


Abstract
The concept of target-specific covalent enzyme inhibitors appears attractive from both an efficacy and a selectivity viewpoint considering the potential for enhanced biochemical efficiency associated with an irreversible mechanism. Aside from potential safety concerns, clearance prediction of covalent inhibitors represents a unique challenge due to the inclusion of nontraditional metabolic pathways of direct conjugation with glutathione (GSH) or via GSH S-transferase–mediated processes. In this article, a novel pharmacokinetic algorithm was developed using a series of Pfizer kinase selective acrylamide covalent inhibitors based on their in vitro-in vivo extrapolation of systemic clearance in rats. The algorithm encompasses the use of hepatocytes as an in vitro model for hepatic clearance due to oxidative metabolism and GSH conjugation, and the use of whole blood as an in vitro surrogate for GSH conjugation in extrahepatic tissues. Initial evaluations with clinical covalent inhibitors suggested that the scaling algorithm developed from rats may also be useful for human clearance prediction when species-specific parameters, such as hepatocyte and blood stability and blood binding, were considered. With careful consideration of clearance mechanisms, the described in vitro-in vivo extrapolation approach may be useful to facilitate candidate optimization, selection, and prediction of human pharmacokinetic clearance during the discovery and development of targeted covalent inhibitors.
Introduction
There has been a renewed interest in covalent inhibitor drugs as therapeutic agents (Singh et al., 2011; Kalgutkar and Dalvie, 2012). The design of targeted covalent enzyme inhibitors may seem attractive, considering the potential for enhanced biochemical efficiency associated with an irreversible or a slow-reversible mechanism that can lead to increased target selectivity and extended pharmacodynamics beyond the pharmacokinetics of the drug. However, the possibility for off-target reactivity and potential direct organelle toxicity or indirect immune responses remain serious issues that need to be considered during the course of drug discovery and development. Although currently there are no well-established methods to completely derisk the toxicity potential of covalent inhibitors, there are some limited methodologies that can help to assess off-target risk, especially with the consideration of total daily body burden (Bauman et al., 2009; Nakayama et al., 2009; Dahal et al., 2013). At the moment, limiting the dose of the covalent inhibitor, although imperfect, appears to be the most reliable method to empirically minimize off-target safety risk (Lammert et al., 2008).
Consideration of target turnover in the context of pharmacokinetic-pharmacodynamic requirements for a covalent inhibitor is particularly important in understanding pharmacokinetic success criteria and ultimate target doses. However, covalent inhibitors present some novel challenges for drug discovery in the absence of reliable methods to predict human clearance (CL). Successful prediction of covalent inhibitor CL will necessarily require a full understanding of the mechanisms involved in the elimination of the compound. For these novel entities, CL pathways can go beyond some of the more traditional cytochrome P450 (P450) mechanisms to include direct conjugation with glutathione (GSH) and/or mediation through GSH S-transferase (GST), as well as potential nonspecific reactivity to tissue proteins.
GSTs are a group of highly diverse cytosolic, microsomal, and mitochondrial enzymes found in all tissues of the body (Marcus et al., 1978; Awasthi et al., 1994; Whalen and Boyer, 1998; Oakley, 2011). In part, they serve a protective function for the cell by using the reduced form of GSH to conjugate with many different types of electrophiles, including typical Michael acceptor covalent inhibitors. The set of GST isoenzymes differs in individual tissue, where liver cytosol contains predominately alpha class enzymes, but also mu class. The pi class of GSTs is particularly important in other tissues, including gastrointestinal tract, heart, kidney, lung, skin, and erythrocytes. The substrate specificity of GSTs is highly varied and overlapping. Polymorphisms in GSTs are believed to be related to susceptibilities to various human diseases and drug-induced toxicities (Lucena et al., 2008).
The current work presents the value of combining both in vitro hepatocyte [i.e., hepatic CL (CLh)] and blood [i.e., extrahepatic CL (CLeh)] stability models to predict the total in vivo CL of a number of covalent inhibitor compounds (Fig. 1). From in vitro and in vivo metabolite profiling work, it was clear that the primary mechanisms of CL included the typical P450-susceptible sites as well as GSH adduction directly on the acrylamide. In this respect, hepatocytes were explored to assess both P450 and GST CLh of covalent inhibitor compounds, whereas whole blood (erythrocytes) was assessed for its ability to predict extrahepatic GST CL. An initial pharmacokinetic algorithm was developed based on the in vitro-in vivo extrapolation (IVIVE) of CL in rats, a common nonclinical species used in drug discovery, for a series of Janus kinase 3 (JAK3)–selective acrylamide covalent inhibitors. Scaling methods were then used to predict CL in other species commonly used in pharmacology and toxicology assessment as well as in humans, the ultimate intended target. Finally, the methodology established to explore JAK3 covalent inhibitors was applied to the prediction of other non-JAK3 clinical covalent inhibitors.

Structures of covalent inhibitor compounds studied. Compounds 1 through 18 are novel JAK3 kinase covalent inhibitors. Compound 12 is the JAK3 clinical lead PF-06651600.
Materials and Methods
Materials.
Novel JAK3 kinase–selective acrylamide covalent inhibitors (Compounds 1–18 prepared as generically described in patents WO 2013085802 and WO 2015083028; Thorarensen et al., 2016), including PF-06651600 (Compound 12; JAK3-selective inhibitor; Clinical trial reg. no. NCT02309827, clinicaltrials.gov) and the clinical covalent inhibitors CI-1033 (canertinib) and PCI-32765 (ibrutinib), were synthesized at Pfizer Inc. (Groton, CT) (Fig. 1). NADPH, reduced GSH, ethacrynic acid, 1-chloro-2,4-dinitrobenzene (CDNB), N-ethylmaleimide were purchased from Sigma-Aldrich (St. Louis, MO). Analytical chemicals were of analytical grade or better, and solvents of high-performance liquid chromatography (HPLC) grade were obtained from Fisher Scientific (Pittsburgh, PA). Phosphate-buffered saline (PBS) was purchased from Lonza (Walkersville, MD). Gibco William’s E Medium was from ThermoFisher Scientific (Grand Island, NY). Cryopreserved human hepatocytes (lot RHT; pooled, n = 5 male livers and n = 5 female livers) were purchased from BioreclamationIVT (Baltimore, MD). Cryopreserved hepatocytes from CD-1 mouse (lot BRZ; n = 50 pooled male), Wistar-Han rat (lot SLA; n = 12 pooled male), beagle dog (lot FNM; n = 5 pooled male), and cynomolgus monkey (lot KNT; n = 5 pooled male) were purchased from Celsis IVT (Baltimore, MD). Human liver microsomes (lot HLM102; n = 50 male/female mix pooled livers) were purchased from BD Gentest (San Jose, CA). Recombinant human GST P1-1 expressed in Escherichia coli (>95% purified) was purchased from Oxford Biochemical Research Inc. (Oxford, MI). Blood from male CD1 mice (n = 6), male Sprague Dawley rats (n = 3), male beagle dogs (n = 2), or male cynomolgus monkeys (n = 2) was collected by Comparative Medicine at Pfizer (Groton, CT) in accordance with regulations and established guidelines, and were reviewed and approved by an Institutional Animal Care and Use Committee. Human blood was collected from one male and one female healthy volunteer at the Occupational Health & Wellness Center at Pfizer (Groton, CT) according to applicable Pfizer policies, including with institutional review board/institutional ethics committee approval. Plasma from human (lot BRH736604; n = 3 each male/female), cynomolgus monkey (lot CYN121225; n = 10 pooled male), beagle dog (lot BGL67132; n = 6 pooled male), Wistar-Han rat (lot RAT222727; n = 70 pooled male), and CD-1 mouse (lot MSE163345; n = 80 pooled male) was purchased from BioreclamationIVT.
Animals.
In vivo pharmacokinetic studies were performed in male C57BL/6 mice (Vital River Laboratories, Beijing, People’s Republic of China), Sprague Dawley rat (Vital River Laboratories), beagle dog (Marshall Farms, North Rose, NY), and cynomolgus monkey (Charles River BRF, Houston, TX). The Pfizer Institutional Animal Care and Use Committee reviewed and approved the animal use in these studies. The Association for Assessment and Accreditation of Laboratory Animal Care International fully accredits the Prizer and BioDuro animal care and use programs.
Plasma Protein Binding.
Plasma was thawed, centrifuged (1500g for 10 minutes) to remove any debris, and pH adjusted to pH 7.4 with CO2 bubbling or 0.1% phosphoric acid. Drug stock (12 µl of 200 µM drug in 50:50 acetonitrile/water) was added to 1188 µl of plasma to give a final concentration of 2 µM test compound (n = 2). Samples were mixed, and 15-µl aliquots (T = 0 hour recovery sample) were taken (n = 2) with immediate addition of 45 µl of Dulbecco’s PBS buffer (Sigma-Aldrich), and precipitation with 180 µl of ice-cold acetonitrile containing an internal standard to enable a recovery calculation. Rapid equilibrium dialysis (RED) devices (96 well plate, catalog #90006; ThermoFisher Scientific) were used to assess plasma protein binding of test compounds according to manufacturer protocol. RED devices were sealed with a gas-permeable membrane (Z380059-1PAK; Sigma-Aldrich) and placed onto a shaking platform (450 rpm) in a CO2 incubator (5% CO2) for 4 hours at 37°C. After incubation, the RED device was removed and a 15-µl aliquot (T = 4-hour recovery sample) was removed (n = 2), with immediate addition of 45 µl of Dulbecco’s PBS buffer and precipitation with 180 µl of ice-cold acetonitrile containing an internal standard. Additional plasma and buffer samples in quadruplicate were taken and precipitated in a fashion similar to that used for the recovery samples. All samples were centrifuged at 3600 rpm at 4°C for 30 minutes. Samples were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) using a Shimadzu quaternary HPLC pump (Shimadzu, Tokyo, Japan) with an Agilent 1100 series membrane degasser and Leap autosampler (LeapTech, Dubai, United Arab Emirates) coupled to a PE Sciex API 4000 QTRAP mass spectrometer (Sciex, Framingham, MA) using electrospray ionization in positive ion mode with multiple reaction monitoring (MRM). Chromatographic separation was achieved using a Opti-Lynx C18, 40 µm, 100 Å, 2.1 × 10 mm column (Optimize Technologies, Oregon City, OR). The mobile phase was 5% acetonitrile and 0.1% formic acid at 2 ml/min for 12 seconds, followed by a 4.5 ml/min gradient flow of an increasing/decreasing percentage of acetonitrile in 0.1% formic acid over 1.5 minutes. Analyst software (Sciex) was used for data collection and analysis.
Microsomal Protein Binding.
Human microsomes were denatured by leaving them on a laboratory benchtop overnight and were diluted to a protein concentration of 0.8 mg/ml with 100 mM phosphate buffer, pH 7.4. The RED device and analytical methodology were used to assess microsomal protein binding in a manner similar to that described above in the Plasma Protein Binding section. Microsomal binding [microsomal fraction unbound (fu mic)] across species was calculated from human microsomal binding with correction for differences in microsomal protein concentration (C) using the following equation:

Blood-to-Plasma Ratio.
Blood was collected fresh into K2-EDTA tubes and kept on ice before use, and was pooled for each species. Test compound (n = 2) was added at a 1 µM final concentration in 400-µl aliquots of blood in a 96-well plate. After mixing, a 20-µl T = 0 hour recovery sample was collected, with subsequent addition of 20 µl of control species plasma and precipitation with 150 µl of ice-cold acetonitrile containing an internal standard. Samples were mixed for 2 minutes and centrifuged (3000 rpm, 10 minutes). Plates were covered in Breathe-Easy gas-permeable membranes (Diversified Biotech, Dedham, MA) and incubated in a chamber at 37°C in an environment of 95% O2/5% CO2 and 90% relative humidity for 1–3 hours at 450 rpm. After 1 and 3 hours, recovery and test samples were collected and processed as described above. Samples were analyzed by LC-MS/MS using an AB Sciex API-5500 Electrospray mass spectrometer with a Jasco PU-1580 Pump (Jasco Inc., Easton, MD) and Apricot Designs (Covina, CA)/Sound Analytics ADDA autosampler in positive ion mode with MRM. Chromatographic separation was achieved using an Optimize Technologies SP Small Molecule Trap column. The mobile phases were 95% 2 mM ammonium acetate/5% 50:50 acetonitrile/methanol and 90% 50:50 acetonitrile/methanol /10% 2 mM ammonium acetate at a flow rate of 1.5 ml/min. Analyst software (Sciex) was used for data collection and analysis.
Hepatocyte Intrinsic CL.
Pooled cryopreserved hepatocytes were suspended in Williams’ medium E supplemented with HEPES and Na2CO3 (91-5233EC; Invitrogen, Grand Island NY) and counted using trypan blue exclusion. Test compounds (n = 2) at a 1 µM final concentration were added into 96-well plates containing 0.5 million hepatocytes/ml [≤0.1% dimethylsulfoxide (DMSO) final concentration]. Plates were covered in Breathe-Easy gas-permeable membranes (Diversified Biotech) and incubated in a chamber at 37°C in an environment of 95% O2/5% CO2 and 95% relative humidity for 4 hours at 150 rpm. Aliquots were taken from 3 to 240 minutes and quenched with two parts ice-cold acetonitrile containing internal standard. Samples were centrifuged at 3000 rpm for 10 minutes at 4°C, and supernatants were subjected to quantitation by general LC-MS/MS methodology using a Shimadzu quaternary HPLC pump with Agilent 1100 series membrane degasser and Leap autosampler (LeapTech) coupled to a PE Sciex API 4000 QTRAP mass spectrometer (Sciex) using electrospray ionization in positive ion mode with MRM. Chromatographic separation was achieved using a Kinetex C18, 2.6 µM, 30 × 2 mm column (Phenomenex, Torrance, CA). The mobile phases were HPLC water (solvent A) and acetonitrile (solvent B), both of which contained 0.1% formic acid. A linear gradient from 5% solvent A to 95% solvent B was applied over 2 minutes at a flow rate of 0.4 ml/min (cycle time, 3 min/injection). Analyst software (Sciex) was used for data collection and analysis. For compounds showing high hepatocyte stability in the standard 4-hour assay described above, a 5-day relay method was used to assess hepatocyte CL as specifically described in the study by Di et al. (2012).
Blood Stability.
Blood was collected fresh into K2-EDTA tubes and kept on ice before use and was pooled for each species. An aliquot of the blood was transferred to microtubes and prewarmed for 10 minutes at 37°C using a heat block. The test compound (n = 2) was then added (1 µM final concentration), and the incubation was continued for 180 minutes at 37°C in duplicate. An aliquot of the incubation mixture was removed at designated time points from 10 to 180 minutes during the course of the incubation, mixed with an aliquot of ice-cold acetonitrile containing an internal standard, vortexed, and centrifuged (3000 rpm, 10 minutes). The resulting supernatants were removed and subjected to quantitation by general LC-MS/MS methodology, as described above in the Hepatocyte Intrinsic CL section.
Stability in Recombinant Human GST Enzymes.
Recombinant human GST (rhGST) P1-1 protein was thawed on ice and then diluted to obtain a final incubation concentration of 53 µg/ml with cold potassium phosphate buffer (100 mM, pH 7.4). A stock solution of GSH was prepared by dissolving GSH in phosphate buffer to obtain a final incubation concentration of 100 mM. The 500-μl incubations were prepared by combining dilute protein (472.5 µl) and GSH (25 µl) and warming them to 37°C in approximately 5 minutes. The reaction was initiated by the addition of 2.5 µl of a 200 µM solution of test compound (n = 2) in DMSO to yield a 1 µM final concentration. Immediately, the incubation was mixed by pipette, and a 50-µl aliquot was transferred to 150 µl of acetonitrile containing 10 mM N-ethylmaleimide (a GSH scavenger) and an internal standard. Additional samples were taken by the same procedure throughout the 4-hour incubation. The positive control CDNB (10 mM) was assessed in a similar manner. After the 4-hour time point, quenched aliquots were mixed and centrifuged (3000 rpm, 10 minutes). Samples were analyzed by LC-MS/MS using a Shimadzu quaternary HPLC pump with an Agilent 1100 series membrane degasser and Leap autosampler (LeapTech) coupled to a PE Sciex API 4000 QTRAP mass spectrometer using electrospray ionization in positive ion mode with MRM. Chromatographic separation was achieved using a Phenomenex Synergy Polar-RP 4 µm 50 × 2.0 mm column. The mobile phases were water/acetonitrile (95%/5%) (solvent A) and water/acetonitrile (5%/95%) (solvent B), both of which contained 0.1% formic acid. A linear gradient of solvent B from 5% to 95% was applied over 2.1 minutes on the column at a flow rate of 0.5 ml/min. All analytes were eluted between 1.25 and 1.8 minutes. CDNB was assessed using UV light detection at wavelength λ = 254 nm. Analyst software (Sciex) was used for data collection and analysis. The intrinsic CL (CLint) was calculated using the following equation:

Pharmacokinetics.
Plasma concentrations of select test compounds were determined in male mouse (n = 3), rat (n = 2), dog (n = 2), and monkey (n = 2) after a single 1 mg/kg i.v. dose. Studies were conducted at BioDuro (Beijing, People’s Republic of China) (mouse, rat) or Pfizer (dog, monkey) according to standard protocols. Briefly, compounds were formulated in standard solubilizing vehicles (rat, 10% DMSO/30% PEG400/60% water; mouse and dog, 5% DMSO/12% sulfobutylether-β-cyclodextrin in sterile water; monkey, normal saline) at 1 mg/ml (mouse, rat) or 2 mg/ml (dog, monkey) and dosed at 1 ml/kg i.v. (mouse, rat) or 0.5 ml/kg i.v. (dog, monkey). Serial blood samples were collected at various times from 5 minutes to 24 hours. Plasma was harvested from blood samples after centrifugation. Plasma samples (50 µl) and standards were prepared by protein precipitation with acetonitrile (1:1 v/v) containing an internal standard, buspirone or metoprolol. Samples were vortexed and centrifuged to obtain supernatant, which was analyzed using general LC-MS/MS methodology, as described above (API 4000; Sciex). Pharmacokinetic parameters were determined using data from individual animals, using noncompartmental analysis in Watson LIMS version 7.4.1 (ThermoFisher Scientific).
Data Analyses.
CLh was determined from scaled intrinsic CL (CLh int; eq. 1) using the well-stirred model of hepatic drug CL (eq. 2), taking into consideration in vitro intrinsic hepatocyte stability (CLh int, in vitro = 0.693/half-life (t1/2) (min) × assay volume (µl)/million cells), hepatocyte yield (130 × 106 cells/g liver), liver weight (human, 21 g/kg; dog/monkey, 32 g/kg; rat, 40 g/kg; mouse, 90 g/kg), blood binding (fub), hepatocyte binding (fu mic), and hepatic blood flow (Q, in ml/min/kg: human, 20; dog, 35; monkey, 44; rat, 70; mouse, 90). Blood binding (fub) was derived from plasma protein binding (fup) and the blood-to-plasma ratio (BPR) concentration (i.e., fub = fup/BPR). Hepatocyte binding (fu mic) was considered to be equal to liver microsomal binding under the experimental conditions used. Unbound intrinsic CLh (CLh int, u) can be derived from in vitro hepatocyte stability and microsomal binding (i.e., CLh int, u = CLh int, in vitro/fu mic.  (1)
(1) (2)To understand the relationship between CLeh and unbound in vitro blood CL (CLb), the apparent CLeh in rats (eq. 3) for 18 Pfizer JAK3 acrylamide covalent inhibitors (Fig. 1) was calculated from the difference between observed CLb [derived from measured plasma CL (CLp) in rats after a single intravenous dose of compound; i.e., CLb = CLp/BPR] and the corresponding CLh scaled from rat hepatocytes (eq. 2). Calculated unbound CLeh (CLeh, u) in rats was derived from the calculated apparent CLeh (eq. 3), fup, and BPR (i.e., CLeh, u = CLeh × BPR/fup). In vitro unbound blood intrinsic CL (CLb int, u) was derived from in vitro blood stability t1/2 and blood binding (fub) [i.e., CLb int, u (µl/min/g) = (0.693/t1/2)/fub]. A linear regression equation from a plot of CLb int, u versus CLeh, u was determined as CLeh, u = (CLb int, u + b)/a, where the slope was represented by a and the y-intercept was represented by b (eq. 4):
(2)To understand the relationship between CLeh and unbound in vitro blood CL (CLb), the apparent CLeh in rats (eq. 3) for 18 Pfizer JAK3 acrylamide covalent inhibitors (Fig. 1) was calculated from the difference between observed CLb [derived from measured plasma CL (CLp) in rats after a single intravenous dose of compound; i.e., CLb = CLp/BPR] and the corresponding CLh scaled from rat hepatocytes (eq. 2). Calculated unbound CLeh (CLeh, u) in rats was derived from the calculated apparent CLeh (eq. 3), fup, and BPR (i.e., CLeh, u = CLeh × BPR/fup). In vitro unbound blood intrinsic CL (CLb int, u) was derived from in vitro blood stability t1/2 and blood binding (fub) [i.e., CLb int, u (µl/min/g) = (0.693/t1/2)/fub]. A linear regression equation from a plot of CLb int, u versus CLeh, u was determined as CLeh, u = (CLb int, u + b)/a, where the slope was represented by a and the y-intercept was represented by b (eq. 4): (3)
(3) (4)Prior to in vivo study, rat extrahepatic CL (Scaled CLeh) for unknown compounds could then be predicted measuring in vitro blood stability for each compound and using the empirically derived extrahepatic scaling equation (eq. 5). Total rat CLb would subsequently be predicted by combining scaled in vitro hepatic CL (CLh; eq. 2) with scaled CLeh (eq. 6).
(4)Prior to in vivo study, rat extrahepatic CL (Scaled CLeh) for unknown compounds could then be predicted measuring in vitro blood stability for each compound and using the empirically derived extrahepatic scaling equation (eq. 5). Total rat CLb would subsequently be predicted by combining scaled in vitro hepatic CL (CLh; eq. 2) with scaled CLeh (eq. 6). (5)
(5) (6)For cross-species CLeh IVIVE, the scaling algorithm derived in the rat was used with species-specific parameters to calculate the extrahepatic CL in the species of interest (eq. 7), where CO is cardiac output (CO; in ml/min/kg: mouse, 400; rat, 296; monkey, 217; dog, 120; and human, 80; Davies and Morris, 1993).
(6)For cross-species CLeh IVIVE, the scaling algorithm derived in the rat was used with species-specific parameters to calculate the extrahepatic CL in the species of interest (eq. 7), where CO is cardiac output (CO; in ml/min/kg: mouse, 400; rat, 296; monkey, 217; dog, 120; and human, 80; Davies and Morris, 1993).
 (7)
(7)Results
IVIVE Model of CL for Covalent Inhibitors in Rats.
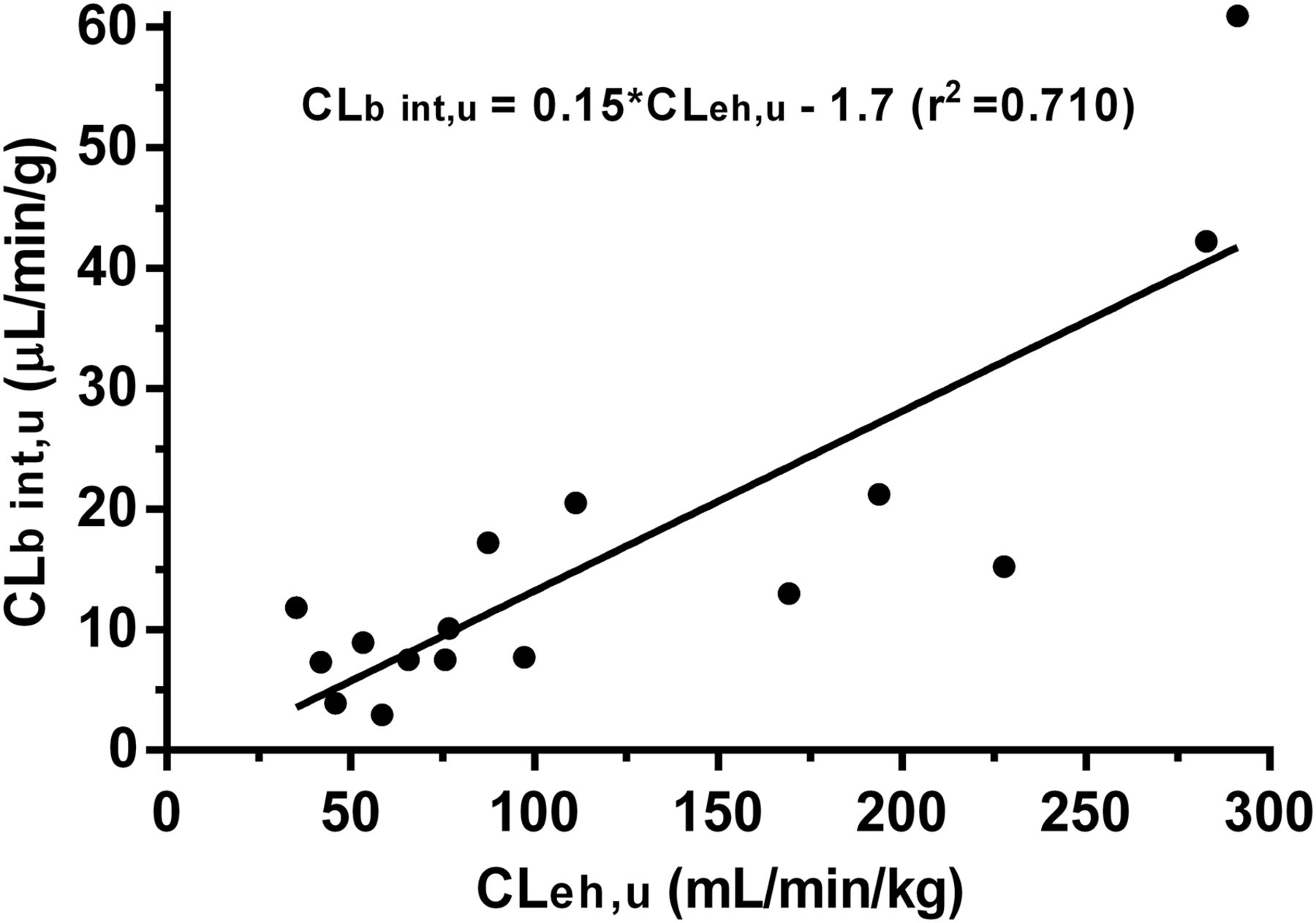
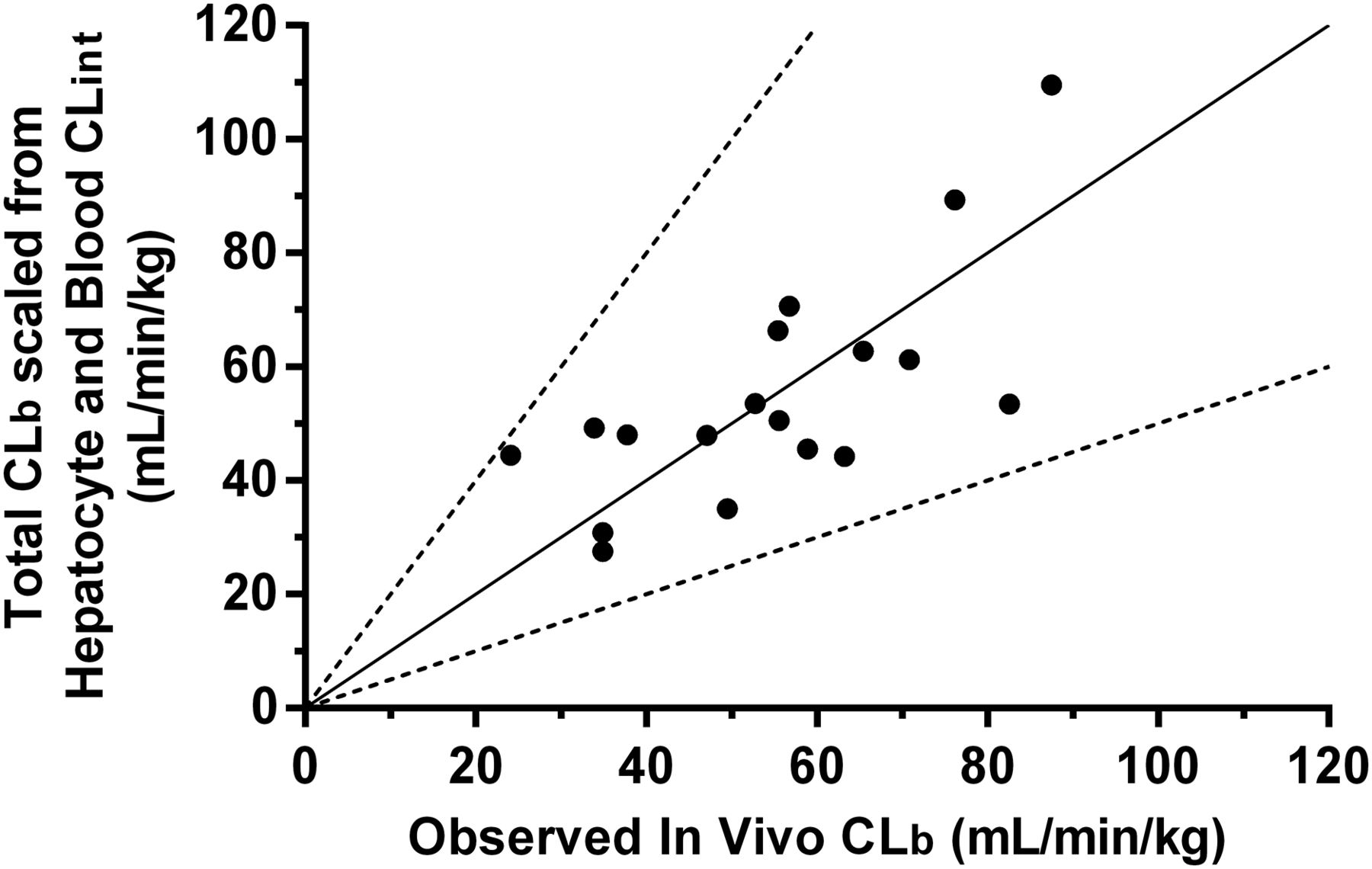
The total CLb for a series of 18 novel JAK3 kinase–selective acrylamide covalent inhibitors was studied in rats after a single intravenous dose, and their corresponding in vitro CLh int, u and in vitro CLb int, u in rats were determined and summarized in Table 1. The blood CLeh of these compounds was calculated by the difference between their corresponding in vivo total CLb and scaled CLh from hepatocyte stability (i.e., CLeh = CLb − CLh) (Fig. 2). The calculated CLeh, u value for each compound was found to correlate acceptably (r2 = 0.710) with their respective in vitro CLb int, u value. Linear regression yielded a best-fit equation with a slope of 0.15 and a y-intercept of 1.7, which enabled a prediction of in vivo CLeh for unknown compounds based on their in vitro intrinsic blood CL values. Combining scaled rat CLh with the empirically scaled rat CLeh yielded total CLb values that were within 2-fold of actual in vivo rat total CLb values for all compounds (Table 1, columns 8 and 9; Fig. 3).
Properties of novel acrylamide irreversible kinase inhibitors used to establish IVIVE in rat

Correlation between calculated CLeh, u values and in vitro CLb int, u values in rats.

Correlation between total CLb scaled from rat hepatocyte intrinsic CL and rat blood CLint, and observed in vivo CLb in rat for acrylamide covalent inhibitors (n = 18). Solid line, line of unity; segmented lines, 2-fold boundaries versus unity.
Cross-Species Prediction of CL.
It was discovered that the total CLb in other species, including mouse, monkey, dog, and human, could be reasonably derived from scaling their respective species hepatocyte and blood CLint values, using the extrahepatic algorithm derived from rat, with a correction for CO differences between species (eq. 8). (8)As CLeh occurs throughout the body and total body CL is the product of CO and the drug extraction ratio (Toutain and Bousquet-Mélou, 2004), equation 8 makes sense because CLeh was shown to be a function of both CO and a whole-body scaled GST extraction relative to rat. Two examples of IVIVE across species are shown for NCE 14 and PF-06651600 (NCE 12) in Table 2. CL predictions were within an approximate 2-fold concordance using this approach. For the ultimate JAK3 lead molecule PF-06651600, the clinical first-in-human CL/bioavailability (F) ratio was in good agreement with predicted CL based on likely by mouth F (estimated to be ∼80–90%, assuming CLh first-pass metabolism).
(8)As CLeh occurs throughout the body and total body CL is the product of CO and the drug extraction ratio (Toutain and Bousquet-Mélou, 2004), equation 8 makes sense because CLeh was shown to be a function of both CO and a whole-body scaled GST extraction relative to rat. Two examples of IVIVE across species are shown for NCE 14 and PF-06651600 (NCE 12) in Table 2. CL predictions were within an approximate 2-fold concordance using this approach. For the ultimate JAK3 lead molecule PF-06651600, the clinical first-in-human CL/bioavailability (F) ratio was in good agreement with predicted CL based on likely by mouth F (estimated to be ∼80–90%, assuming CLh first-pass metabolism).
Cross-species prediction of CL
Alternative CLeh Prediction using rhGST P1-1.
A subset of the JAK3 kinase–selective acrylamide covalent inhibitors with varying CLb int, u values were run in the human rhGST P1-1 stability assay to establish their correlation (Fig. 4). Considering the importance of GST P1-1 in blood, the good correlation (r2 = 0.955) was not surprising. The internal control for GST activity, CDNB, depleted substantially in the presence of rhGST P1-1, with a t1/2 of approximately 2–4 minutes, suggesting good enzyme activity. This method of CLeh prediction was useful for compounds with high blood stability like PF-06551600 (Table 2).

Correlation between human rGST P1-1 CLint (rGST P1-1 CLint) and in vitro CLb int, u.
CL Prediction of Other Covalent Inhibitors.
Further confidence in covalent inhibitor CL prediction comes from applying this scaling approach to other clinical covalent inhibitors, including canertinib (an acrylamide pan-erbB receptor tyrosine kinase inhibitor, CI-1033), ethacrynic acid (methylenebutanoyl sodium-potassium-chloride cotransport inhibitor), and ibrutinib (an acrylamide Bruton tyrosine kinase inhibitor, PCI-32765), all of which are outside the JAK3 kinase inhibitor chemotype (Fig. 1). In most cases, using the methodology established with JAK3 kinase inhibitors, both rat and human CL predictions for these other acrylamide molecules were in very good agreement with observed in vivo CL (Table 3). Of note is the directional overprediction for canertinib, which in part may be related to its direct reactivity to albumin protein and low recovery (25%) in the blood assay, highlighting the importance of understanding all relevant CL mechanisms when applying this methodology.
Scaled and observed CL of clinical acrylamide covalent inhibitors in human and rat
Discussion
With a good understanding of P450- and GST-mediated CL mechanisms, a novel cross-species predictive pharmacokinetic algorithm was developed using a series of JAK3 kinase–selective acrylamide covalent inhibitors based on their in vitro to in vivo translation of systemic CL in rats. The approach encompasses the use of the following: (1) hepatocyte stability as an in vitro model for CLh representing both P450 metabolism and GSH conjugation catalyzed through, presumably, GST alpha and/or mu; and (2) whole-blood stability as an in vitro surrogate for CLeh primarily involving GSH conjugation catalyzed primarily through GST pi. Results thus far have suggested that blood (or rhGST P1-1) may adequately serve as an in vitro surrogate for CLeh via GSH conjugation, in part due to the dominant presence of pi class GST in erythrocytes and in many extrahepatic organs (Marcus et al., 1978; Awasthi et al., 1994; Whalen and Boyer, 1998), as well as the promiscuous substrate characteristic of GST enzymes. Initial evaluations suggested that the algorithm developed in rats may also be useful to predict pharmacokinetics across species when species-specific parameters (i.e., hepatocyte and blood stability and blood binding) were included. In addition, the CL of covalent inhibitors outside of the JAK3 inhibitor chemotype could be predicted with this pharmacokinetic algorithm. An approximate 2-fold concordance between predicted and observed CL is consistent with an acceptable success criterion for well-precedented CL mechanisms like P450 (Lavé et al., 2009). With renewed attention on targeted covalent inhibitors, this IVIVE approach may be useful to facilitate candidate optimization, selection, and prediction of human pharmacokinetic CL during discovery and development.
A similar approach for the hepatic and extrahepatic clearance prediction of the covalent inhibitors afatinib, ibrutinib, and neratinib was recently reported by Shibata and Chiba (2015). However, power-based simple allometric scaling rather than an in vitro blood approach was used for the extrahepatic component of the overall CL prediction. The predicted oral drug exposure in humans of afatinib and neratinib showed within 3-fold agreement with observed values in Phase 1 dose-escalation studies. In contrast, the CLeh of ibrutinib failed to follow simple allometric scaling, and, hence, human CL or oral exposure of this drug could not be predicted well by this approach. However, the methodology discussed here showed good predictability of clinical ibrutinib data. Since species differences in GST activity exist, the use of allometric scaling may be problematic for human CL prediction involving GST-mediated reactions. It is possible that simple allometry may work better for compounds where GSH-mediated CL is high (e.g., afatinib and neratinib) or perhaps where there are other CL mechanisms in play (e.g., nonspecific tissue binding). The current study supports a species-based algorithm strategy to assess the contribution of GST-mediated CLeh. Species differences in GSTs in rat and human are reasonably well documented (Commandeur et al., 1995). Experience with this manuscript's authors’ laboratories also suggested large species differences in GST-mediated conjugation in blood of mouse, rat, dog, monkey, and human, and was illustrated well with the observed differences seen with PF-06651600.
Target-mediated disposition is an important consideration as a potential CL mechanism. However, in cases where the molar amount of drug in the body is significantly larger than the molar amount of target, the CL related to target adduction is typically trivial and can be safely ignored in the IVIVE. This is true in the case of JAK3, as it is limited in its expression to immune cells and intestinal epithelial tissue, and is likely in the subnanomolar range. In support of this estimate, CL was shown to be independent of therapeutically relevant doses for the JAK3 covalent inhibitors studied (IC50 values in the range of ∼100–1000 nM).
It remains to be determined whether the algorithm developed in the present study may apply to covalent inhibitors consisting of different chemotypes and reactive moieties, besides acrylamides, that are typically being considered in covalent inhibitor design (Flanagan et al., 2014). It is highly recommended that researchers establish their own rat CL IVIVE with the covalent inhibitor chemotype of interest to assess intraspecies and interspecies relationships, with a particular eye toward understanding CL mechanisms. The work reported here can serve as a rational framework to ultimately predict CL of novel covalent inhibitors in humans.
To further expand on the mechanistic understanding of the present methodology, other approaches may be worth exploring as well. These include physiologic-based pharmacokinetic scaling methodology, using conjugation activity in different recombinant GST enzymes and enzyme abundance in blood and in hepatic and extrahepatic organs or tissues (Marcus et al., 1978; Whalen and Boyer, 1998). Such a physiologic-based pharmacokinetic approach may have the added benefit of evaluating the potential effect of genetic polymorphisms of different GST enzymes, such as GSTM1 in liver and P1 in blood in extrahepatic tissues (Ginsberg et al., 2009), on the pharmacokinetics and pharmacodynamics of targeted covalent drugs.
Authorship Contributions
Participated in research design: Leung, Yang, Strelevitz, Montgomery, Brown, Zientek, Gilbert, Thorarensen, and Dowty
Conducted experiments: Yang and Strelevitz
Contributed new reagents or analytic tools: Yang, Strelevitz, Montgomery, Brown, Gilbert, and Thorarensen
Performed data analysis: Leung, Yang, Strelevitz, Banfield, and Dowty
Wrote or contributed to the writing of the manuscript: Leung, Zientek, and Dowty
Footnotes
- Received August 3, 2016.
- Accepted October 25, 2016.
Abbreviations
- BPR
- blood-to-plasma ratio
- C
- concentration
- CDNB
- 1-chloro-2,4-dinitrobenzene
- CL
- clearance
- CLb
- blood clearance
- CLb int
- u, unbound intrinsic blood clearance
- CLeh
- extrahepatic clearance
- CLeh
- u, unbound extrahepatic clearance
- CLh
- hepatic clearance
- CLh int
- intrinsic hepatic clearance
- CLh int
- u, unbound intrinsic hepatic clearance
- CLint
- intrinsic clearance
- CLp
- plasma clearance
- CO
- cardiac output
- DMSO
- dimethylsulfoxide
- F
- bioavailability
- fu mic
- microsomal fraction unbound
- fub
- blood fraction unbound
- fup
- plasma fraction unbound
- GSH
- glutathione
- GST
- glutathione S-transferase
- HPLC
- high-performance liquid chromatography
- IVIVE
- in vitro-in vivo extrapolation
- JAK3
- Janus kinase 3
- LC-MS/MS
- liquid chromatography-tandem mass spectrometry
- MRM
- multiple reaction monitoring
- NCE
- novel chemical entity
- P450
- cytochrome 450
- PBS
- phosphate-buffered saline
- Q
- hepatic blood flow
- RED
- rapid equilibrium dialysis
- rhGST
- recombinant human glutathione S-transferase
- t1/2
- half-life
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}