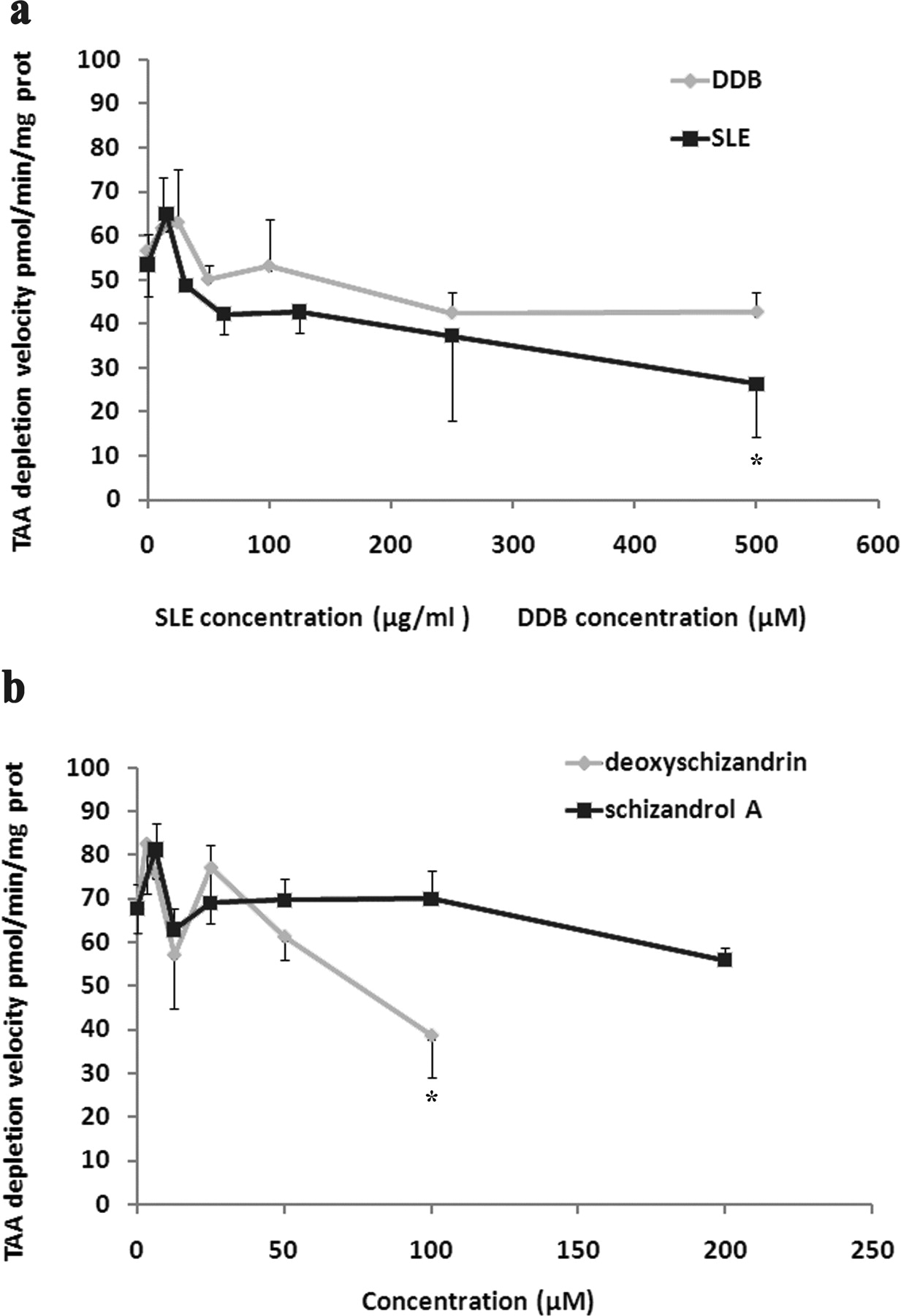
To determine whether SLE and DDB could influence TAA disposition, the NADPH-dependent metabolic depletion of TAA was determined in microsomal incubation systems in vitro. The influences of DDB and schizandrol A on the metabolic elimination of TAA were very limited. SLE and its lignan component deoxyschizandrin at high concentrations exhibited slight inhibitory effects on the metabolic elimination of TAA in microsomes; however, such a high concentration would be difficult to achieve at in vivo conditions (Fig. 9).

Effects of DDB and SLE and its major components on the metabolic depletion of TAA in microsomal incubation systems. a, effects of DDB and SLE; b, effects of deoxyschizandrin and schizandrol A. TAA (0.1 mM) was incubated in liver microsome incubation systems containing NADPH regenerating system for 0.5 h at 37°C. Velocity of TAA metabolic depletion was calculated by measuring the loss of TAA in the incubation media. Data are expressed as mean ± S.D. of five incubations. *, P < 0.05 compared with control incubations (without the addition of hepatoprotective agents).
Discussion
As a typical hepatotoxicant, TAA induces a progressive liver fibrosis and cirrhosis in experimental animals that mimics the pathological processes and consequences of liver cirrhosis in human beings. The study presented here contributes to disclose that the mRNA and protein expression levels of the major UGT isoforms in rats, including UGT1A1, UGT1A6, and UGT2B1, are adaptively upregulated in a TAA-induced rat liver fibrosis model, whereas the enzyme activities of UGTs remain largely unchanged. The dissociation of protein expression and enzyme activity is potentially attributed to the inactivating effects of TAA upon NADPH-dependent bioactivation on UGTs. The hepatoprotective agents SLE and DDB elicit a significant counteracting effect on the adaptive change of UGT expressions, accompanied with powerful hepatoprotective effects as evidenced from the examinations of serum biomarkers and histopathology.
In accordance with previous reports (Karantonis et al., 2010; Shaker et al., 2010), the study presented here confirmed that TAA treatment resulted in a significant retardation of natural body growth, liver hypertrophy, elevated serum biomarkers of liver function, and the progressive liver fibrosis. As expected, SLE and DDB treatments exhibited powerful hepatoprotective effects, supported by the significant recovery of most indexes determined. The hepatoprotective effect of SLE (and its lignans components), a well known herbal medicine not only widely used in China but also used as a component of Kampo medicines and dietary supplements (Mu et al., 2006), had been previously proven in many other experimental models of liver injury. DDB, an intermediate produced from the chemical synthesis of schisandrin C, had been clinically used for more than 30 years in the treatment of viral and chemical hepatitis (Gao and Kang, 2006; Abdel-Hameid, 2007). The model-independent hepatoprotective effects of SLE and DDB may be explained by their powerful antioxidative effects (Chiu et al., 2003; Ko and Chiu, 2005).
Although the pathological processes and underlying mechanisms of TAA-induced liver fibrosis/cirrhosis have been extensively studied, little is known about the potential dysregulation of drug metabolizing enzymes in this model. The study presented here found that the major rat UGT isoforms, including UGT1A1, UGT1A6, and UGT2B1, were transcriptionally upregulated after an 8-consecutive-week TAA intoxication. Likewise, the protein levels of all three isoforms were increased in parallel with the mRNA up-regulation. The references in the current literature concerning the transcriptional regulation of UGTs in liver injury are largely controversial. Some reports showed that most UGT isoforms were transcriptionally downregulated in the liver samples from humans with liver diseases, which was found to correlate with inflammation but not with fibrosis scores (Congiu et al., 2002), and from the liver samples of mice treated with lipopolysaccharides or bacterial infections (Richardson et al., 2006). However, another set of reports witnessed an up-regulation of most UGT isoforms in humans with cirrhosis (Debinski et al., 1995) and rat liver samples (Debinski et al., 1996), as well as in regenerated rat liver tissues after partial hepatectomy (Pellizzer et al., 1996). Such discrepancies across different researches may be explained by the diverse severity and property of liver diseases, various pathological factors, and different animal models applied. The large individual variability of UGT isoforms in human beings caused by potential genetic and environmental factors and the exposures to drugs and dietary components further complicated the determination of liver injury per se on regulating UGTs.
It is important to note that most previous studies concerning UGT regulations in liver injury have not performed a concomitant assay of mRNA, protein, and enzyme activity, which may constitute another cause for the controversial results obtained in different researches. We found that despite a 2- to 3-fold up-regulation of mRNA and protein levels after 8 weeks of TAA treatment, the enzyme activities of all three UGT isoforms remain unchanged. A similar phenomenon had been found in a previous study, in which the influence of octachlorostyrene treatment on inducing UGT mRNA levels was much more prominent than that on the glucuronidation activity toward 1-naphthol (Yanagiba et al., 2009). It is now well known that TAA is bioactivated mainly by CYP2E1 to produce reactive intermediates and the metabolite di-S-oxide, which is highly reactive against macromolecules by covalent binding and/or oxidative modifications (Kang et al., 2008). We hypothesized that the enzyme activity of UGTs may be inactivated by the reactive metabolites and/or reactive oxygen species generated from the process of TAA bioactivation. To test this hypothesis, microsomes obtained from healthy rats were preincubated with TAA or hydrogen peroxide for 0.5 h, and then the remaining enzyme activities of UGTs were determined. The results confirmed that the enzyme activity of all three UGT isoforms was dramatically inactivated by TAA treatment, providing a good explanation of TAA-induced unparallel change between the expressions and activities of UGTs. It is important to note that the inactivating effect of TAA on the enzyme activities of UGTs is NADPH-dependent, suggesting that the metabolic bioactivation is necessary for TAA to inactivate UGTs. Hydrogen peroxide can also inactivate the enzyme activities of UGTs; however, a relatively high concentration is necessary to take effect. This evidence suggests that the reactive metabolites produced from TAA bioactivation may play an important role in inactivating UGTs. However, the contribution of TAA-induced oxidative stress cannot be excluded, considering that GSH and DDB pretreatment partially abolished TAA-induced inactivation of UGTs. Because DDB has little effect on the metabolic depletion of TAA in liver microsomes (Fig. 9), its effect on protecting against the TAA inactivating effect on UGTs is unlikely to have resulted from its influence on TAA bioactivation. Further research is necessary to determine whether such effects of DDB and GSH are attributed to their antioxidant activities and/or capacities of trapping TAA reactive metabolites.
The potential influences of single-dose treatment of TAA to rats were further evaluated to determine whether the long-term TAA-intoxication-induced transcriptional up-regulation of UGTs was associated with liver fibrosis progression or merely an acute effect of TAA. The results showed that single-dose treatment of TAA had little effect on regulating the mRNA levels and enzyme activities of UGTs. This seems controversial between the in vivo and in vitro assay in terms of the inactivating effect of TAA on UGTs. However, it would be understandable considering that some in vivo protective machinery such as antioxidant and the trapping of reactive metabolites might prevent the enzyme from inactivation by a single dose of TAA treatment. Together, these results suggest that the transcriptional up-regulation of UGTs after long-term TAA treatment is more likely an adaptive response from the progression of liver fibrosis, and that repeated doses of TAA intoxication may be necessary to inactivate UGTs in vivo. The major limitation of this study is that the underlying molecular mechanisms of TAA-induced up-regulation of UGTs have not been addressed. TAA-induced liver injury was typically characterized with oxidative stress and lipid peroxidation (Low et al., 2004; Natarajan et al., 2006; Aller et al., 2008), and the antioxidant treatment was very effective in preventing TAA-induced liver injury (Hyoudou et al., 2007; Tsai et al., 2010). In addition, we found in the study presented here that SLE and DDB, both proven as good antioxidants, could significantly attenuate TAA-induced liver injury and transcriptional up-regulation of UGTs. These lines of evidence suggest that TAA-triggered oxidative stress might be an underlying factor contributing to the transcriptional up-regulation of UGTs. Therefore, it will be of interest to determine in future studies whether nuclear factor-like-2, an oxidative stress-responsive transcription factor, is involved in TAA-induced up-regulation of UGTs considering that some reports documented an important role of nuclear factor-like-2 on regulating UGTs (Enomoto et al., 2001; Umemura et al., 2006; Yeager et al., 2009).
In conclusion, TAA-induced liver fibrosis leads to a transcriptional up-regulation of UGTs; however, the enzyme activities of UGTs remain largely unchanged. The dissociation between enzyme expression and activity is possibly attributed to the UGT inactivating effects of TAA upon bioactivation. SLE and DDB can largely abolish TAA-induced transcriptional up-regulation of UGTs despite the hepatoprotective agents themselves possessing little effect on regulating UGTs. Results obtained from the study presented here may provide novel explanations on the preserved UGT activities in liver injury and may shed a light on understanding the influence of hepatoprotective agents on regulating UGTs in liver injury.
Authorship Contributions
Participated in research design: Hao and Wang.
Conducted experiments: Hao, Zhang, Jiang, Sun, Xie, and Zhou.
Performed data analysis: Hao, Zhang, Jiang, and Gong.
Wrote or contributed to the writing of the manuscript: Hao, Zhang, Jiang, and Wang.
Footnotes
This work was supported by the National Natural Science Foundation of China [Grants 91029746, 30801422, 30973583]; the Program for New Century Excellent Talents in University of China [Grant NCET-09-0770]; and a Foundation for the Author of National Excellent Doctoral Dissertation of China [Grant 200979].
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.111.039172.
↵
 The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.-
ABBREVIATIONS:
- UGTs
- UDP-glucuronosyltransferases
- TAA
- thioacetamide
- SLE
- Schisandra lignans extract
- DDB
- dimethyl diphenyl bicarboxylate
- TES
- testosterone
- 4-MU
- 4-methylumbelliferone
- UDPGA
- UDP-glucuronic acid
- HPLC
- high-performance liquid chromatography
- ALT
- alanine aminotransferase
- AST
- aspartate aminotransferase
- AP
- alkaline phosphatase
- ALB
- albumin
- γ-GT
- γ-glutamyl transpeptidase
- T-Bil
- total bilirubin
- RT-PCR
- reverse transcriptase-polymerase chain reaction
- PCR
- polymerase chain reaction
- TBST
- Tris-buffered saline/Tween 20 buffer
- CMC-Na
- sodium carboxymethyl cellulose
- bp
- base pair(s).

- Received March 1, 2011.
- Accepted July 6, 2011.
- Copyright © 2011 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}