


Abstract
During the development of any PEGylated protein or peptide, toxicology in relevant species will be conducted prior to human exposure. Normally, comprehensive metabolism data accompany the toxicity studies for a small molecule. We have examined whether such studies would be relevant in the safety assessment of PEGylated material. Literature data indicate that the polyethylene glycol (PEG) associated with a biological molecule should provide no extra concern because the exposure-toxicity relationship of PEG in animals and humans has been thoroughly investigated and metabolism/excretion of PEG is well understood. Based on the comparisons of PEG exposure from PEGylated biological products and the exposure of PEG associated with toxicity in humans, the therapeutic index is large (approximately 600-fold or greater). Therefore, assuming that toxicological evaluation of a biological molecule of interest is complete and satisfactory therapeutic windows are achieved, the data contained in this review indicate that the PEG associated with a protein or other biological molecule does not represent an additional unquantified risk to humans.
The conjugation of small proteins, peptides, and oligonucleotides with polyethylene glycol (PEG), or PEGylation, has become an increasingly common method of improving the half-life of biological products, mainly through reducing the urinary excretion of the molecule (Yang et al., 2004), but also by reducing the enzymic degradation due to the increased steric bulk (Veronese and Pasut, 2005). In addition, PEGylated biological products often exhibit a reduced affinity for the target receptor compared with the native precursor. This reduced affinity can lead to a lower clearance by target-mediated clearance mechanisms. Finally, the addition of the PEG moiety can have beneficial effects on the immunological profile of a molecule by reducing the ability of the compound to raise antibodies in humans (Mehvar, 2000).
PEG is a polymer made up of identical ethylene glycol subunits. PEGs have a descriptor associated with them that represents the mean molecular weight of the molecule (i.e., PEG200 has a molecular weight of 200) (Smyth et al., 1955). The PEG molecules conjugated to proteins can also have the terminal hydroxyl group capped with a methyl group (Molineux, 2003). The structures of these PEGs are detailed in Fig. 1. Higher molecular weight PEGs can have some degree of branching. The PEGs used to conjugate biologicals are polydispersed in nature (i.e., have a range of molecular weights), and this can to lead to a range of drug molecules with potentially subtly different biological properties. The impact of polydispersity must be considered when dealing with these conjugated biological agents (Veronese and Pasut, 2005).
To couple the PEG to the protein, peptide, or oligonucleotide, the PEG (generally monomethoxy PEG) is first activated. Several methods can be used to achieve this activation and coupling, including cyanuric chloride, 1,1′-carbonyldiimidazole, phenylchloroformate, or succidinimidyl active ester (Delgado et al., 1992; Mehvar, 2000). The mechanisms by which PEGs are coupled to proteins have also been discussed more recently by Veronese and Pasut (2005).
Toxicology studies with the PEGylated proteins are normally carried out before studies in humans, and these provide excellent evidence of the safety of these PEG-containing molecules. Such safety studies, when performed on small organic molecules, are normally accompanied with corresponding metabolism studies exploring the biological fate of the molecule in the various toxicology species and human. These studies are very difficult to conduct on the PEGylated material, and this leaves a possible concern about the PEGylated portion of the molecule and its impact on human safety. In their nonconjugated form, polyethylene glycols are widely used as excipients for a large variety of drugs and are also regularly used in children for the treatment of pediatric constipation or for colonoscopy (Pashankar et al., 2003, 2004; Bell and Wall, 2004).
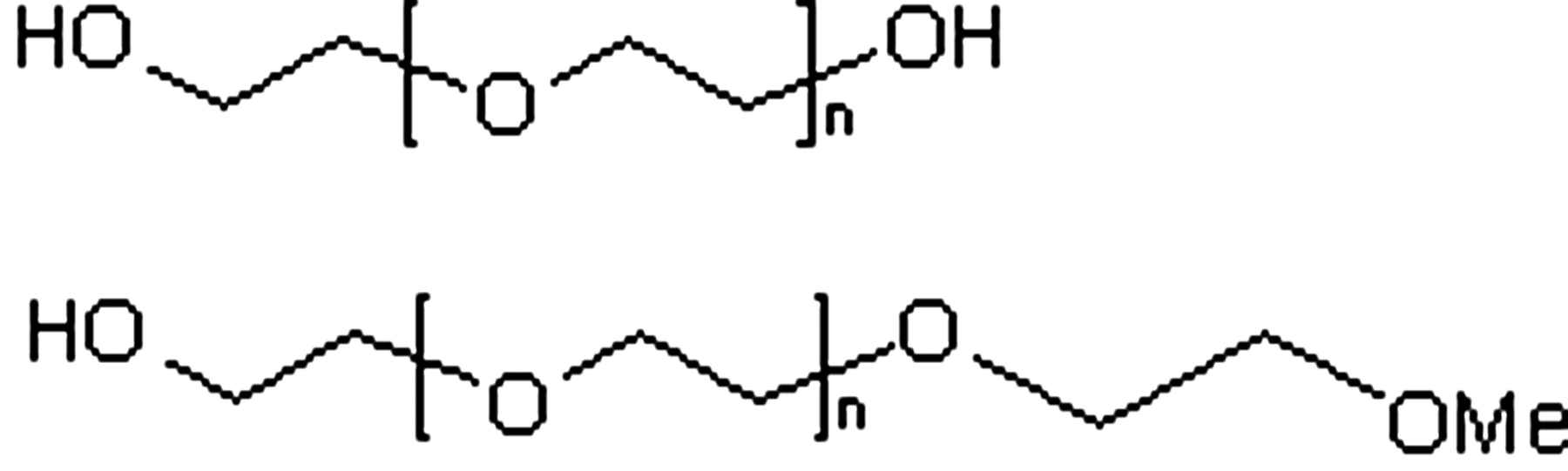
Structure of PEG.
The purpose of this review is to consider the toxicology (animal and human), metabolism, excretion, and exposures of humans to PEG and to evaluate the safety of the PEG on PEGylated proteins to those exposed to these biological products. The feasibility of conducting ADME studies will also be considered, as well as any value they would bring to the risk assessment.
Toxicology of Polyethylene Glycol in Animals
Acute, short-, and long-term toxicology studies with PEGs administered by the oral, intraperitoneal, and intravenous routes in a wide range of animal species have been carried out with PEGs with molecular weights of up to 10,000. The toxicity of PEG has been thoroughly reviewed (Fruijtier-Polloth, 2005); in light of this, a brief summary of the toxicology of PEG is given below. After acute administration, the LD50 values of PEG are generally in the region of 10g/kg or higher. The clinical signs associated with this acute exposure include jumping, tremors, convulsions, piloerections, and dyspnea. Postmortem examination showed pulmonary hyperemia and edema (Fruijtier-Polloth, 2005). Acute poisoning in rabbits suggests that poisoning occurs at plasma concentrations in excess of 30 to 70 mM (Herold et al., 1989).
In chronic oral toxicology studies in the rat, PEG1500 (0.06 g/kg/day) and PEG4000 (0.02 g/kg/day) did not cause any significant adverse effects after 2 years' administration (Smyth et al., 1955). When PEG1540 and 4000 were administered as part of the diet (4%) to rats, no effects were observed (Smyth et al., 1955). Experiments with PEG400 showed no effect at doses of 2% in diet, with higher doses showing nonspecific effects on growth and cloudy swellings in the liver (Smyth et al., 1955). These data indicate that after chronic administration to the rat PEG is not a significant toxicological concern.
In chronic toxicology studies in nonrodent species there were no adverse events in dogs that received PEGs ranging from 400 to 4000 molecular weight (2% in diet) for a year (Smyth et al., 1955). In the monkey, PEG200 caused pathological lesions during a 13-week study in the kidney at an oral dose of 2.2 to 4.4 g/kg. The renal lesions consisted of intratubular deposition of oxalate crystals in the renal cortex but were not associated with other clinical or pathological findings (Prentice and Majeed, 1978). Adverse effects have also been observed in monkeys treated by intravenous infusion (1 ml/h) for up to 1 month with 60% PEG400 in water (approximately 3 g/kg/day). At this dose, the monkeys had reduced appetite, a greasy texture of their lower extremities, edema of their genitalia and legs, and deteriorating infusion sites (Working et al., 1971; Fruijtier-Polloth, 2005). These data indicate the excellent safety profile of PEG when administered chronically to nonrodent species. No adverse reproductive or teratogenic effects are reported with PEGs. Polyethylene glycols are neither mutagenic nor carcinogenic (Working et al., 1971; Fruijtier-Polloth, 2005).
Toxicology of PEGylated Molecules in Animals
Preclinical toxicology studies performed with PEGylated proteins have also not revealed any PEG-specific toxic findings. For example, with PEG-interferon, the toxicity profile was evaluated in cynomolgus monkeys dosed subcutaneously for 4 weeks either twice weekly (dose up to 562.5 μg/kg) or daily (doses up to 600 μg/kg) and for 13 weeks administered twice weekly (doses up to 150 μg/kg). PEG-interferon was well tolerated. The characteristic pattern of interferon α toxicity was observed with PEG-interferon. These effects included suppressive effects on the hematopoietic system and increases in liver enzymes [Pegasys (peginterferon α2a); EPAR].
With Peg-Intron (PEGylated interferon α2a), repeated dose toxicity studies were performed in cynomolgus monkeys using subcutaneous doses administered every other day for 1 month. Important findings included decreases in all types of blood cells, serum proteins, calcium, phosphorus, and potassium. The findings observed in PEG-Intron-dosed monkeys were similar to those produced by Intron A. There was no unique toxicity due to the PEGylation. Greater incidence and/or severity of the findings were noted in the high-dosed monkeys given PEG-Intron compared with those given Intron A. This is in accordance with the prolonged exposure and higher AUC values obtained using PEG-Intron (PEG-Intron; EPAR). Treatments with PEGylated proteins such as PEG-interferon α2a (40 kDa; Pegasys) and PEGylated asparaginase do not reveal any specific adverse events linked to the PEG moiety (Avramis et al., 2002; Schwarz et al., 2003).
Overall, the acute or chronic administration of PEG with a range of molecular weights by a range of routes has not led to any major toxicities, and signs of toxicity that do occur are only apparent at high dose. In light of this information, PEG can be considered to have a toxicological profile of very low concern in animals.
Clearance of Polyethylene Glycol by Metabolism
The metabolism of PEG has been investigated in animals and in humans. A summary of the results is presented in Table 1, along with data on the excretion of PEG.
Summary of PEG metabolism and excretion data (for more, detail see text)
Metabolism of PEG. The metabolism of PEG itself is simple and involves the oxidation of the alcohol groups present on the PEG to a carboxylic acid. For example, the diacid and hydroxyl acid metabolites of PEG have been observed in the plasma and urine of burn patients and rabbits and in the bile of cats (Hunt et al., 1982; Friman et al., 1990, 1993). In the isolated guinea pig liver and in rat/guinea pig in vitro liver preparations, PEG has been shown to be sulfated (Roy et al., 1987, 1988). Evidence from studies with PEG400 indicate that ethylene glycol is not formed as a metabolite of PEG in humans (Schaffer et al., 1950). Minor amounts of oxalic acid are liberated after the metabolism of PEG (Fruijtier-Polloth, 2005).
The phase 1 metabolism of PEG in mammalian systems is mediated by alcohol dehydrogenase (Herold et al., 1989). P450s may also play a role in the oxidation of PEG (Veronese and Pasut, 2005), although the evidence for this is not clear. Also, PEG has been shown to be metabolized by sulfotransferases (Roy et al., 1987, 1988). Although there is evidence that PEG can be metabolized to a series of phase 1 and phase 2 metabolites, the toxicology data presented above indicate that these metabolites are of very little toxicological concern. However, metabolism of PEG to the acid metabolite(s) has been implicated in the acidosis and hypercalcemia observed in patients after overdose (Bruns et al., 1982). It is clear that these metabolites can be formed in multiple toxicology species and that the phase 1 metabolites are seen in animals and humans. These data indicate that humans and animals will be exposed to similar metabolites after administration of PEG.
Impact of Molecular Weight on PEG Metabolic Clearance. Excretion balance studies have been carried out in humans with PEG. Assuming that any PEG that has not been recovered intact in these studies has been metabolized prior to secretion, it is apparent that metabolic clearance of PEG decreases markedly as molecular weight increases. For PEG400, up to 25% of the dose may be metabolized in humans (Schaffer et al., 1950); similar results are also seen in the rabbit (Schaffer et al., 1950). For PEG1000 and 6000, a maximum of 15% and 4% of the doses is cleared by metabolism, respectively (Schaffer et al., 1950). The human result on PEG1000 is supported by cat data with PEG900 that indicates that 75% of the biliary eliminated material (biliary elimination is likely to be a minor clearance pathway; see below) was unchanged, the rest representing oxidized metabolites (Friman et al., 1993).
Sulfation in the rat and guinea pig also shows a molecular weight dependence with PEG200 showing the highest rate and PEG6000 showing no detectable turnover (PEG200, 400, 1000, and 6000 investigated in the isolated perfused guinea pig liver) (Roy et al., 1987, 1988).
Overall, the metabolic pattern of PEG in vivo is simple and involves the oxidation of the alcohol groups to carboxylic acids. This is likely to be mediated by alcohol dehydrogenase. Also, there is the potential to form a sulfate conjugate by attachment at the same alcohol group by sulfotransferases. PEG metabolism appears to be similar in animals and humans, and the metabolism is dependent on molecular weight, with high molecular weight PEGs (>5000; typical of those used to PEGylate proteins) showing little or no metabolism.
Is Metabolism Likely to be a Major Route of Clearance of PEG Used to PEGylate Proteins? There are no data presented on the metabolism of the PEG associated with PEGylated proteins. The data presented in the previous paragraph indicate that for low molecular weight PEGs, clearance by metabolism is a minor pathway. These data also demonstrate that clearance via metabolism of the PEGs, which are typically used to alter the pharmacokinetics of biological products [molecular weight of 5000 or greater (Molineux, 2003; Veronese and Pasut, 2005)], is likely to be insignificant. This is because the metabolism of PEG is molecular weight-dependent, with high molecular weight PEGs showing less metabolism. The primary sites of metabolic attack on PEG are frequently chemically modified, rendering metabolism unlikely (Molineux, 2003).
Excretion of Unchanged Polyethylene Glycol in the Urine and Bile: The Major Route of PEG Clearance?
Studies have demonstrated that PEG is eliminated in both the bile and urine in mammals; a summary of these data can be found in Table 1.
Urinary Excretion. Urinary excretion of PEG occurs through passive glomerular filtration and is therefore dependent on molecular weight. Studies in the mouse have shown the molecular weight dependence of the urinary elimination, with markedly reduced urinary clearance observed as the molecular weight exceeds 20,000 (Yamaoka et al., 1994). Human data also show urinary clearance as the major excretory pathway for PEGs up to 4000 molecular weight. Human excretion balance studies have shown that 86% and 96% of PEG1000 and 6000 were excreted in the urine 12 h after intravenous administration (Schaffer and Critchfield, 1947; Schaffer et al., 1950). In rats, urinary recovery of PEG1000 is essentially 100% (Friman and Svanvik, 1997). In the dog, PEGs with molecular weights in the range 400 to 4000 are cleared from plasma at a rate identical to the glomerular filtration rate (Schaffer and Critchfield, 1947), which suggests a passive filtration process.
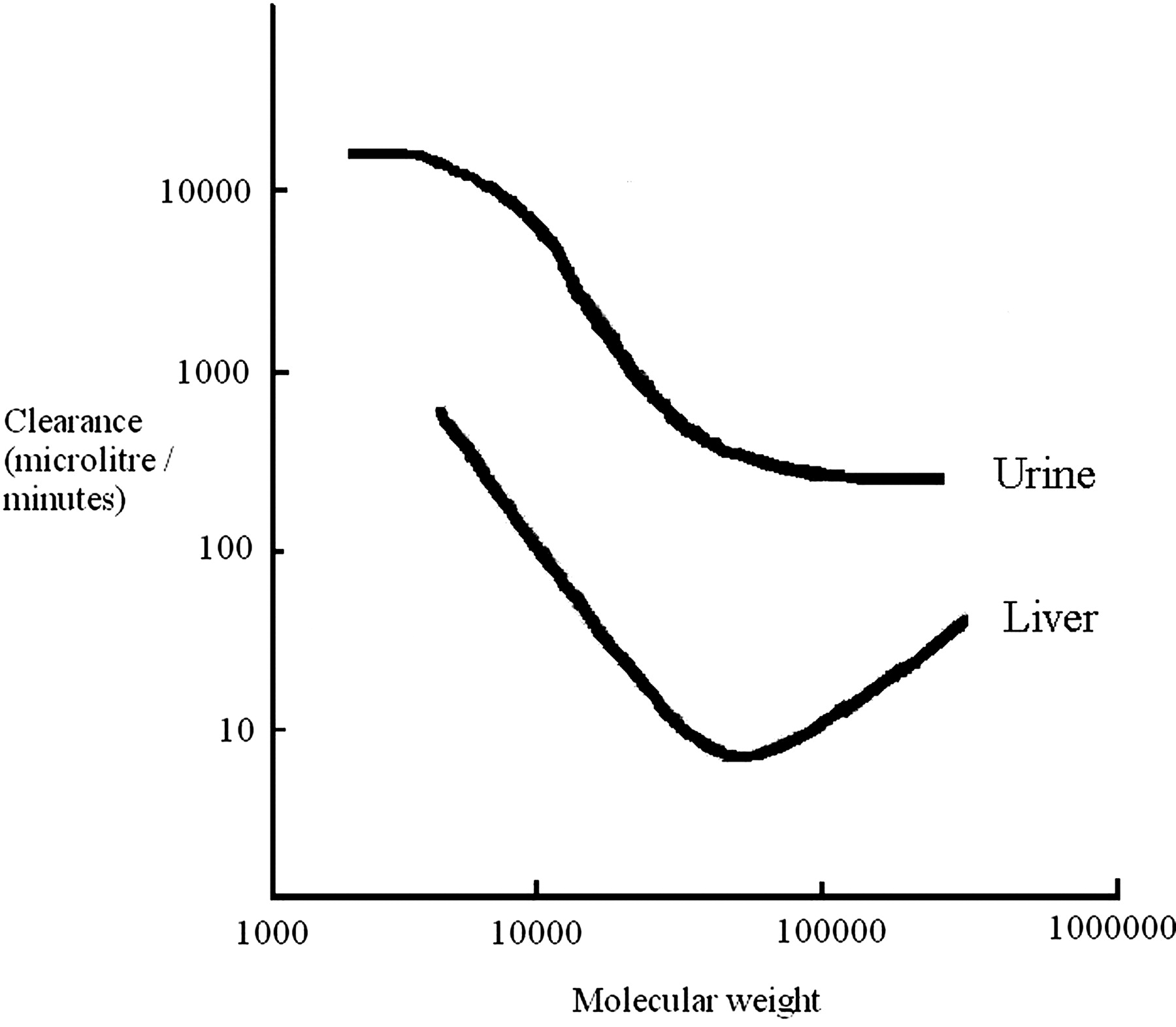
Biliary Excretion. PEG also undergoes biliary excretion, and this process is molecular weight-dependent, with hepatic clearance reaching a minimum at about 50 kDa molecular mass (mouse). Lower and higher molecular weight PEGs show increased hepatobiliary clearance (Yamaoka et al., 1994). The impact of molecular weight on the hepatic and renal clearance of PEG in the mouse is shown in Fig. 2. In vitro studies have demonstrated that the uptake of PEG by Kupffer cells increases when molecular weights increase beyond 50,000. These observations may explain the increase in hepatic clearance seen when PEG molecular weight increases over 50,000 (Yamaoka et al., 1994). Excretion studies in the bile duct-cannulated dog after intravenous administration of PEG400 indicated that no PEG400 was eliminated by this route (Schaffer et al., 1950).

The effects of the molecular weight of PEG on urinary and hepatic clearance (Yamaoka et al., 1994).
High biliary clearance of PEG900 in humans has been reported in patients with T-tube cholecystectomy (Friman et al., 1995). The concentration of PEG in the bile in these studies was 31-fold higher than those seen in plasma. A similar high bile/plasma ratio has been seen in both pigs and cats with molecular weights ranging from 450 to 4000 (Friman et al., 1988, 1990). For PEG900, the biliary clearance may be a passive process. However, other authors have suggested an active component in the biliary elimination of PEG900 involving vesicular transport via the lysosome (Roma et al., 1991). None of these studies report the urinary excretion component of PEG clearance. The data presented above indicate that for PEG, urinary clearance is likely to be the major clearance mechanism (Friman and Svanvik, 1997). In the cat, the maximum biliary clearance (20–25 ml/h/kg) was seen with PEG900 [PEG450 had a biliary clearance of 5–10 ml/h/kg, and for PEG2500 and 4000 it was <5 ml/h/kg (Friman et al., 1990)].
Excretion balance studies with PEG4000 administered intravenously to rats demonstrated that although urinary excretion was the major route of clearance (61% of the dose), 20% of the dose administered was present in the feces (Carpenter et al., 1971). These data indicate that for PEGs of molecular weights in the region of 4000, biliary excretion may represent a significant, but not major, clearance mechanism.
The data presented above indicate that both the liver and kidney can play a role in the excretion of PEG. However, from these studies, it is clear that for molecular weights up to and including 190,000, urinary excretion of PEG is likely to be the major route of elimination, with hepatobiliary clearance representing a minor pathway.
Likely Clearance Mechanisms for PEG Associated with PEGylated Proteins
There are no data presented on the excretion of PEG associated with PEGylated protein conjugates. Metabolic clearance of high molecular weight PEGs used for PEGylation is likely to be insignificant. The data presented above also suggest that urinary excretion of unchanged material will be the major route of clearance of any PEG released by degradation of the conjugate. Biliary excretion of unchanged material would be expected to be a minor route.
Protein conjugates that are extensively metabolized in the hepatocyte may be an exception to this since biliary clearance may prove more a convenient clearance mechanism due to the proximity to the bile duct canaliculus in the hepatocyte.
Based on the information summarized above, clearance of PEG by the biliary, urinary, and metabolic routes is precedented in humans for nonconjugated PEGs and, therefore, is unlikely to be of any extra toxicological consequence.
PEG Exposure in Humans
PEGylated Proteins Versus Exposure by Other Products
Products That Contain PEG. PEG is widely used as an excipient in medicines that are administered by the intravenous, oral, rectal, and topical routes. PEG is also found in nonpharmaceutical products, meaning that humans are routinely exposed to PEG. PEG-containing nonpharmaceutical products include: toothpaste, shampoo, moisturizers, colorants, foods, drinks, and deodorants. Exposure to PEG via these products will be by the oral and topical routes.
The absorption of PEG by the oral route is molecular weight-dependent. Urinary recovery data for PEG400 indicate that 50 to 60% of PEG with this molecular weight is absorbed from the intestine (Schaffer et al., 1950). For PEG1000, oral absorption in humans is approximately 10%. The oral absorption of PEG6000 is very low, because no PEG could be found in urine after oral administration. Therefore, the oral exposure to PEG is dependent on the PEG molecular weight used in a product and its dose; systemic exposure after oral administration can vary from high to very low, depending upon the source of the PEG.
Topical absorption of PEG will also occur and, again, will show a molecular weight dependence similar to that seen orally. Absorption by this route is likely to be poor (Tsai et al., 2003). However, in burn patients administered PEG-containing ointments, PEG exposure can reach significant (70 μM) levels (PEG300, 1000, and 4000) (Herold et al., 1989). A review of the toxicology information has suggested an acceptable daily intake of PEG for human. This estimate is up to 10 mg/kg or 0.7 g/70-kg human/day (Fruijtier-Polloth, 2005). For low molecular weight PEGs, this acceptable dose could, in theory, give rise to a systemic (absorbed) dose of approximately 400 mg/day.
Exposure to PEG from Intravenous Medicines: Comparison to Those Seen for PEGylated Proteins. To put into context the potential risk associated with the administration of PEG conjugated to a protein (or other molecule), the dose of PEG received as an excipient during administration of intravenous drugs was compared with those obtained when administering a PEGylated protein at its therapeutic dose.
The data obtained in this evaluation are summarized in Table 2 and have been expressed in terms of the mass administered, to provide an understanding of the ethylene glycol units exposure. The body burden has also been expressed as a molar amount because this represents the number of PEG molecules. These two exposure estimates allow a realistic comparison of PEG burden in humans.
Comparison of the estimated PEG exposures in humans resulting from PEGylated protein or as an excipient in intravenous medicines
Other intravenous products and PEGylated proteins are available, but insufficient data could be found to allow estimation of exposure. These included Adagen (pegadamase bovine), Oncaspar (pegaspargase), and torsemide. Assumptions were: 1) data were estimated from product package inserts or labels (see www.fda.gov), 2) the highest dose presented in the insert was used in the estimate, and 3) specific gravity of PEG is 1.1 g/ml.
The data collated for this analysis demonstrate that the amount of PEG a human is exposed to from a PEGylated biological product is significantly less than the acceptable daily intake. Even for the highest-dose PEGylated protein, Somavert (pegvisomant), the exposure of PEG in humans is approximately 25-fold lower than the estimate of acceptable human dose, based on mass. Comparison of the exposure (based on mass) of the PEGylated protein to that seen when PEG is used as an intravenous excipient, again indicates that the exposure is in all cases equal to or lower than intravenous PEG exposure. If exposure is considered on a molar basis, exposure from PEGylated biologics is at least 10-fold lower than that seen for the lowest PEG intravenous excipient dose. It should also be noted that using lorazepam for this lower-end comparison is misleading because this estimate is based on a single dose and lorazepam can be administered on multiple occasions (see below). Finally, exposure to PEG from PE-Gylated biological products lies in a range similar to the potential exposure seen in humans for PEG impurities detailed in the blood product, Venoglobin S (both in terms of mass and molar amounts).
The data presented above demonstrate that the PEG exposure resulting from PEGylated biologicals is comparable to or lower than the intravenous exposure of PEG as an excipient. In addition to this evaluation, evidence of adverse events in humans associated with PEG were sought, and these data are summarized in Table 2. There have been three literature reports of PEG-related adverse events in humans; these incidences are summarized below.
Intravenous nitrofurantoin (McCabe et al., 1959) has been shown to cause acute renal tubular necrosis, oliguria, and azotemia in 6 of 32 patients dosed for 3 to 5 days with a cumulative PEG dose of 121 to 220 g (PEG300: 400,000 to 750,000 μmol). Topical nitrofurazone has shown a similar toxicity (Hunt et al., 1982). The topical cream consist of 63% PEG300/5% PEG1000/32% PEG4000, and estimates of the concentrations in plasma associated with toxicity are in the region of 30 to 70 mM (Herold et al., 1989). One patient who received multiple administrations of lorazepam presented a similar toxicity (acute renal tubular necrosis) after a cumulative dose of approximately 240 g of PEG400 (600,000 μmol) (Laine et al., 1995).
Comparison of the exposures associated with PEG-related adverse events in humans with those seen from PEGylated protein indicate that exposures from PEGylated proteins are at least 600-fold lower. The major human adverse event related to high PEG exposure is renal toxicity. Renal toxicity has also been observed in the monkey at high dose, indicating that similar toxicities occur across species (Fruijtier-Polloth, 2005).
Overall, the exposures of PEG from PEGylated biologicals that lie below the estimate of the acceptable daily PEG dose are no higher (and often substantially lower) than the exposures of PEG seen when PEG is used as an excipient in humans and are substantially lower (>600-fold) than the exposure of PEG that is associated with toxicity (acute renal tubular necrosis) in humans. These data indicate that given the low doses used for PEGylated biological products and the PEG toxicological profile, of very low concern, it is unlikely that adverse events will occur in humans specifically as a result of the PEG used to PEGylate a biological product.
Feasibility of Metabolism Studies with PEGylated Materials
Studying the metabolism of PEGylated proteins represents a significant challenge, especially in vivo. For small organic molecules, radiolabeling is typically used to provide a selective method to identify the fate of a compound when administered to animals and human. Labeling the PEG associated with a biological molecule to elucidate the routes of elimination is not a viable option. Several methods exist to label PEG; these include tritium exchange, labeling the PEG with the addition of a 14C-ethylene oxide or 1-bromo-3-hydroxy-propan-2-one to the hydroxyl terminus of the PEG (Shemilt et al., 2004), iodination (Yamaoka et al., 1994), or using gamma irradiation (Leung et al., 2000). Although tritium exchange is a simple method of radiolabeling PEG, this method is nonspecific and the tritium may be lost through exchange with water. Gamma irradiation is a destructive process that leads to a lower molecular weight species than the original PEG. The most frequent method for radiolabeling PEG requires modification of the terminal hydroxyl group. Depending on the modification of the PEG, a range of radiolabels can be incorporated onto the PEG molecule; these include 125I, 14C, 18F, and 111In (Yamaoka et al., 1994; Wen et al., 2003; Chen et al., 2004; Shemilt et al., 2004). These methods are generally not applicable to PEGylated proteins because radiolabeling is performed at the terminal hydroxy groups of the molecules, and these groups are frequently either methylated or used to link to the protein or linker. These methods also result in the modification of the PEG structure, limiting their usefulness for this type of investigation. This is particularly the case since the PEG hydroxyl group represents both the site of radiolabeling and the site of metabolism of the unconjugated PEGs. The addition of an extra 14C-labeled ethylene unit to the terminus of the PEG chain seems the most attractive method of labeling for PEGs. However, this frequently increases the polydispersity of the PEG, and because this is a modification at the terminus, it is possible that this radiolabel could be lost, leaving unlabeled PEG metabolites (Shemilt et al., 2004). Overall, unlike a small molecule, there is no readily accessible method of radiolabeling the PEG used to PEGylate proteins. Although radiolabeling is possible, any chosen method has sufficient issues to mean that any results obtained may be difficult to interpret.
Using a specific assay to detect PEG and its potential metabolites could be considered. To address the metabolism question, such an assay would have to have a broad cross-reactivity with PEG and the acid and sulfate metabolites, ensuring that all metabolites are detectable. However, humans and animals are commonly exposed to PEG via a variety of sources, which means that it is likely that human plasma, urine, etc., contain a range of PEGs and PEG metabolites. When the trace doses of most PEGylated biological products are combined with the ubiquitous “contamination” likely to be seen in animals and humans, it is unlikely that this approach to metabolite identification would be successful.
Finally, identifying metabolites of PEG will also be difficult because the PEGs used have polydispersed molecular weight (Veronese and Pasut, 2005). Subtle changes in mass due to metabolism will be undetectable by mass spectrometry because of the range of molecular weight present in the PEG. Also, work has shown that PEG can suppress ionization in a mass spectrometer, and this may increase the uncertainty surrounding metabolite identification and quantification (Weaver and Riley, 2006). These data indicate that the technical challenges facing an experimenter will make any experimentation on the metabolism of PEG of little value.
Conclusions
As for any other therapeutic protein being developed for administration to humans, a full toxicology package in relevant species will be required for a PEGylated protein prior to commercialization. This toxicology examination will provide a satisfactory evaluation of the toxicology associated with this molecule.
PEG has a toxicological profile of very low concern and is well tolerated at high doses after chronic and acute administration. The PEG associated with a biological molecule itself should provide no extra concern because the toxicity versus exposure relationship in animals and humand has been thoroughly investigated and metabolism/excretion is well understood. Based on the comparisons of PEG exposure from PEGylated biological products and the exposures of PEG associated with toxicity, the therapeutic index is large (≥600-fold). The metabolism of PEG is limited to metabolic modification of the hydroxyl group, and the data available suggest that the metabolites seen in humans are seen in animals. Also, for PEGs typically used on biologicals, metabolism will not play a major role in PEG elimination. In light of these data, PEG metabolites do not represent a significant issue, especially when combined with the low overall exposure to PEG discussed above.
Studying the metabolism of PEGylated biologics will represent a significant challenge. First, radiolabeling of PEG associated with a biological molecule is not a viable option. Second, the doses of these PEGylated biologicals are usually very low. Third, PEG is present in a range of products that humans are routinely exposed to. The detection of trace exposures of PEG metabolites produced from PEGylated biologicals will be impossible against the background of PEG and its metabolites present as a result of routine exposure. Moreover, because the products of metabolism are the same regardless of the route of administration, because metabolism represents a minor route of clearance, and because data demonstrate that PEG exposures considerably higher than those possible from PEGylated biologicals are required for toxicity, any additional experiments seem unjustified and of very limited value.
The data presented in this article indicate that, assuming toxicological evaluation of a biological molecule of interest is completed in an appropriate species and satisfactory therapeutic windows are achieved, the PEG associated with a protein or other biological molecule does not represent a significant additional unquantified risk to humans, because of 1) the low exposures involved, 2) the low toxicity profile of PEG, and 3) the similarity of the metabolites that are formed in all species.
Further studies to elucidate the metabolism of the PEG associated with a biological molecule in humans will not provide any more information to place into context the safety of PEG, and such studies may not even be possible.
Footnotes
-
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
-
doi:10.1124/dmd.106.012419.
-
ABBREVIATIONS: PEG, polyethylene glycol; ADME, absorption, distribution, metabolism, and excretion; EPAR, European Public Assessment Report; AUC, area under the curve.
-
 Eric Didier is a Docteur en Pharmacie, graduating from the University of Auvergne, France in 1995. He worked for the Institut de Recherche Pierre Fabre (Castres and Toulouse, France) as a Clinical Pharmacokineticist (1996–2001), before joining the Clinical Pharmacology Department of Pharmacia, High Wycombe, UK, in 2001. He is currently Associate Director in the Clinical Pharmacology Department of Pfizer Global R&D in Sandwich, UK.
Eric Didier is a Docteur en Pharmacie, graduating from the University of Auvergne, France in 1995. He worked for the Institut de Recherche Pierre Fabre (Castres and Toulouse, France) as a Clinical Pharmacokineticist (1996–2001), before joining the Clinical Pharmacology Department of Pharmacia, High Wycombe, UK, in 2001. He is currently Associate Director in the Clinical Pharmacology Department of Pfizer Global R&D in Sandwich, UK. -
Dr. Philip Harris trained as an adult endocrinologist. He has written numerous publications in neuroendocrinology, endocrine oncology, and thyroid disease. Currently he is Senior Director in Clinical Development, Pfizer, Sandwich, UK, and is an Honorary Consultant Endocrinologist at St Bartholomew's Hospital, London, UK.
-
Ned Siegel received B.S. and M.S. degrees in Biology and Botany, respectively, from the University of Illinois in 1972–1973. He began his professional career with the Monsanto Company in 1974, where he worked in the Agricultural Research department, before subsequently assuming basic research activities for the former G.D. Searle and Pharmacia Corporations, with responsibilities involving the analytical characterization of proteins and peptides. His work has included preformulation development support for the clinical development of protein therapeutics and, more recently, project leadership responsibilities for Pfizer Inc. in the area of novel formulation and delivery strategies, including expertise in the area of protein and peptide PEGylation. He retired in 2006 from Pfizer Global Research and Development as a Research Fellow at the Chesterfield, MO facility.
-
Dr. Dennis Smith has worked in the pharmaceutical industry for the past 30 years since gaining his Ph.D. from the University of Manchester. For the last 18 years he has been at Pfizer Global Research and Development, Sandwich, UK, where he is Vice President-Pharmacokinetics, Dynamics and Metabolism. His research interests and publications span all aspects of drug discovery and development, particularly where drug metabolism knowledge can have an impact on the design of more efficacious and safer drugs. During this 30-year span he has directly helped in the discovery and development of eight marketed new clinical entities (NCEs) with, hopefully, several more to come. He has authored over 100 publications including two textbooks. He is active in a teaching role, holding appointments as Visiting Professor at the University of Liverpool and Honorary Senior Lecturer at the University of Aberdeen, and lectures widely to students at several other universities.
-
His wish in much of his work now is “to inspire another generation to take up the cudgel against disease”.
-
Jeanne Stadler received a degree in Pharmacy from the Faculté de Pharmacie, Université de Paris, in 1972. She joined Pfizer the same year and, as a toxicologist, became a specialist in reproduction and developmental toxicology.
-
She is a member of the Teratology Society and the European Teratology Society, and is a Council Member of the AFARED (Francophone Association for Research in Reproduction and Development). After a more than 30-year career in preclinical research, she is currently an independent consultant in toxicology.
-
Dr. Lorraine Tilbury joined Pfizer Global Research and Development in April 2005 as an Associate Research Fellow in their Regulatory Submissions department, and leads the Regulatory Submissions team. Prior to joining Pfizer, she devoted 15 years to the E.I. DuPont de Nemours Europe/Middle East/Africa business, as a toxicologist specializing in human health risk assessment of chemicals and, subsequently, as leader of the French and European Registration and Regulatory Affairs group. Dr. Tilbury's background is in veterinary medicine with postdoctoral work in toxicology carried out at New Mexico State University and Research Triangle Park (NC), and working with CIIT, a toxicology research institute.
-
Rob Webster joined Pfizer Global Research and Development in 1989 in their Pharmacokinetics, Dynamics and Metabolism department Currently, Rob is a Director in this department, with responsibilities for providing guidance and direction on drug metabolism questions/issues, from lead discovery through to and beyond filing. Prior to joining Pfizer, Rob studied Biochemistry at Liverpool Polytechnic.
- Received August 9, 2006.
- Accepted September 29, 2006.







- The American Society for Pharmacology and Experimental Therapeutics
References
In this issue





{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- Toxicology of Polyethylene Glycol in Animals
- Toxicology of PEGylated Molecules in Animals
- Clearance of Polyethylene Glycol by Metabolism
- Excretion of Unchanged Polyethylene Glycol in the Urine and Bile: The Major Route of PEG Clearance?
- Likely Clearance Mechanisms for PEG Associated with PEGylated Proteins
- PEG Exposure in Humans
- Feasibility of Metabolism Studies with PEGylated Materials
- Conclusions
- Footnotes
- References
- Figures & Data
- Info & Metrics
- eLetters