


Abstract
Bernard B. Brodie's laboratory was the first to examine the mechanisms of drug-induced toxicity at the molecular level. They found that acetaminophen hepatotoxicity was due to the metabolic activation of the drug to a highly reactive toxic metabolite that depleted cellular glutathione and covalently bound to protein. Subsequent studies revealed that activation of acetaminophen to an active metabolite is primarily carried out by CYP2E1, an ethanol-inducible cytochrome P450 that was first suggested by characterization of the microsomal ethanol oxidation system. CYP2E1 is developmentally regulated, under liver-specific control, and undergoes substrate-induced protein stabilization. It is also regulated by starvation and diabetes through insulin-dependent mRNA stabilization. In addition to acetaminophen, CYP2E1 metabolically activates a large number of low Mr toxicants and carcinogens and thus is of great toxicological importance. The mechanism of regulation CYP2E1 and its role in acetaminophen toxicity will be discussed.
I am delighted and honored to be a recipient of the Bernard B. Brodie award. Like Brodie, I have spent my career at the National Institutes of Health. Other than our mutual interest in xenobiotic metabolism, the similarities end. Brodie was a pioneer in drug metabolism and toxicity and the concepts he generated during the course of his career are still of major importance in the development and use of drugs, and in the fields of dietary and environmental toxicology. Brodie led the transition of the field of classical physiological-based pharmacology into biochemical pharmacology where mechanism of drug action and toxicity could be determined. In particular, he noted that drug-induced toxicities can be due to dose-dependent production of highly reactive and unstable intermediates that covalently bind to cellular macromolecules. These concepts laid the foundations for subsequent studies including the enzymology, purification, and cloning of drug-metabolizing enzymes and, more recently, the production of genetically modified mice that were used to reveal the in vivo role of P450s and other drug-metabolizing enzymes in chemical toxicities. Among the most important P450s involved in chemical toxicities is CYP2E1.
Ethanol-Inducible CYP2E1
CYP2E1 has been extensively studied for many years because of its relevance to chemical toxicity and carcinogenicity. It is also conserved in mammals; although there are a number of single nucleotide polymorphisms in the CYP2E1 gene, there are no known polymorphisms that are the result of inactive CYP2E1 genes in humans or any animal models, and there are no large differences in catalytic activity among humans, rabbits, rats, and mice. This suggests that CYP2E1 has an important function in mammals.
The Microsomal Ethanol Oxidation System and CYP2E1. The finding that microsomal membrane fractions are capable of catalyzing the oxidation of ethanol, designated the microsomal ethanol-oxidizing system (MEOS), was the first indication of the presence of an ethanol oxidizing P450 since it was membrane-bound, required NADPH, and was inhibited by CO, properties that are distinct from those of alcohol dehydrogenase (Lieber et al., 1970). This study also found that ethanol administration to rats resulted in marked induction of the MEOS activity and was associated with enhanced ethanol clearance that was not blocked by pyrazole, an inhibitor of alcohol dehydrogenase. This paper was a major advance in the alcohol field with substantial clinical implications that would increase upon characterization of the P450, CYP2E1, which is responsible, in large part, for MEOS activity.
An ethanol-induced P450 capable of ethanol oxidation was first purified from rabbits (Koop et al., 1982). This enzyme was shown to be unique from other previously purified P450s by N-terminal sequencing of the protein. The rat ortholog was subsequently isolated and found to have high catalytic activity toward aniline p-hydroxylation (Ryan et al., 1985) and N-nitrosomethylethylamine N-demethylation (Patten et al., 1986), and to be inducible by ethanol (Ryan et al., 1986) and acetone (Patten et al., 1986). Subsequent studies using purified and recombinant P450 revealed that CYP2E1 metabolizes a large number of low Mr chemicals including acetaldehyde, acetaminophen, acrylamide, aniline, benzene, butanol, carbon tetrachloride, diethyl ether, dimethyl sulfoxide, ethyl carbamate, ethylene chloride, halothane, glycerol, ethylene glycol, N-nitrosodimethylamine, 4-nitrophenol, pyrazole, pyridine, and vinyl chloride, many of which are of low Mr and cancer suspect agents used as solvents (Guengerich et al., 1991). Thus, CYP2E1 is of enormous toxicological and carcinogenic importance.

Stabilization of CYP2E1. A, rats were fed acetone in the drinking water for 10 days and were injected i.p. with a bolus dose of NaH14CO3. Liver microsomes were isolated and CYP2E1 protein was purified by immunoabsorption, subjected to SDS-polyacrylamide gel electrophoresis, and the bands were excised and counted by liquid scintillation. B, Coomassie-stained gel, autoradiograph, and plot of CYP2E1-specific radioactivity from which the half-lives (t1/2) of protein decay were calculated at 7 h and 37 h.
A later study revealed that CYP2E1 was capable of carrying out the oxidation of acetone, a product of fatty acid oxidation, to acetol, and acetol to 1,2-propanediol in a pathway that leads to the production of glucose, termed the propane diol pathway of gluconeogenesis (Koop and Casazza, 1985). This suggested an important physiological function for CYP2E1 in a secondary pathway of gluconeogenesis. These results also explain the marked induction of CYP2E1 seen in fasted and diabetic rats, because both conditions result in an increase in serum acetone that could serve to stabilize the enzyme (see below).
Cloning of the CYP2E1 cDNA and Gene. A polyclonal antibody against CYP2E1 was used to clone the rat and human CYP2E1 cDNAs by screening λ phage expression libraries in which specific clones express fragments of the P450 protein that are captured on filters (Song et al., 1986). The DNA-derived protein sequence further established CYP2E1 as a distinct member of a new CYP2 subfamily. Unlike other CYP2 genes in which there are up to five multiple closely related members that duplicated and diverged from a common ancestor, CYP2E1 is the only member of the CYP2 subfamily in humans, rats and mice, although rabbits have a second P450, CYP2E2. CYP2E1 was also found to be constitutively expressed in liver and developmentally regulated in this tissue; its expression in liver is markedly elevated within 1 day after birth (Song et al., 1986). To this date, the mechanism of this rapid developmental activation of CYP2E1 gene expression has not been established. The induction of CYP2E1 by various agents such as pyrazole, 4-methylpyrazole, and acetone, as shown by other investigators, was found to be through a post-transcriptional mechanism inasmuch as there was no concomitant induction of mRNA with protein (Song et al., 1986). The rat and human CYP2E1 genes were subsequently cloned and sequenced, revealing the presence of nine exons typical of all CYP2 family genes (Umeno et al., 1988a,b).
Control of Cyp2e1 Transcription. Studies using in vitro transcription extracts suggested the existence of factors that control developmental activation of rat CYP2E1 gene transcription and the location of transcription factor binding sites (Ueno and Gonzalez, 1990) in the promoter of this gene. Later experimentation established that the mouse Cyp2e1 and rat and human CYP2E1 genes are under control of the liver enriched homeodomain-containing transcription factor, hepatocyte nuclear factor 1α (Hnf1α) (Akiyama and Gonzalez, 2003). This was determined by in vitro transactivation transfection studies using reporter gene constructs (Liu and Gonzalez, 1995) and by Hnf1α-null mice in which the Cyp2e1 gene was down-regulated in the absence of Hnf1α (Cheung et al., 2003). Control by Hnf1α accounts in large part for the liver-specific expression of the Cyp2e1 gene. However, recent studies have revealed that control of Cyp2e1 expression in mice is not solely dependent on Hnf1α expression. For example, liver-specific disruption of the β-catenin gene in mice revealed almost complete loss of CYP2E1 mRNA in liver, where expression of Hnf1α was not affected (Sekine et al., 2006). This finding suggests another mode of control of Cyp2e1 that overrides the presence of Hnf1α. Among the possibilities is the potential influence of β-catenin on a transcriptional coactivator required for Cyp2e1 activation by Hnf1α or a micro RNA that destabilizes the CYP2E1 mRNA or alters its translation. Others observed an effect of leptin on constitutive levels of CYP2E1 mRNA in ob/ob mice, although Hnf1α expression was not determined (Leclercq et al., 2000b).
Substrate-Induced Stabilization of CYP2E1. As noted above, CYP2E1 protein and activity are induced by acetone, ethanol, and pyrazole, all of which are substrates for the enzyme. Induction proceeds via a post-transcriptional mechanism of protein stabilization as revealed by in vivo labeling of proteins with 14C-sodium bicarbonate in rats treated with acetone, according to the experimental scheme shown in Fig. 1A. In this experiment, each rat is administered 1 mCi of isotope followed by immunopurification and counting the radioactivity in CYP2E1 protein excised from polyacrylamide gels (Song et al., 1989). In untreated animals, CYP2E1 exhibits biphasic turnover with t1/2 of 7 h and 32 h; when rats are administered acetone in the drinking water, the 7-h component is lost (Fig. 1B), thus accounting for the increase in microsomal content of CYP2E1. These data suggest that the protein is stabilized by the presence of the substrate as a result of loss of the rapid turnover component. However, in contrast, others found that electron transfer actually increases proteosomal degradation of CYP2E1 (Goasduff and Cederbaum, 1999; Zhukov and Ingelman-Sundberg, 1999). These studies were done using cultured cell lines that do not have the extensive endoplasmic reticulum (ER) network found in haptocytes.
Two pathways, the lysosomal pathway and the proteosome pathway, can participate in the degradation of membrane proteins (Correia et al., 2005). Membrane proteins, in particular, are degraded by the lysosomes through fusion of ER with lysosomal membranes, a process that is slower than that of the proteosome pathway. Indeed, it has been shown in cell lines that CYP2E1 degradation can occur via a ubiquitin-independent proteosomal pathway (Goasduff and Cederbaum, 1999; Huan et al., 2004). In the context of the established biphasic nature of CYP2E1 turnover found in vivo, it is tempting to speculate that, in the presence of substrate, CYP2E1 may not be subject to the more rapid phase of turnover via proteosomes due either to preferential sequestration of the enzyme in certain regions of the ER or to altered conformation of the protein as a result of substrate binding, or a combination of both. The lysosomal pathway would continue to function in the presence of substrate. The trigger for degradation via this latter pathway is not known but could be a phosphorylation event (Oesch-Bartlmowicz and Oesch, 2004) or altered folding of the protein. In any case, additional experimentation is required to establish a role for the proteosome pathway in CYP2E1 substrate-induced stabilization, especially in view of studies in cultured cells that do not support this hypothesis and suggest that substrate and catalytic activity would increase proteosomal degradation (Goasduff and Cederbaum, 1999; Zhukov and Ingelman-Sundberg, 1999). It should also be noted that substrate-induced stabilization has not been demonstrated for other P450s, but it would not be surprising if this mechanism is more common than previously thought. To achieve significant P450 stabilization, long-term in vivo exposure to the substrate would be required, a condition that could be achieved with chronic drug treatment.
Stabilization of CYP2E1 mRNA. When rats are made diabetic by destruction of pancreatic islet cells with streptozotocin, a DNA-alkylating agent specifically taken up by pancreatic β cells, CYP2E1 protein, and mRNA are highly induced (Song et al., 1987). This is in contrast to acetone- or pyrazole-treated animals in which only the protein is induced by the protein stabilization mechanism discussed above. The increased mRNA is not accompanied by an enhanced rate of transcription of the CYP2E1 gene, suggesting that the CYP2E1 mRNA is stabilized. Because diabetes is associated with high serum levels of ketone bodies, substrates for CYP2E1, the increase in protein may be due in large part to protein stabilization. The stabilization of the CYP2E1 mRNA that is reversed by insulin (Woodcroft et al., 2002) is less well understood. Recent studies revealed the presence of a 16-nucleotide sequence in the 5′ region of the CYP2E1 mRNA that is responsible for insulin-mediated destabilization of the mRNA (Truong et al., 2005). This sequence bound a 60-kDa cytosolic protein; however, the identity of this protein and the mechanism by which it is activated by insulin and destabilizes the CYP2E1 mRNA are not known. The induction of CYP2E1 through mRNA and protein stabilization is an ideal mechanism for increasing CYP2E1 activity since it would require less energy than activation of transcription, an important consideration under conditions of starvation and glucose deprivation. More recently, others provided evidence, using cultures of rat hepatocytes, that both post-transcriptional and transcription events mediate the induction of CYP2E1 by diabetes (Woodcroft et al., 2002). Transcriptional activity was assessed by analysis of heterogeneous nuclear RNA. It also cannot be ruled out that the peroxisome proliferator-activated receptor α (PPARα) coactivator 1α, which is induced under glucose deprivation (Lin et al., 2005), potentiates CYP2E1 transcription.
The Cyp2e1-null Mouse. There was a priori evidence that CYP2E1 has an important role in mammals. It is constitutively expressed in liver and participates in gluconeogenesis. The enzyme and its activities are conserved in humans and other experimental animals such as rabbits, rats, and mice. In addition, there is no functional polymorphism in humans or any animal model that substantially alters expression or catalytic activity of CYP2E1. Thus, it was anticipated that any important function for this P450 would be evident by phenotypes obtained from Cyp2e1 gene knockout mice. Quite surprisingly, the Cyp2e1-null mouse was not different from its wild-type counterpart (Lee et al., 1996). These mice had no apparent phenotypes suggestive of a role for CYP2E1 in development or physiological homeostasis. However, this animal model has been of great value in determining the role of CYP2E1 in chemical toxicity and carcinogenicity.
Role of CYP2E1 in Chemical Carcinogenesis. Azoxymethane (AOM) is a colon-specific carcinogen that is metabolically activated by CYP2E1 to methylazoxymethanol (MAM) (Shone et al., 1991) (Fig. 2A). The level of 7-methylguanine (Fig. 2B) and O6-methylguanine adducts in colon and other extrahepatic tissues was lower in Cyp2e1-null mice compared with wild-type treated with AOM (Sohn et al., 2001). In contrast, colon DNA adducts were increased in Cyp2e1-null when the stable MAM derivative MAMAc was administered; the reason for this increase is not known but could be due to lack of hepatic metabolism. In extrahepatic tissues, including colon, non-P450 enzymes such as alcohol dehydrogenase can activate MAM. Colonic aberrant crypt foci, precursors to colon polyps, were also lower in the null mice treated with AOM, in agreement with the DNA adduct data (Fig. 2B). This study revealed an important role for CYP2E1 in AOM activation and colon carcinogenesis. Most interestingly, CYP2E1, a highly liver-enriched enzyme, is not significantly expressed in the colon. These data suggest that MAM, produced in the liver, is transported to the colon where it is further converted to the methyldiazonium-alkylating agent responsible for producing the DNA adducts, which are likely precursors of the colon polyps. Indeed, it is known that MAM is stable with a half-life of approximately 12 h. This study reveals the importance of organotropism, as implied by the transport of MAM from the liver to the colon, in AOM carcinogenesis. In addition, this is one of the few examples where levels of DNA adducts are correlated with cellular responses, in this case, the production of aberrant crypt foci.
Role of CYP2E1 in Alcohol-Induced Liver Disease. It was suggested that CYP2E1 may play a role in alcohol-induced liver damage (Lieber, 1999). During ethanol oxidation, CYP2E1, as a result of uncoupling of oxygen consumption with NADPH oxidation, produces O2–· and H2O2 that can deplete glutathione and cause cellular damage. In the presence of iron catalysts, these reactive oxygen species (ROS) can produce powerful oxidants such as the hydroxyl radical (Gonzalez, 2005; Cederbaum, 2006). ROS can also result in elevated lipid peroxides that can form adducts with cellular nucleophiles such as proteins and nucleic acids, resulting in cell damage. Indeed, a number of studies using cultured cells have established the importance of CYP2E1 in oxidative stress (Cederbaum, 2006). In human hepatoma-derived HepG2 cells overexpressing CYP2E1, there is an elevation in oxidative stress, as revealed by depletion of glutathione and activation of the p38 mitogen-activated protein kinase pathway and induction of the transcription factor nuclear factor-erythroid 2-related factor 2 (Gong and Cederbaum, 2006). There is also evidence that ethanol oxidation by CYP2E1 can produce elevated oxidative stress and liver damage in vivo. Correlative analysis revealed that CYP2E1 is expressed in the perivenular zone, where liver damage is noted in alcohol-induced cirrhosis (Lieber, 1999). Alcohol-treated wild-type mice exhibited an increase in oxidative DNA damage and an induction of base excision DNA repair genes; this response was not found in similarly treated Cyp2e1-null mice, thus suggesting a role for CYP2E1 in ethanol-induced liver damage (Bradford et al., 2005). However, another study found no significant differences in several end points of ethanol-induced liver damage, such as steatosis, inflammation, and necrosis, between wild-type and Cyp2e1-null mice (Kono et al., 1999). Thus, although subtle differences in DNA damage may occur as a result of CYP2E1 metabolism of ethanol, it may be of little consequence to liver toxicity. This is not surprising, since the liver has several mechanisms to protect against increased ROS, such as the enzymes that scavenge radicals, superoxide dismutase, catalase, glutathione peroxidase, and induction of target genes under control of nuclear factor-erythroid 2-related factor 2 such as NADPH-quinone oxidoreductase 1. However, it should be recognized, that alcohol-induced liver damage in humans may differ from that in mice and other rodent models because of species difference in liver architecture, nonparenchymal cell populations, and other mediators and modifiers of ROS and stress and, thus, a potential role for CYP2E1 in humans cannot be ruled out.

A, metabolism of AOM by CYP2E1. AOM is converted to MAM in the liver. MAM can be further oxidized in extrahepatic tissue to the methylazoxyformaldehyde, which rearranges and loses formic acid, resulting in the formation of an unstable methyldiazonium ion that can bind to cellular nucleophiles. MAM can also spontaneously rearrange to the methyldiazonium ion after loss of formaldehyde that can alkylate DNA. B, 7-methylguanine (7-MeG) formation in liver and colon of AOM- and MAMAc-treated wild-type and Cyp2e1-null mice (left panels). Aberrant crypt foci are found in the colons of AOM- and MAMAc-treated wild-type and Cyp2e1-null mice (right). C, schematic illustration of AOM metabolism to MAM in the liver and transport of the latter to the colon for further conversion to the methyldiazonium ion. Data replotted from Sohn et al. (1991).
Nonalcoholic steatohepatitis (NASH), induced by treating animals with a methionine- and choline-deficient diet, yields liver damage similar to that in alcoholic liver disease. Similar to ethanol treatment, CYP2E1 deficiency did not influence the development of NASH, nor did it abrogate the increased microsomal NADPH-dependent lipid peroxidation, one of the mediators of cellular destruction (Leclercq et al., 2000a). However, quite surprisingly, CYP4A10 and CYP4A14 were up-regulated in the NASH model, possibly because of the production of endogenous ligands for PPARα. Both of these P450s were associated with the production of lipid peroxides. This study again demonstrates that CYP2E1 alone may not be of major consequence in alcohol-induced liver damage and that other P450s should be considered.
Despite a potential modest effect on liver toxicity induced by alcohol consumption, a recent study revealed that in ethanol-treated mice, CYP2E1 does have an impact on the central nervous system, where CYP2E1 is expressed, albeit at considerably lower levels than in liver (Joshi and Tyndale, 2006). Cyp2e1-null mice had longer ethanol-induced sleep times, especially at higher ethanol doses, than wild-type mice, suggesting an influence of CYP2E1 in the brain (Vasiliou et al., 2006). CYP2E1 status did not significantly influence the clearance of ethanol; however, the product of ethanol oxidation, acetaldehyde, was lower in Cyp2e1-null compared with wild-type mice. Although it is still not certain whether the hepatic or brain CYP2E1 mediates the CNS effects, this study suggested that CYP2E1 might have a more important impact on the brain under conditions of chronic alcohol exposure than on the liver.
Acetaminophen Hepatotoxicity
Acetaminophen (APAP) overdose is a common cause of drug-induced hepatotoxicity and ranks 1st in acute liver failure in the United States (Ostapowicz et al., 2002). APAP-induced acute toxicity is a clinically important model of drug-induced liver injury. Increased formation of the reactive intermediate metabolite N-acetyl-p-benzoquinone-imine (NAPQI) and depletion of cellular glutathione have been proposed as the major reasons for the hepatotoxicity of APAP overdose (Hinson et al., 2004). Hallmark events after APAP overdosing have been characterized as metabolic bioactivation, covalent binding of NAPQI to proteins, glutathione depletion, and hepatic necrosis (James et al., 2003a); however, the ultimate cellular events that lead to cell death are still not well understood. Among the first comprehensive studies on the mechanism of APAP hepatotoxicity was conducted by Bernard Brodie's laboratory and reviewed in the first issue of Drug Metabolism and Disposition (Mitchell et al., 1973a).
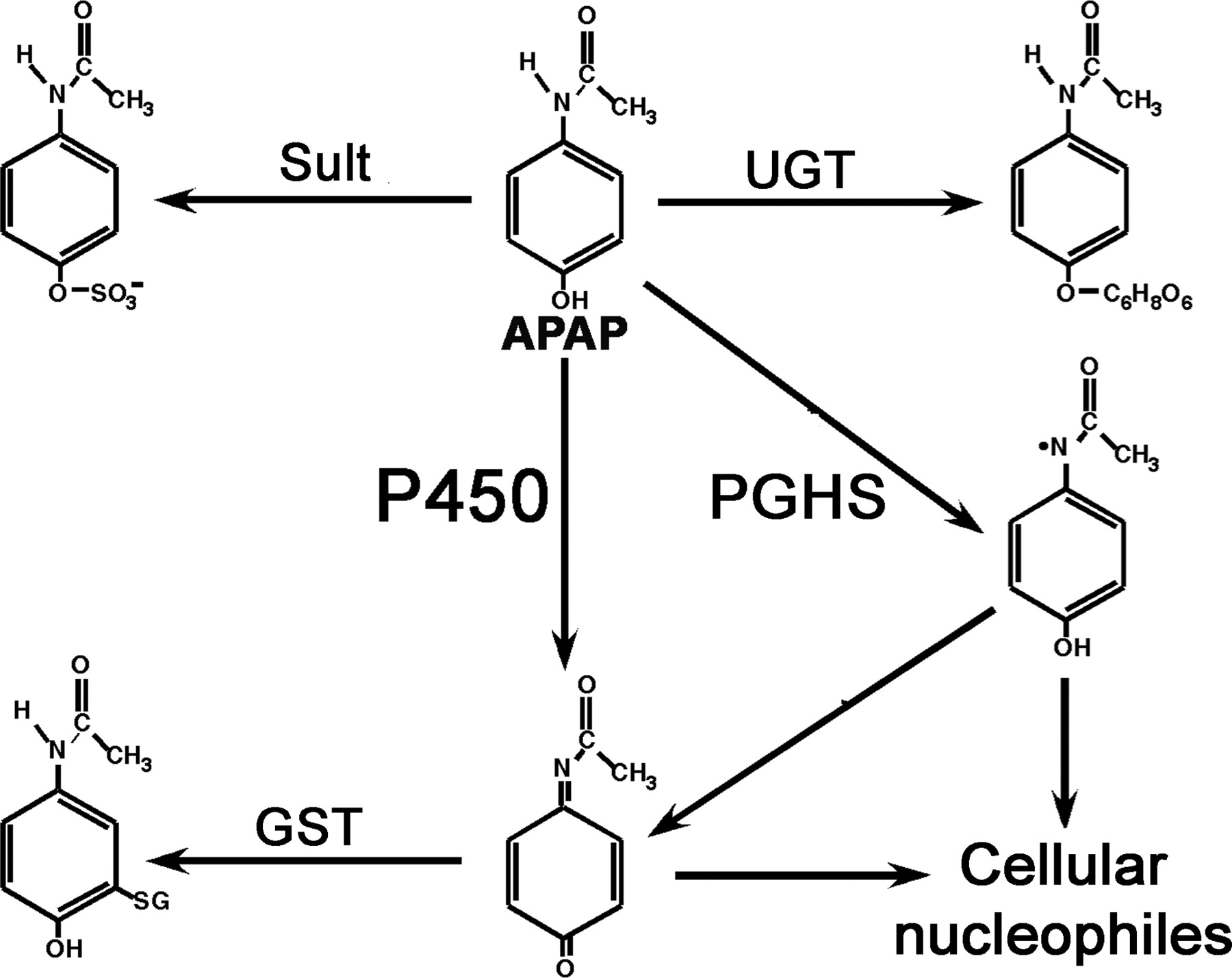
Metabolism of APAP. APAP can be directly conjugated by sulfotransferase (Sult) and UDP-glucuronosyltransferase (UGT) or oxidized by P450 to NAPQI. In extrahepatic tissues, APAP can be metabolized by PGHS, leading to a cation that can spontaneously decompose to NAPQI. The latter compound, a reactive quinone, can bind to cellular macromolecules. NAPQI can be conjugated by glutathione S-transferase (GST) and excreted in urine.
Studies of Bernard B. Brodie's Laboratory. Brodie's contributions to the field of drug metabolism and toxicity were enormous, as detailed in a review written by some of his former coworkers (Costa et al., 1989). With Axelrod, he found that phenacetin, a widely used analgesic and antipyretic agent, was converted to the biologically active metabolite APAP in the liver, which was responsible for its therapeutic efficacy (Brodie and Axelrod, 1949) and which is thought to be due to inhibition of prostaglandin H synthase (PGHS) 1 and 2 in endothelial cells and neurons. Under conditions of high hepatic APAP concentrations, metabolism can result in hepatotoxicity, which is frequently fatal. Among the last series of studies published by the Brodie group were four concurrent papers in Journal of Pharmacology and Experimental Therapeutics, which dealt with the mechanism of hepatotoxicity of acetaminophen (Jollow et al., 1973; Mitchell et al., 1973b,c; Potter et al., 1973). These studies revealed that APAP was metabolically activated to a reactive metabolite(s) that covalently bound to protein and depleted glutathione (GSH); the latter finding led to the use of N-acetylcysteine as an antidote for APAP poisoning. Among the many seminal findings was that treatment with phenobarbital, a P450 inducer, enhances APAP hepatotoxicity (Mitchell et al., 1973c).
APAP is metabolized by direct conjugation of the parent compound by sulfotransferase and UDP-glucuronosyltransferase, and conversion by P450 oxidation to the reactive intermediate metabolite NAPQI, as shown in Fig. 3 (Hinson et al., 2004). The conjugation reactions are in competition with the P450 oxidation. NAPQI can then either bind to cellular nucleophiles or be inactivated by conjugation with glutathione by the activity of glutathione S-transferase (GST). In extrahepatic tissues, APAP can be oxidized to a reactive intermediate by PGHS. Although the mechanism of APAP-induced cytotoxicity has been the subject of intense research for many years, it has become clear that P450 production of NAPQI is the initial event.

APAP hepatotoxicity. A, mice of the indicated genotypes were administered various doses of APAP i.p. and the extent of lethality was determined. B, mice of the indicated genotypes were administered different doses of APAP and hepatotoxicity was measured by levels of serum alanine aminotransferase. Data taken from Zaher et al. (1998).
Role of CYP2E1 in APAP Toxicity. CYP2E1 was found to activate APAP to NAPQI, which is measured as a GSH conjugate because of the instability of NAPQI (Morgan et al., 1983; Raucy et al., 1989). This involvement of CYP2E1 is in agreement with earlier findings in which alcohol was found to potentiate fatal APAP hepatotoxicity in rats (McClain et al., 1980) and in humans as revealed by use of the CYP2E1 inhibitor disulfiram, which resulted in decreased clearance of APAP (Manyike et al., 2000). To investigate the role of CYP2E1 in APAP hepatotoxicity, wild-type and Cyp2e1-null mice were administered APAP. Cyp2e1-null mice were markedly resistant to lethal APAP toxicity (Lee et al., 1996), and double null mice lacking both CYP1A2 and CYP2E1 were almost totally resistant to toxicity (Fig. 4A). The single mouse in the double null group exhibited normal liver pathology but had evidence of kidney toxicity. Both Cyp2e1- and Cyp1a2-and-Cyp2e1-double null mice also had no detectable APAP protein adducts and a lower extent of GSH depletion (Zaher et al., 1998). To determine how the human CYP2E1 influences APAP-induced hepatotoxicity, the human CYP2E1 gene was introduced as a bacterial artificial chromosome genomic clone into Cyp2e1-null mice (Cheung et al., 2005). The CYP2E1-humanized mice were actually less sensitive to APAP than were wild-type mice (Fig. 4B), indicating a potential species difference in the extent of activation of APAP by CYP2E1.
In addition to CYP2E1 and CYP1A2, as noted above, the CYP3A P450s have been implicated in the formation of NAPQI in human liver microsomes. However, the role of human CYP3A4, the most abundant P450 in human liver, in APAP hepatotoxicity is unclear. Pregnenolone 16α-carbonitrile, a rodent-specific pregnane X receptor ligand that induces CYP3A P450s, significantly enhanced APAP hepatotoxicity in mice (Guo et al., 2004). Phenytoin, a widely used anticonvulsant that can induce CYP2C and CYP3A P450s, but not CYP2E1, also potentiated APAP hepatotoxicity (Brackett and Bloch, 2000). Pretreatment with ethanol increases acetaminophen hepatotoxicity in Cyp2e1-null mice (Sinclair et al., 2000), and induction of CYP3A by ethanol was confirmed both in vitro and in vivo (Feierman et al., 2003; Liangpunsakul et al., 2005). Thus, although most evidence suggests that CYP2E1 is the major P450 responsible for APAP hepatotoxicity, other P450s may also contribute, depending on the circumstances.
In addition to the P450s, GST also influences the sensitivity of animals to APAP toxicity. Brodie and colleagues were the first to note that significant protein binding did not occur until more than 60% of APAP was eliminated from the liver and attributed this observation to glutathione depletion (Mitchell et al., 1973a). To investigate the role of GST in sensitivity to APAP, GstP1-null mice were analyzed. However, surprisingly, mice that lack GST-Pi are actually resistant to APAP hepatotoxicity (Henderson et al., 2000). This result is counterintuitive to the role of GST in NAPQI conjugation; however, it was suggest that this form of GST does not carry out direct conjugation of the metabolite. Instead, it may serve to modulate GSH levels since in GstP1-null mice, GSH levels depleted by APAP metabolism rapidly recover. The mechanism(s) by which GST-Pi modulates GSH levels are not known, but several possibilities including its potential role in redox cycling have been discussed (Henderson et al., 2000).
Activation of the nuclear receptor, constitutive androstane receptor (CAR), increased the sensitivity of mice to phenobarbital (Zhang et al., 2002) in agreement with the earlier studies from the Brodie laboratory (Mitchell et al., 1973c). Since GstP1 is a CAR target gene, its induction may be the means by which phenobarbital increases the sensitivity to APAP toxicity. CAR also induced the CYP2B P450s that can metabolize APAP to NAPQI. It was suggested that the involvement of CAR could be used as a therapeutic approach to APAP poisoning since androstanedione, a CAR inhibitor, was found to prevent toxicity (Zhang et al., 2002).
Mechanism of APAP-Induced Hepatotoxicity
The mechanism by which APAP and its electrophilic derivative NAPQI cause cell death has been the subject of intense investigation (Hinson et al., 2004). Among the critical events are production of protein adducts, increases of superoxide that, upon reacting with nitric oxide, produces the highly reactive peroxynitrite, and elevation in proinflammatory cytokines such as interleukin-1β. However, Kupffer cells, a main source of cytokines in the liver, do not appear to be involved in the process of these events as revealed by the independence of NADPH oxidase, expressed in Kupffer cells, on APAP hepatotoxicity (James et al., 2003b). Considered among the possible terminal events in the toxic response to APAP is mitochondrial damage that could result from protein adduction by NAPQI, peroxynitrite, or both. Increased mitochondrial membrane permeability is also another source of cellular superoxide generated during APAP poisoning (Hinson et al., 2004).
With all available evidence taken together, APAP-induced toxicity likely results from initial APAP oxidation to NAPQI, followed by rapid depletion of GSH, and increased protein and tyrosine adduction as shown by the Brodie group. This then leads to mitochondrial damage and cell death that is independent of the presence of APAP; removal of APAP early after exposure of hepatocytes and before cell toxicity still results in cell death. This scenario has been termed the two-phase induction of toxicity, the metabolic phase followed by the oxidative phase (Boobis et al., 1986; Hinson et al., 2004). However, the precise molecular event that results in the mitochondrial alterations remains unclear.
It is noteworthy that activation of PPARα results in resistance to APAP-induced toxicity; this effect was not found in Ppara-null mice (Chen et al., 2000). PPARα controls genes encoding proteins involved in fatty acid transport and metabolism, including the fatty acid β-oxidation systems of peroxisomes and mitochondria. Thus, in the event of APAP toxicity, peroxisomal fatty acid oxidation induced by activation of PPARα could compensate for mitochondrial damage by production of NADPH and ATP required for cell survival.
Future Studies on CYP2E1
Metabolomics. Metabolomics is the analysis of small molecules in cells, tissues, serum, and urine to determine the result of altered physiological-based metabolism or the response to pathophysiological stimuli such as drugs or toxicants. These measurements, made on small molecules generally under 800 Da, can be carried out by using 1H NMR, high-performance liquid chromatography, gas chromatography-mass spectrometry, and liquid chromatography-tandem mass spectrometry. 1H NMR, which allows the precise determination of the structure and quantity of chemicals in a biological sample, has been used in most studies to date. By use of animal models, biomarkers can be identified that determine organ-specific drug toxicity; this field has been termed metabonomics (Nicholson and Wilson, 2003). 1H NMR metabonomics may also be of value to diagnose diseases such as atherosclerosis, cancer, and neurological disorders (Robertson, 2005). Metabolomics is only possible in combination with powerful data analysis software. Data from multiple samples is subjected to chemometric and multivariate analysis to extract relevant information showing differences between samples or groups under study. In particular, principal components analysis (PCA) is commonly used in metabonomics/metabolomics studies. PCA exploits the redundancy in multivariate data, enabling the reduction in the dimensionality of the data and analysis of patterns or relationships in the variables. PCA allows the simultaneous analysis and comparison of thousands of ions from two or more groups.
Although 1H NMR yields the exact quantity and structure of chemicals to be determined, for the most common 400- and 600-mHz instruments used in academic research, it lacks the sensitivity and resolution power to study low-abundance metabolites in complex mixtures. Metabolomics can also be carried out by use of the more accessible LC-MS technology which, although it does not give precise chemical structures and quantification in the absence of standards, allows the analysis and comparison of thousands of compounds. Recently, an ultra performance liquid chromatography-coupled time-of-flight mass spectrometry (UPLC-TOFMS), along with a software package, was developed for use in metabolomics (Plumb et al., 2005). This instrumentation allows the resolution and exact mass determinations of thousands of molecules in a biological sample and is ideally suited for studies in rodent model systems, where the amounts of biological materials such as serum and urine are limited. A typical urine specimen can typically yield 5 to 10,000 ions that are detected by the MS and can be analyzed by PCA.
Mouse Metabolomics. By use of genetically modified mice, metabolomics can find novel endogenous and dietary substrates and, conversely, biomarkers for CYP2E1 expression. Urine, serum, and tissue from wild-type and Cyp2e1-null mice can be compared by UPLC-TOFMS, followed by chemometric analysis of data. Compounds that are found in wild-type and not in null mice could represent the end-products of CYP2E1 metabolism, whereas compounds that are present in Cyp2e1-null mice but not in wild-type mice could represent potential CYP2E1 substrates. Biomarkers for expression of individual P450s can be used to determine expression in humans by analysis of urine. These biomarkers, once validated, would be of value for molecular epidemiology studies to measure CYP2E1 expression in subjects to determine the role of CYP2E1 in risk assessment to chemical toxicity, cancer, and liver disease.
Metabolomics can also be used for metabolite profiling or metabolite mapping, as recently demonstrated in studies with the areca nut alkaloids arecoline, where 11 metabolites were discovered (Giri et al., 2006), and aminoflavone, where 13 metabolites were identified (Chen et al., 2006) by multivariate data analysis on the ions produced by UPLC-TOFMS analysis of urine. Metabolite profiling could also be brought to bear on the analysis of APAP-induced hepatotoxicity. Wild-type and Cyp2e1-null mice could be administered APAP and the relative levels of APAP-derived metabolites determined in liver, serum, and urine using the metabolomic approach. Other metabolites that correlate with liver damage could simultaneously be monitored to potentially identify biomarkers for toxicity. These analyses may yield additional insights into the role of metabolism in APAP hepatotoxicity and the resultant metabolic disturbances within the liver, as assessed by endogenous metabolites, that lead to hepatocyte death and liver failure.
Conclusions
Although there has been considerable progress made in understanding the role of CYP2E1 in alcohol and chemical toxicity, and on chemical carcinogenesis, much remains to be done to determine the mechanisms of regulation of the CYP2E1 gene during development, the stabilization of the mRNA by diabetes, and the substrate-induced stabilization of the protein. The role of CYP2E1 in liver diseases such as alcohol-associated fibrosis and fatty liver disease in humans also require further studies. Metabolomics offers the hope to find new endogenous and dietary biomarkers for the enzyme that could be used as probes to measure enzyme levels in humans, as well as potential biomarkers for CYP2E1-mediated chemical toxicity.
Acknowledgments
I wish to acknowledge my former mentors Carleton Garrett, Charles Kasper, Daniel Nebert, and Harry Gelboin and the many postdoctoral fellows who have worked in my laboratory over the past 22 years, most notably Byoung-Joon Song, my first fellow at NCI who did the CYP2E1 cDNA cloning, Morio Umeno, my second fellow who cloned the CYP2E1 gene, and Susanna Lee who generated the Cyp2e1-null mice. I also thank my long-time collaborator and colleague Shioko Kimura and my friend and collaborator Jeff Idle for many years of support and for review of the manuscript.
Footnotes
-
This work was supported by the National Cancer Institute Intramural Research Program.
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.012492.
-
ABBREVIATIONS: P450, cytochrome P450; MEOS, microsomal ethanol-oxidizing system; Hnf1α, hepatocyte nuclear factor 1α; ER, endoplasmic reticulum; PPARα, peroxisome proliferator-activated receptor α; AOM, azoxymethane; MAM, methylazoxymethanol; MAMAc, methylazoxymethanol acetate; ROS, reactive oxygen species; NASH, nonalcoholic steatohepatitis; APAP, acetaminophen; NAPQI, N-acetyl-p-benzoquinone-imine; PGHS, prostaglandin H synthase; GST, glutathione S-transferase; GSH, glutathione; CAR, constitutive androstane receptor; PCA, principal components analysis; UPLC-TOFMS, an ultra performance liquid chromatography-coupled time-of-flight mass spectrometry.
-
 Frank J. Gonzalez received a B.A. in Biology and M.A. in Microbiology from the University of South Florida, Tampa, and a Ph.D. in Oncology from the University of Wisconsin, Madison. He was a postdoctoral at the National Institute of Child Health and Human Development prior to joining the National Cancer Institute. His studies focus on xenobiotic-metabolizing enzymes, notably the cytochromes P450, and xenobiotics receptors, the molecular interfaces between the chemical environment and the body. Recently, his group has been developing and characterizing P450 and xenobiotic receptor humanized mice and has combined these and other gene knockout models with the use of LC-MS-based metabolomics to study xenobiotic metabolism and to develop biomarkers for P450 expression and receptor activation.
Frank J. Gonzalez received a B.A. in Biology and M.A. in Microbiology from the University of South Florida, Tampa, and a Ph.D. in Oncology from the University of Wisconsin, Madison. He was a postdoctoral at the National Institute of Child Health and Human Development prior to joining the National Cancer Institute. His studies focus on xenobiotic-metabolizing enzymes, notably the cytochromes P450, and xenobiotics receptors, the molecular interfaces between the chemical environment and the body. Recently, his group has been developing and characterizing P450 and xenobiotic receptor humanized mice and has combined these and other gene knockout models with the use of LC-MS-based metabolomics to study xenobiotic metabolism and to develop biomarkers for P450 expression and receptor activation. - Received August 18, 2006.
- Accepted September 29, 2006.

- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}