


Abstract
Compound LY335979 is a P-glycoprotein inhibitor currently entering phase I clinical trials for potential reversal of multidrug resistance to cancer chemotherapy. In early exploratory studies, LY335979 was found to be rapidly transformed in incubations with liver microsomes from rats, dogs, monkeys, and humans. Although the parent compound was completely metabolized, no prominent metabolite peaks were observed. One peak did appear early in the time course, but it did not increase over time. In another preliminary experiment, rats were treated iv with [3H]LY335979 (prepared for pharmacology studies), and urine and bile fractions were collected. Analysis of the urine by reverse-phase HPLC with UV and radioactivity detection revealed that almost all of the material eluted with the solvent front. More than half the radioactivity in bile was accounted for by two peaks eluting earlier than the parent compound (the rest eluted at the solvent front). With both bile and the incubations with microsomes, initial attempts to isolate metabolites were not successful. There was also evidence in both systems of products derived from cleavage of LY335979 (by both further metabolism and degradation). LC/NMR was thus used to analyze materials directly in their respective matrices. AnN-oxide metabolite (LY389551) formed by oxidation of the quinoline nitrogen was identified in the microsomal incubations; in bile, three glucuronide metabolites were identified, all of which were conjugates of products formed by oxidation of the quinoline ring of LY335979. There have been few reports in the literature of LC/NMR analysis of bile, which is a more complex matrix than either urine or microsomal suspensions. However, the HPLC techniques developed in this work for the HPLC/UV and LC/MS analyses of LY335979 metabolites in the microsomal matrix and in bile proved readily adaptable for LC/NMR. Using a 500-MHz instrument, basic 1H NMR spectra could be obtained in 2–3 hr with approximately 100 ng of material in the LC/NMR microprobe. With approximately 1.5 μg of material injected onto the column,1H-1H correlation spectroscopy spectra could be acquired overnight. Along with LC/MS data, the LC/NMR technique facilitated direct identification of a number of metabolites of LY335979 at a point at which their identification by traditional methods would not have been pursued.
LY335979 (I) was developed as a potent inhibitor of P-glycoprotein, which has been implicated in the multidrug resistance phenomenon observed in cancer chemotherapy (Dantzig et al., 1996; Starling et al., 1997). This compound has been shown to effectively reverse the specific multidrug resistance phenotype in a number of in vitro and in vivo models (Dantzig et al., 1996; Starling et al., 1997) and has recently entered clinical trials. No metabolism information was known for this compound, or for any structurally similar analog. However, it was noted very early in development that the compound appeared to be rapidly metabolized. In incubations containing liver microsomes, the parent drug disappeared rapidly, but no prominent metabolites were observed. One ephemeral metabolite was noted, but LC/MS characterization of this compound was not definitive.
Integrated LC/NMR systems are now commercially available, enabling the direct analysis of very complex mixtures, and this technique is now being actively applied to drug metabolism problems (Lindon et al., 1997). There have been a number of interesting examples using LC/NMR to identify endogenous compounds and drug metabolites in human and rat urine (Lenz et al., 1996; Bollard et al., 1996; Shockor et al., 1996b,c; Sidelmann et al., 1997) and in simpler matrices such as liver microsomal incubations (Shockor et al., 1996a) and cell culture media (Cannellet al., 1997). Urine is composed of many compounds of endogenous and metabolic origin that are of low molecular weight, making it readily amenable to analysis using LC/NMR. Bile, however, is a more complex matrix than urine, possessing high bile salt concentrations, micellar components, and detergent properties. These properties of bile make it a more difficult matrix to analyze, and only a few reports described the characterization of metabolites from bile, using 1H NMR spectroscopy (Gartland et al., 1989) and coupled LC/NMR (Spraul et al., 1995) in cases where the metabolism was already largely known. In another thorough metabolism study, a number of iloperidone metabolites found in urine, bile, and microsomal incubations were characterized, in many cases with the aid of LC/NMR (Mutlib et al., 1995). That example, published in 1995, illustrates the rapid technological development of LC/NMR, because that group extracted and concentrated a number of metabolites from bile after very large doses (1000 mg/kg) were administered to rats to facilitate LC/NMR analysis. Our use of LC/NMR had previously been limited to evaluating LC/NMR performance and limits of detection for a well-known metabolism case involving urinary metabolites.1 This report describes our first attempt to use LC/NMR for the rapid and definitive characterization of metabolites in bile and human liver microsomal incubations. Specifically, the identification of four novel metabolites of LY335979 by direct LC/NMR analysis of bile or liver microsomal suspensions is described.
Materials and Methods
HPLC, LC/MS, and LC/NMR Analyses.
HPLC was performed using a Waters Instrument Co. model 600 pump, model 990 photo-diode array UV detector, and model 712 WISP autosampler. Mass spectral data were obtained with a Finnigan TSQ 7000 mass spectrometer operating in positive ion spray mode, set with a spray voltage of 4.5 kV, column temperature of 270°C, sheath gas of 80 psi N2, and auxiliary gas flow of 30 ml/min. The mass range of m/z 135–750 was scanned at 1 sec/scan. For LC/MS analysis, the HPLC eluent was split to allow a flow of 20 μl/min into the ion spray source. Collision-activated dissociation used collisional energy of −30 V, with argon set at 1–1.5 mtorr.
LC/NMR analyses were performed using a Varian Inova 500-MHz instrument equipped with a1H{13C}, pulsed-field gradient, indirect detection, LC/NMR microprobe having an active volume of about 60 μl. The LC/NMR microprobe was interfaced to a Varian model 9012 solvent delivery system, and a Varian model 9065 programmable photo-diode array UV detector was used to provide UV spectrum peak detection. The 2H resonance of D2O provided the field-frequency lock, and the spectra were centered on the methyl resonance of acetonitrile. Suppression of resonances from HOD and the methyl of acetonitrile was accomplished using a train of four selective WET2 pulses, followed by a B0 gradient pulse (Ogg et al., 1994;Smallcombe et al., 1995). This solvent suppression scheme was followed by a 90° composite read pulse. 1H NMR spectra were acquired in a stop-flow mode using the UV maximum to trigger peak detection. After peak detection and a time delay of about 13.5 sec, the HPLC pump was stopped, trapping the peak of interest in the LC/NMR microprobe. 1H NMR stop-flow spectra were acquired using an acquisition time of 1.64 sec, a spectral width of 10,000 Hz, and 32,000 time-domain data points. WET pulsed-field gradient COSY data were obtained using the same conditions as for the one-dimensional 1H spectra, except for an acquisition time of 0.21 sec, 4000 time-domain data points, and 256 increments with 8 scans each. For convenience, the residual methyl resonance of acetonitrile was referenced to 2.0 ppm.
Materials.
[3H]LY335979·3HCl [1-[4-(trans-11,11-difluorodibenzo[b,e]-bicyclo[5.1.0]oct-5-yl)piperazin-1-yl]-3-(6,8-ditritioquinolin-5-yl)oxy-R-2-propanol, trihydrochloride salt] (specific activity, 51 Ci/mmol) was prepared at Amersham (lot TRQ7711), and its radiochemical purity was found to be 99.3% by HPLC analysis. Unlabeled LY335979·3HCl (lot PA-18366059; 98.1% by HPLC analysis) was prepared at Eli Lilly and Co.
Independent synthesis of the N-oxide metabolite LY389551 was accomplished according to the procedure of Pfister et al.(1995). The intermediate 5-(2,3-epoxypropoxy)quinoline was oxidized using standard conditions (1 equivalent ofm-chloroperbenzoic acid in methylene chloride at 25°C) to yield the expected N-oxide. This material was subsequently reacted with difluorocyclopropyldibenzosubarylpiperazine to yield LY389551. See Results (especially fig. 3) for LC/NMR characterization data compared with data for the metabolite identified as LY389551.
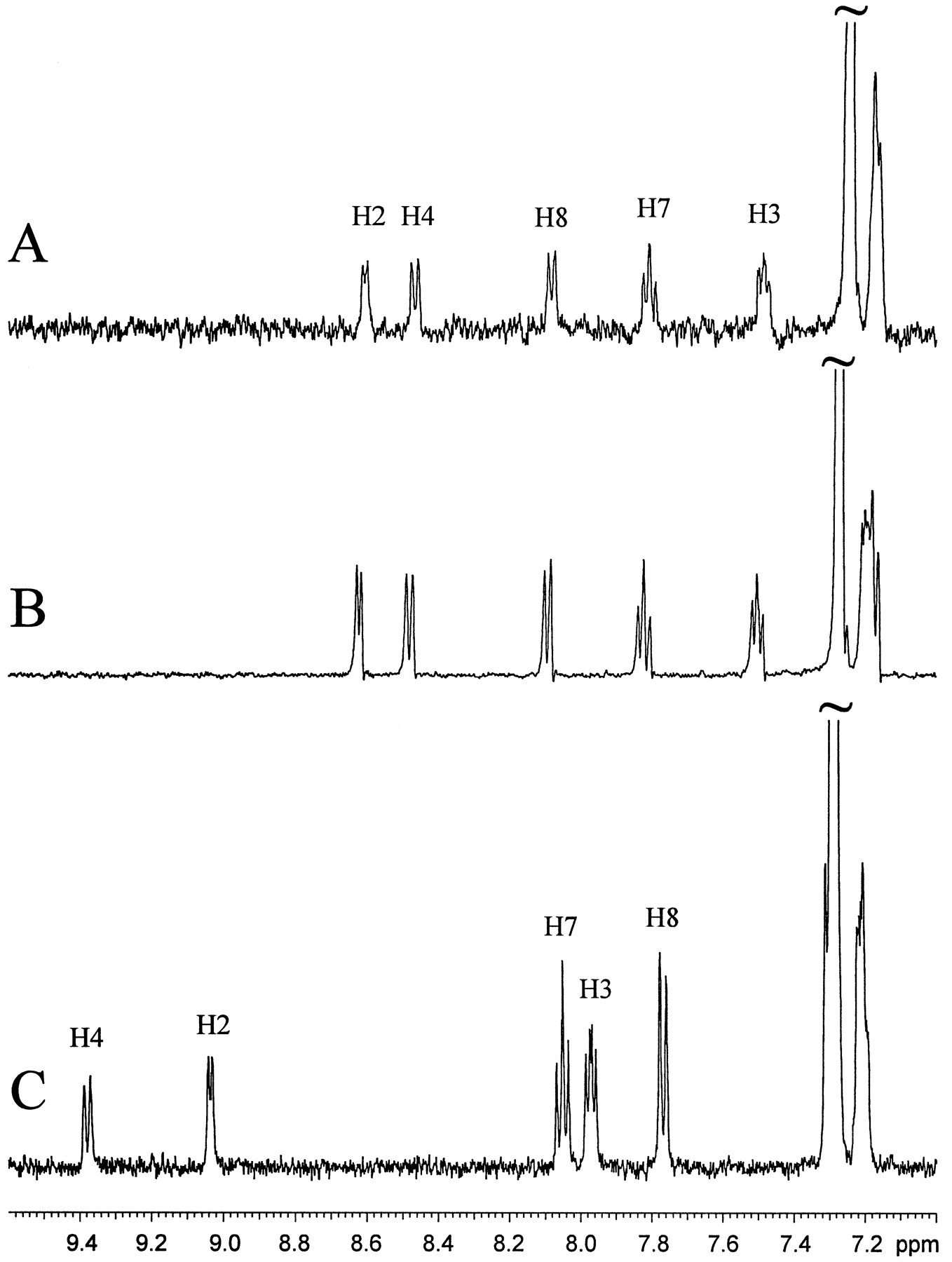
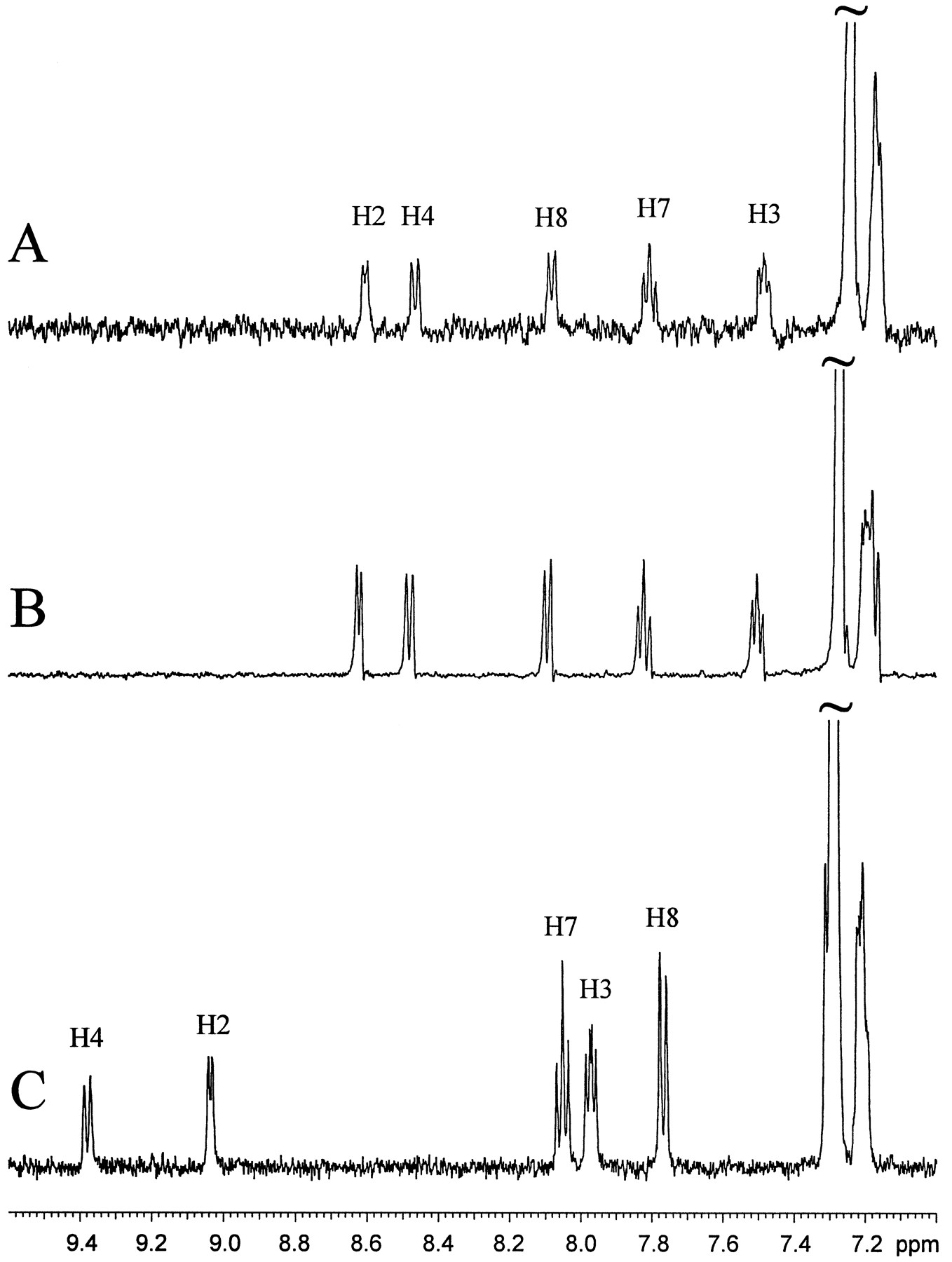
LC/NMR stop-flow 1H spectra of LY389551 generated from a human liver microsomal incubation of LY335979 (A), synthesized LY389551 (B), and the parent compound LY335979 (C).
LC/NMR analysis was carried out as described in Materials and Methods.
Microsomal Incubations.
Liver microsomes (final concentration of 1 mg/ml, as determined colorimetrically) (Lowry et al., 1951) were prepared by standard methods (van der Hoeven and Coon, 1974) and preincubated at 37°C with final concentrations of 100 mM sodium phosphate, pH 7.4, 5 mM magnesium chloride, 2 mM NADPH, 10 mM glucose-6-phosphate, and 2 units of glucose-6-phosphate dehydrogenase, in a 1-ml incubation volume. Reactions were initiated by the addition of LY335979. Incubation times for the measurement of disappearance of LY335979 over time for rat microsomes were 5, 15, and 30 min, for dog microsomes were 10, 20, and 40 min, and for monkey and human microsomes were 1, 2, and 3 hr. The final concentration of LY335979 was 5 μM. Incubations were stopped by placement in an ice or dry ice/acetone bath. An equal volume of acetonitrile was added to each sample, which was then centrifuged before an aliquot (typically 150 μl) was subjected to analysis.
Analysis of these incubations was performed by HPLC using both UV detection (λmax, 240 nm) and fluorescence detection (excitation, 240 nm; emission, 415 nm). A Tosohaas Super ODS HPLC column (4.6 × 250 mm; pore size, 5 μm) was eluted, at 0.8 ml/min, with 30% acetonitrile/35 mM sodium acetate buffer, pH 7, for 2 min, followed by a linear change to 60% acetonitrile in 20 min. The results shown in fig. 1 are based on these chromatographic analyses (typical traces shown in fig.2).

Disappearance of LY335979 after incubation with rat, dog, monkey (Cyn., cynomolgous;Rh., rhesus), or human liver microsomes.
LY335979 (2.6 μg/ml) was incubated with a liver microsomal suspension from the listed species, and its concentration at the times shown was estimated using the HPLC/fluorescence assay described inMaterials and Methods.

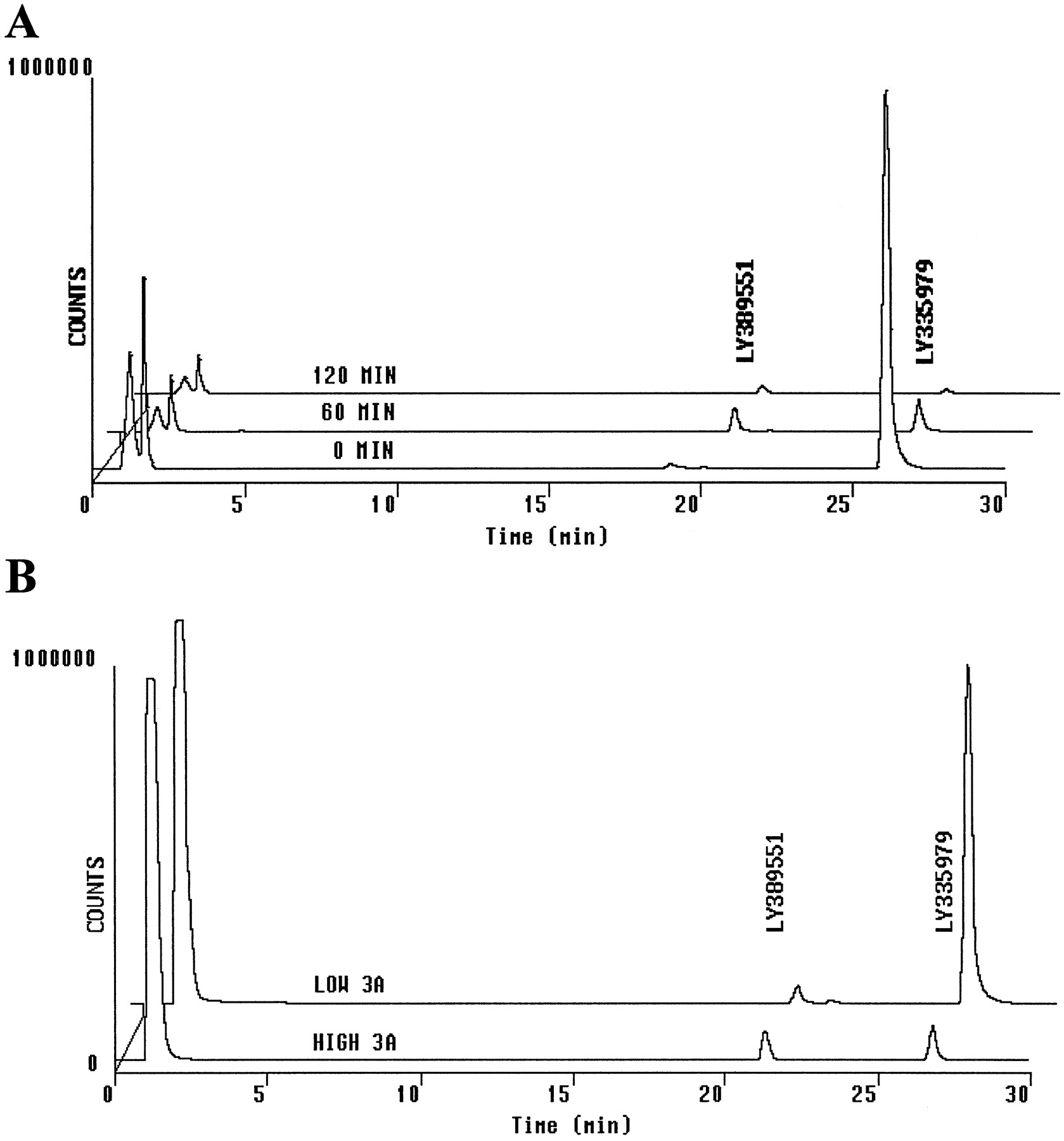
HPLC/fluorescence analysis of human liver microsomal incubations with LY335979.
A, HPLC/fluorescence analysis of an incubation of LY335979 (2.6 μg/ml) with a pooled (N = 14) human liver microsomal suspension. Analysis of selected time points from fig.1 is shown. B, HPLC/fluorescence analysis of samples after a 1-hr incubation of LY335979 with human liver microsomes from samples relatively high (HIGH 3A) or low (LOW 3A) in CYP3A catalytic activity.
Preparation and Use of Human Liver Microsomes.
Human liver samples were obtained from the liver transplant units at the Medical College of Wisconsin and the Medical College of Virginia, under protocols approved by their committees for the conduct of human research. Microsomes were prepared from the human liver specimens by differential centrifugation (van der Hoeven and Coon, 1974), and protein concentrations were determined colorimetrically (Lowry et al., 1951). These human liver microsomes have been phenotyped for 11 human liver CYP forms (Wrighton and Stevens, 1992). After preliminary screening of LY335979 metabolism using these microsomes, members of the CYP3A subfamily were implicated as the primary catalyst for metabolism of the compound. Microsomes prepared from liver HL-O of this bank (Wrighton and Stevens, 1992), which are known to possess relatively high CYP3A4 catalytic activity (7897 pmol of 1′-hydroxymidazolam formed/min/mg of protein), converted substantially more LY335979 to oxidized species, compared with microsomes prepared from liver HL-A, which possess relatively low CYP3A4 catalytic activity (988 pmol of 1′-hydroxymidazolam formed/min/mg of protein), as illustrated in fig. 2. Subsequently, microsomes with the highest levels of CYP3A4 (HL-O) were used to generate larger quantities of anN-oxide-metabolite of LY335979 than would be obtainable from microsomes with low or average levels of CYP3A4.
Rat Metabolism Studies.
Male Fischer 344 rats (204 ± 4 g) were prepared with a bile cannula surgically implanted through a tether system (Johnson and Rising, 1978), to allow movement of the rats in the Nalgene metabolism cage, which was equipped with a refrigerated collection system to keep bile and urine samples frozen during the collection period. Doses of 20 mg/kg [3H]LY335979 (2.5 μCi/mg) were given iv to four rats, and urine and bile were collected for the time periods listed in table 1. Animals were allowed food and water ad libitum (Purina chow 5002) after the dose. Tritium in aliquots of the dose solutions (before and after dosing) and in bile and urine samples was determined by scintillation counting (ReadySolv HP; Beckman) using a TriCarb system (Packard Instrument Co.). Efficiencies were ≥50% for 3H, using an external standard. To assess the loss of tritium in each sample, presumably by exchange with water, samples of bile and urine were evaporated to dryness under a nitrogen stream and redissolved in water, and the residual radioactivity was again measured by scintillation counting. Residual tritium in one rat carcass was determined by its dissolution in 2.5 volumes (v/w) of boiling 10% KOH in absolute ethanol. To avoid loss of tritium as3H2O, the carcass was sectioned in small pieces to allow placement in a round-bottomed flask, to which a reflux condenser was attached. Aliquots of 0.2 ml were then treated with 200 μl of hydrogen peroxide (30%, w/v)/glacial acetic acid and counted using 20 ml of scintillation cocktail.
Excretion of [3H]LY335979 into bile and urine in male Fischer 344 rats
For profiling of bile, each 0–6-hr sample was diluted in an equal volume of methanol and filtered (0.2-μm filters; Gelman); 150-μl injections were analyzed by HPLC with UV and radioactivity detection, using a Zorbax RX-C8 column (4.6 × 250 mm; pore size, 5 μm; Du Pont) at 30°C eluted with 20% acetonitrile/25 mM sodium phosphate, pH 2, buffer at 1.5 ml/min for 5 min, followed by a linear change in 25 min to 70% acetonitrile. Column effluent was collected in 0.5-min fractions, and 12 ml of Aquasure scintillation cocktail (DuPont) was added to each fraction for subsequent radioscintillation counting.
Analysis of Bile Metabolites by LC/MS.
A Zorbax RX-C8 HPLC column (4.6 × 250 mm; pore size, 5 μm) operating at 25°C and a flow rate of 1.5 ml/min, split 1:3 (375 μl to the electrospray source and 1125 μl to the radiochemical detector), was used to separate two major preparative HPLC-isolated radiopeaks separated under these same HPLC conditions from a 0–6-hr fraction of bile isolated from rats dosed with 20 mg/kg [3H]LY335979. Using a 25 mM ammonium acetate buffer containing 10% (v/v) methanol and acetonitrile, an initial mobile phase composition of 17% acetonitrile/83% buffer was maintained for 5 min before linear ramping to 50% acetonitrile in 25 min and maintenance of that composition for 10 min. Mass spectral conditions are given above.
Analysis of Microsomal Incubations and Bile by LC/NMR.
For the human microsomal incubations used for the generation of the metabolite of LY335979 for the LC/NMR analysis, the incubation conditions described above in Microsomal Incubations were used, except that an NADPH-generating system was not used and a 2 mg/ml concentration of the high-CYP3A microsomal protein was incubated for 1 hr at 37°C. The final concentration of LY335979 was 50 μM in the incubation mixture, and the reaction was terminated by freezing the sample in a dry ice/acetone bath. After lyophilization, the fluffy beige residue was resuspended in 0.5 ml of a 1:1 mixture of acetonitrile/deuterium oxide for LC/NMR analysis.
A sample of the reconstituted human microsomal suspension containing approximately 1.1 μg of the metabolite LY389551 (based on the assumption that this metabolite possesses a UV molar extinction coefficient comparable to that of the parent compound) was injected onto a Waters Symmetry ODS HPLC column (3.9 × 150 mm; pore size, 5 μm) operating at 25°C, at a flow rate of 1 ml/min. Initial mobile phase conditions were 20:80 acetonitrile (Omnisolve)/20 mM sodium phosphate (prepared using Aldrich D3PO4, 85%, w/v, in D2O with 99.9 atom % deuterium) in D2O. The composition was maintained for 2 min before a linear gradient change in 20 min to 75% acetonitrile, which was then maintained for an additional 5 min. Under these conditions, the 1H NMR spectrum of metabolite LY389551 shown in fig. 3A was derived from an initial experiment (involving 7700 acquisitions in 3.5 hr) with approximately 100–200 ng of material in the flow cell (table2). Using a synthetic standard of LY389551, acquisition of basic 1H NMR and WET gradient COSY spectral data was possible with approximately 20 μg of the material in the LC/NMR microprobe (table 2).
NMR data for LY335979 and the metabolite LY389551 ( II)
Additional bile samples from surgically cannulated male Fischer 344 rats dosed iv with 30 mg/kg LY335979·3HCl were collected over 0–6 hr for LC/NMR analysis. Neat bile that had been filtered through 0.45-μm Z-spin filters was injected onto a Zorbax RX-C8HPLC column (4.6 × 250 mm; pore size, 5 μm) operating at 25°C, with a flow rate of 1 ml/min. The HPLC conditions were similar to those used for analysis of radiolabeled material in bile (fig.4), here using an initial mobile phase composition of 20% acetonitrile (Omnisolve) in 25 mM sodium phosphate, pH 2 (prepared using Aldrich D3PO4, 85%, w/v, with 99.9 atom % deuterium), in D2O, followed by a linear change to 70% acetonitrile in 25 min, maintenance of this composition for 2 min, and then a linear change to 80% acetonitrile in 8 min. Comparison of this HPLC analysis with both UV absorption spectra and radiochromatograms obtained from the tritiated LY335979 bile analysis (fig. 4) enabled two distinct peaks to be analyzed by LC/NMR in the stop-flow mode. Conditions for the NMR analyses are indicated above.
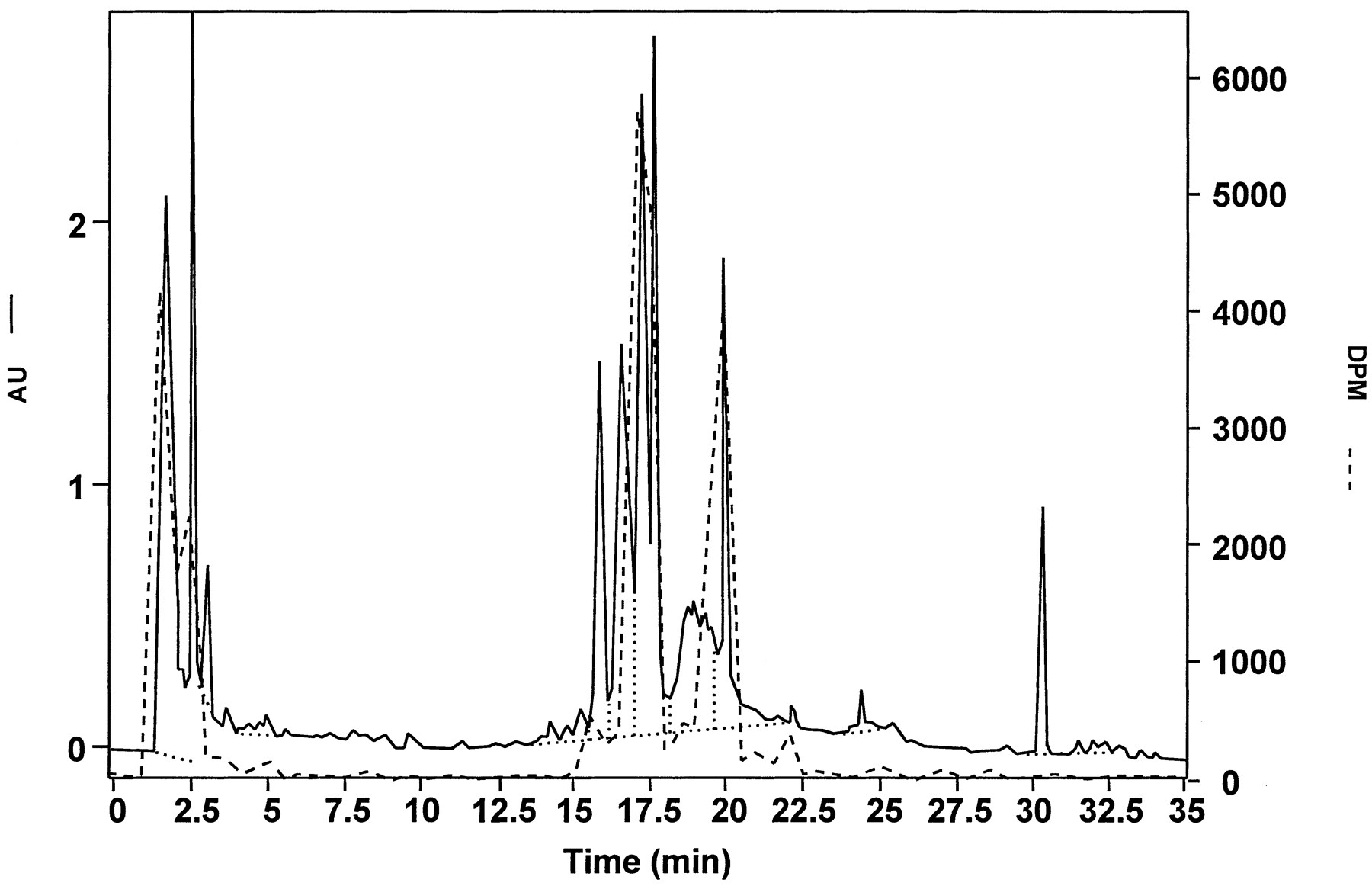
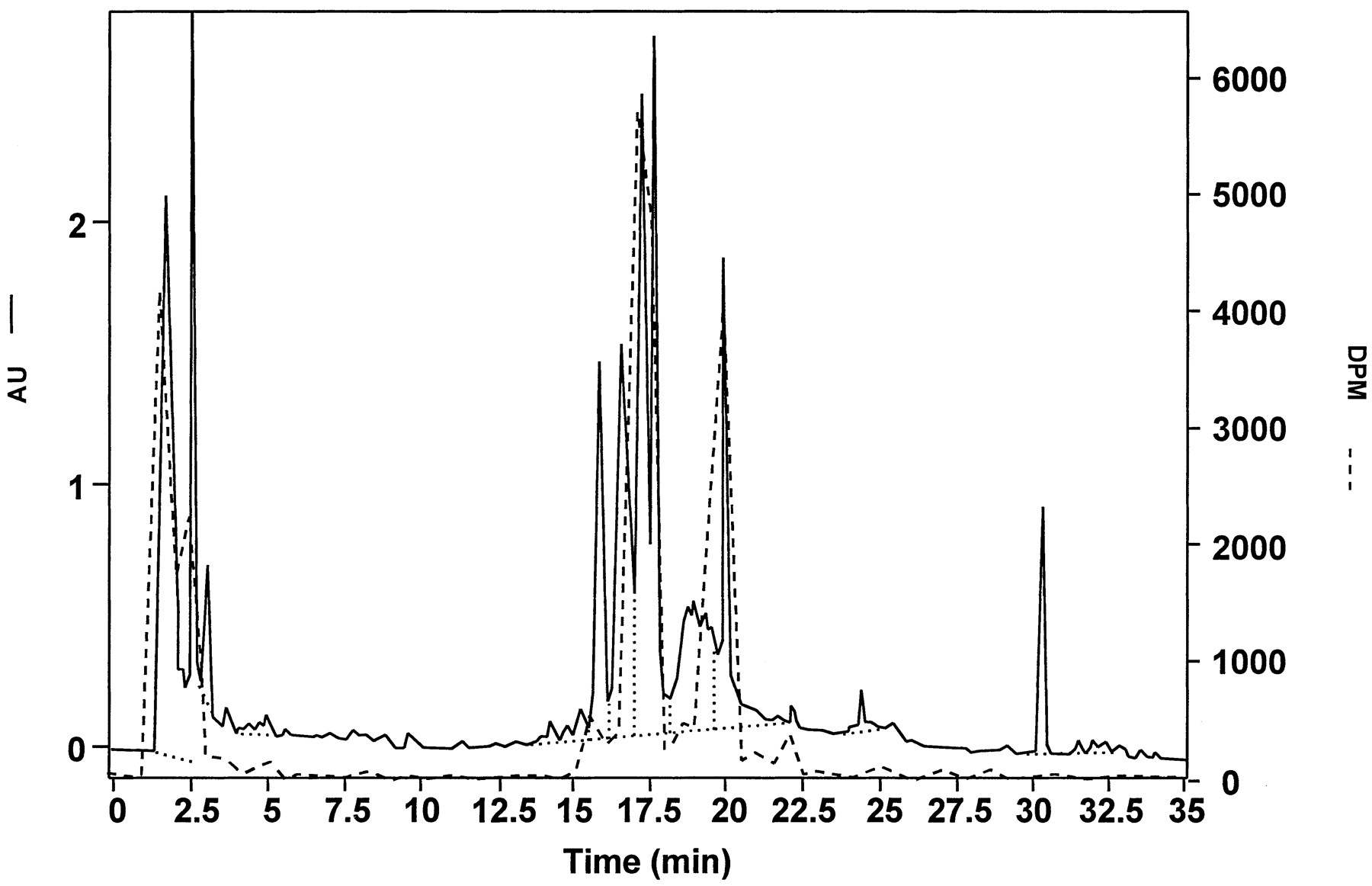
Comparison of HPLC/UV (solid line) and radiochromatographic (dashed line) analyses of bile collected in 0–6 hr from a male Fischer 344 rat (table 1, rat 1) given an iv dose of 20 mg/kg [3H]LY335979.
Bile was filtered (0.2-μm filter) and then diluted 1:1 with methanol before analysis. Scales are in absorbance units (AU) for UV detection and in decompositions per min (DPM) for radioactivity detection. Note that the materials corresponding to the peaks at RT values of 17–18 and 20 min in this radiochromatogram were subjected to LC/NMR analysis of bile, in which filtered (0.2-μm filter) bile was analyzed directly. Dotted lines define baseline for peak integration.
Results
Microsomal Incubations.
Incubations of LY335979 (I in fig.5) with liver microsomes were carried out to provide an assessment of the relative extent of metabolism in different species, compared with humans, and to possibly facilitate characterization of metabolites. As shown in fig. 1, the disappearance of LY335979 was quite rapid with liver microsomes from rats and monkeys (half-lives under these conditions of <10 min), as well as with those from dogs (half-life of approximately 10 min). The experiment indicated that metabolism with liver microsomes from humans might be slower but still extensive, with an observed half-life of roughly 20 min.
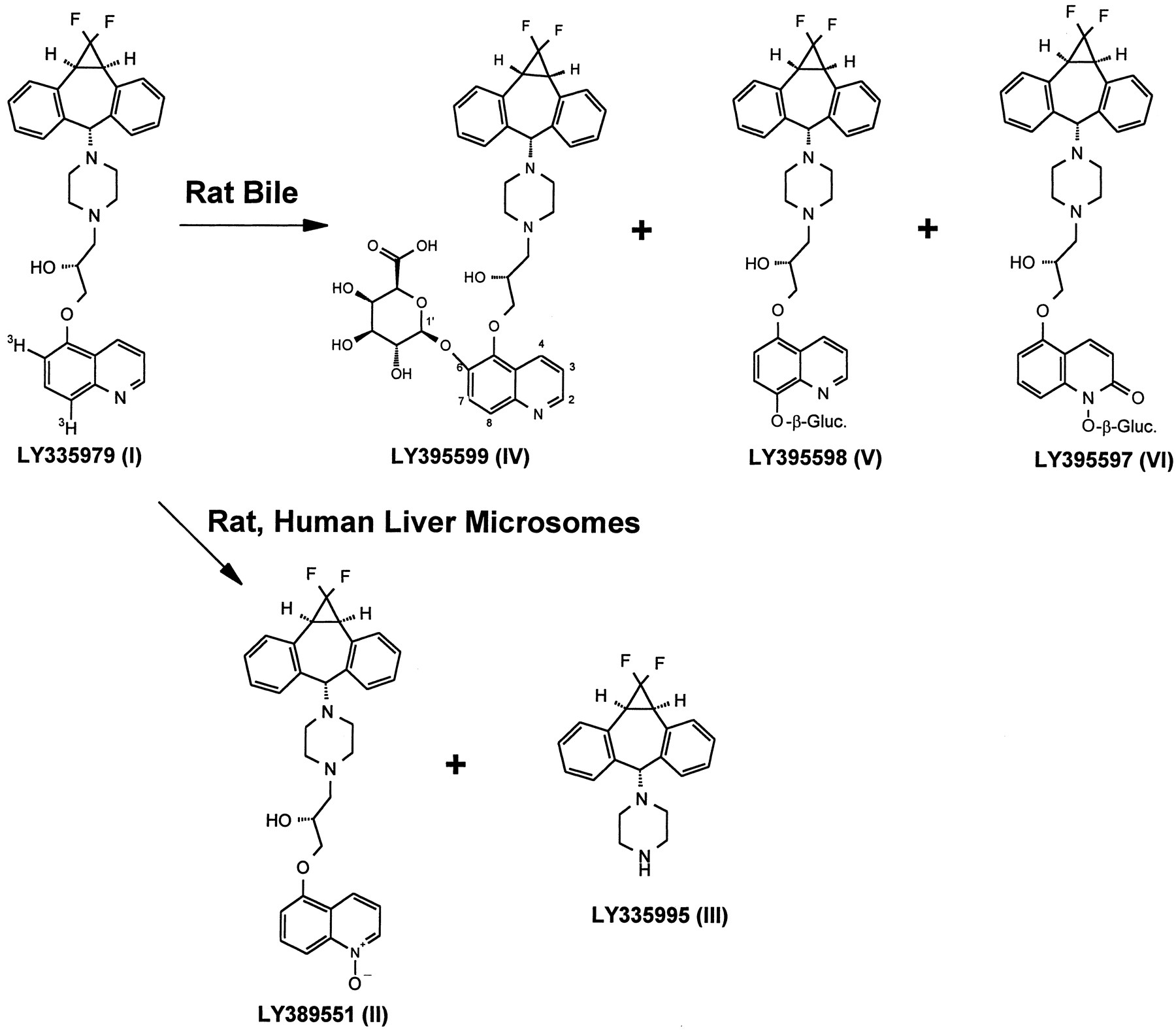
Metabolism scheme for LY335979.
Radiolabeled material was prepared with 3H in the quinoline ring, as shown.
Despite the rapid disappearance of LY335979 in vitro for all of these species, profiling of these incubations by reverse-phase HPLC did not reveal formation of any prominent metabolites. The pattern seen for pooled human liver microsomes from 14 donors (Wrighton and Stevens, 1992) (fig. 2A) was typical of all species tested. One minor metabolite (RT, 21–22 min) was observed, which eluted just before the parent compound LY335979 (RT, 26–27 min). Chromatograms with fluorescence detection (excitation, 240 nm; emission, 415 nm) are shown in fig. 2. Analysis of these incubations with UV detection at 250 nm was carried out as well but did not reveal any additional peaks. Although there appeared to be rapid formation of this metabolite (area about 5% of the initial area for the parent compound after 5 min, as judged by HPLC/UV and HPLC/fluorescence peak integration), this peak did not increase with time even though the parent peak decreased rapidly (fig.2A, 60-min chromatogram). Furthermore, as the parent compound was depleted in these incubations, the peak at 21–22 min itself decreased in area (fig. 2A, 120-min chromatogram). Also observed in these incubations, although not readily discernible in the scale reproduction of fig. 2A, was the presence of four to six other metabolites formed in much smaller amounts.
Because a loose association has been noted between substrate specificity for P-glycoprotein and that for CYP3A (Wacher et al., 1995; Scheutz et al., 1996), incubations with LY335979 were carried out with human liver microsomes possessing relatively high or low specific activities for midazolam 1′-hydroxylation, which is a measure of CYP3A activity (Wrighton and Stevens, 1992). As shown in fig. 2B, there was indeed a dramatic difference in the rate of LY335979 disappearance, with practically no conversion of LY335979 being seen after 60 min with microsomes with low levels of CYP3A. Consistent with a faster rate of LY335979 conversion with microsomes containing high levels of CYP3A, there was a higher steady-state concentration of the metabolite at 21–22 min in incubations using these microsomes. The microsomes containing high levels of CYP3A were thus used for the LC/NMR characterization work described below.
Characterization of Metabolites Produced In Vitro.
With slight modification of the reverse-phase HPLC conditions described above, LC/MS analysis of both rat and human in vitroincubations was carried out. Two notable peaks related to the parent drug were found in the LC/MS analysis. One was a very prominent peak, with a RT corresponding to about 15–17 min for the system shown in fig. 2; it did not, however, correspond to any major peak observed in the HPLC/UV or HPLC/fluorescence analyses. Its base peak was at m/z 327, indicating that it might contain the substructure corresponding to LY335995 (III in fig. 5). It was shown to be LY335995 by LC/MS comparison with the authentic material, which is an intermediate used in the synthesis of LY335979. Note that the main chromophore for LY335979 is the quinoline ring, which is lost in LY335995. Despite this, a small peak corresponding to LY335995 could be observed in the HPLC/UV analysis, and the peak was present only in incubations carried out in the presence of NADPH.
For the metabolite eluting at 21–22 min (fig. 2), a base peak ofm/z 544 was observed by LC/MS. As shown in fig.6, collision-activated decomposition led to fragments at m/z 191, 221, 241, and 383, which are the same as those for LY335979. It was thus postulated that this biotransformation involved addition of oxygen somewhere on the quinoline ring. However, LC/MS could not provide any indication of the position of substitution. Initial attempts to isolate material by preparative HPLC for additional analysis by NMR failed. The material did not appear to survive a procedure that involved collection of the eluent, evaporation under gentle conditions (by either rotary evaporation or lyophilization), and extraction or resolubilization of the residue.
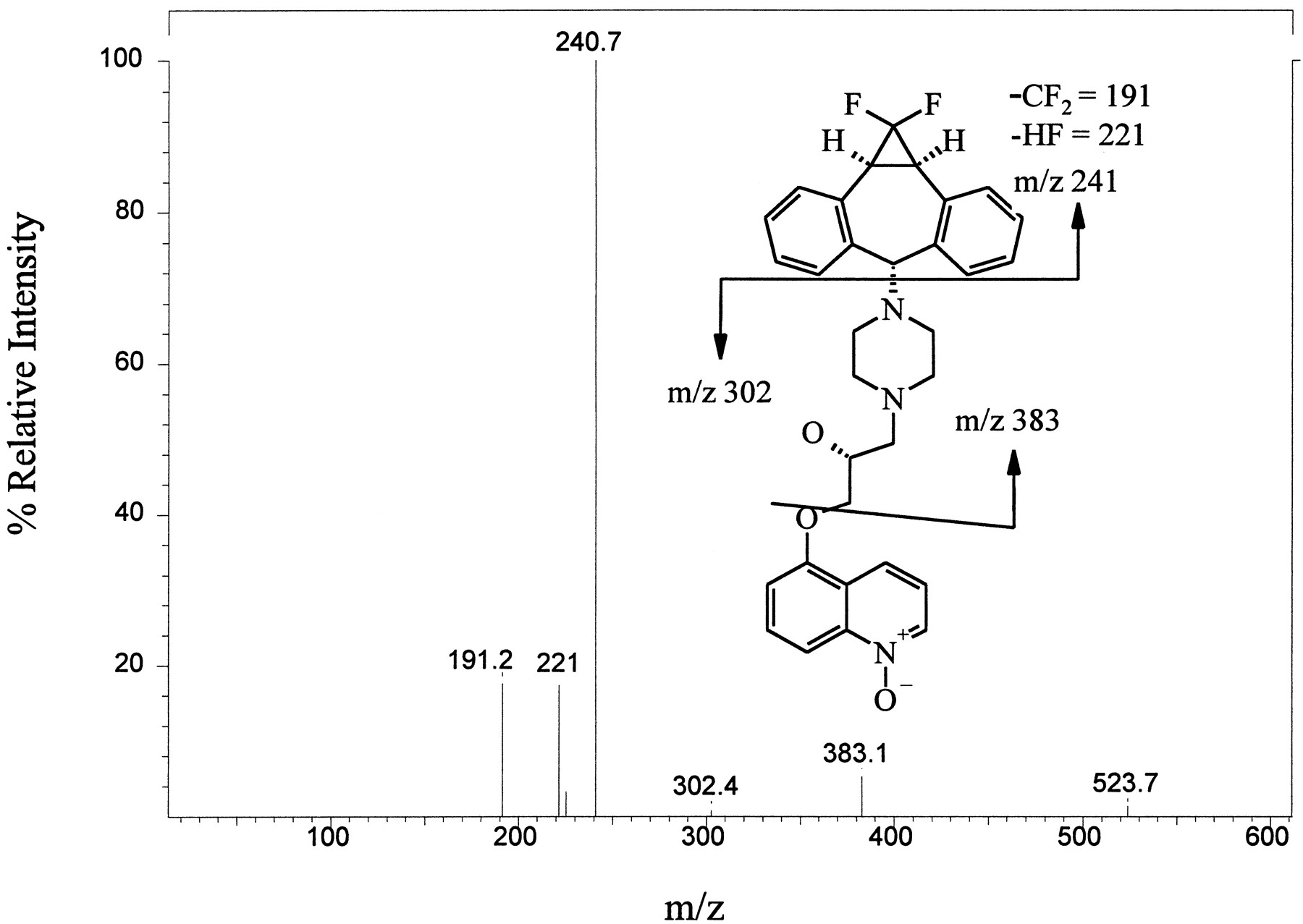
Daughter-ion spectrum of LY389551 (m/z 544) found in incubations of LY335979 with rat liver microsomes.
LC/MS/MS analysis of the material corresponding to a RT of 21–22 min in the HPLC/fluorescence analysis (fig. 2) was carried out by collision-activated decomposition of the m/z 544 base peak. See Materials and Methods for details.
Analysis of liver microsomal incubations by LC/NMR was thus attempted. Our initial evaluation of LC/NMR1 indicated that, for a molecular weight of about 500, as little as 100 ng of analyte in the LC/NMR microprobe was sufficient to readily yield1H NMR spectra in a few hours. Accordingly, increase of the amount of the in vitro metabolite was accomplished using an incubation of human liver microsomes relatively high in CYP3A, with LY335979 at a relatively high concentration of 50 μM (see Materials and Methods). The lyophilized material was then subjected to LC/NMR. Stopped-flow analysis of the metabolite peak, which eluted just before the parent compound and possessed a similar characteristic UV absorption spectrum, was performed. With 7710 scans in 3.5 hr, a very high-quality 1H NMR spectrum was obtained (fig. 3A). A comparison of the coupling patterns and chemical shifts for the metabolite and the parent compound LY335979 indicated that all proton resonances in the heterocyclic and benzenoid portions of the quinoline ring showed the same coupling patterns, but with slightly different chemical shifts (table 2; fig. 3, A and C). These data indicating an absence of hydrogen substitution in the quinoline ring implicate formation of an N-oxide. The change in chemical shifts for H2 and H8, compared with the parent compound LY335979 (−0.44 and 0.31 ppm, respectively), can be attributed to the magnetic anisotropy and electric field effects of an N—O group, similar to observations for the relative chemical shifts of quinoline vs. quinolineN-oxide (Barbieri et al., 1975). After assignment of the N-oxide structure to this metabolite, this compound was synthesized, as described in Materials and Methods. The stop-flow 1H NMR spectrum of the synthesizedN-oxide is shown in fig. 3B. Because sufficient synthesized material was available, it was possible to assign all of the protons and confirm the location of H6 (table 2). The assignment of this metabolite as the N-oxide metabolite of LY335979 (LY389551 in fig. 5) is also consistent with mass spectral evidence suggesting an increase of 16 mass units to the quinoline portion of the molecule (fig. 6).
Rat Bile Cannulation Study.
At the time of these studies, [3H]LY335979 labeled with tritium in two positions of the quinoline ring system (fig. 5) was available. A metabolism study was thus carried out in four bile duct-cannulated rats dosed iv with 20 mg/kg [3H]LY335979; both urine and bile fractions were collected. As shown in table 1, in 0–6 hr an average of about 6% of the dose was recovered in urine and about 27% in bile. After 72 hr, about 20% of the dose was recovered in urine and 46% in bile, for a total recovery of 66% in this experiment.
Tritium is not ideal for use in metabolism studies, because of a greater possibility of loss of the label through exchange with protons of water and other compounds. Although there was no indication of loss of tritium from the parent compound LY335979 under the conditions used in any of these studies (data not shown), the lability of tritium in metabolites was not predictable. Accordingly, 100-μl aliquots of the urine and bile fractions collected from one rat were evaporated to dryness, and the residue was counted for tritium. The results (table 1) indicated that essentially all of the tritium in the 0–6-hr urine and bile fractions was nonvolatile, which presumably meant that there was little or no 3H2O in those samples. However, in the samples from later times, a significant portion of the tritium was lost upon evaporation of the sample (table1). The residual radioactivity in one rat was also assessed, with a procedure in which the rat carcass was boiled in a KOH/ethanol/water solution. To avoid loss of tritium, this procedure was carried out in a round-bottomed flask with a reflux condenser attached. In that rat carcass, an additional 13% of the dose was found, for a total recovery of 81% for that animal. Of course, if exchange with water occurs, it would be expected that some loss of3H2O would occur throughout the experiment (for example, through breathing). It should also be noted that, because exchange of tritium from metabolites and perhaps the parent compound did occur, the recovered nonvolatile radioactivity would itself not necessarily represent just the parent compound or metabolites of LY335979 but could indicate exchange of protons with other nonvolatile materials. Although a quantitative assessment of mass balance was not possible, the use of [3H]LY335979 did provide an estimate of the overall disposition of LY335979, in addition to establishing early directions for metabolism studies.
Rat Bile Metabolites.
As discussed above, chromatographic analysis of the bile derived from animals dosed iv with 20 mg/kg [3H]LY335979 was used to locate potential metabolites. As shown in fig. 4, HPLC/UV analysis of the 0–6-hr bile sample was quite complex, but analysis by HPLC/radioactivity revealed two peaks containing tritium that were well retained on the column, in addition to tritium eluting at the solvent front. As described above, the tritium eluting at the solvent front did not appear to be primarily3H2O, but, although a number of different columns and chromatographic conditions were tried, this material could not be separated from the solvent front. The two major peaks, eluting at 17–18 and 20 min in this system (fig. 4), were subjected to further analysis.
LC/MS was performed on these bile metabolites isolated by preparative HPLC after multiple injections of the 0–6-hr bile samples (seeMaterials and Methods). An ammonium acetate/methanol buffer used in concert with acetonitrile in the HPLC gradient was developed to optimize HPLC electrospray ionization. Whereas the sodium phosphate, pH 2/acetonitrile mobile phase used to initially identify tritum-containing bile constituents resolved the components better (fig. 4) than did the ammonium acetate system, its nonvolatility precluded use with electrospray mass spectrometry. Accordingly, the region from 17 to 20 min was collected by preparative HPLC and subjected to LC/MS analysis of enriched samples using the ammonium acetate system. Two large peaks, with characteristic ions atm/z 736 (RT, 22.0 min) andm/z 720 (RT, 22.24 min) in full-scan positive-ion mode, corresponded to the materials identified by HPLC with radioactivity detection (fig. 4, peaks at 20 min and 17–18 min, respectively).
Subsequent MS/MS characterization was carried out to obtain product-ion mass spectra of the peaks at m/z 736 and 720 (fig.7); this analysis showed, for both, product ions m/z 191 and 241, which are characteristic of the parent compound LY335979. The ion at m/z 241 indicates that modification of the parent drug occurred somewhere other than the dibenzosuberane moiety. Additionally, for the m/z 720 ion, an ion present at m/z 544 (also observed for LY389551) represented an ion 16 mass units greater than that for the parent compound, indicating possible addition of oxygen to LY335979 itself. The daughter-ion spectrum of the m/z 736 ion showed a product ion present at m/z 560, indicating possible addition of two atoms of oxygen to the parent compound. For both peaks (m/z 720 and 736), the remaining difference between putative mono- and dioxygenated parent adducts and their molecular parent-ion masses was 176 mass units, indicative of possible glucuronide conjugates. However, incubations of these bile metabolites with glucurase at 37°C for 1 hr failed to significantly diminish their starting amounts, thus calling into question (although not precluding) their assignment as glucuronide adducts.

Daughter-ion spectra of bile metabolites in the LC/MS analysis.
Mass spectral analyses of the base peaks corresponding tom/z 720 (RT, 17–18 min) (A) and m/z 736 (RT, 20 min) (B) in the radiochromatographic analysis shown in fig. 4 were performed. LC/MS/MS analyses were carried out by collision-activated decomposition, as described in Materials and Methods.
Using HPLC conditions slightly modified for NMR analysis (slower flow rate, no methanol dilution; see Materials and Methods), bile obtained from additional rats dosed iv with 30 mg/kg LY335979·3HCl produced two UV-distinct peaks at longer RTvalues (24.8 and 26.6 min) than were obtained with the HPLC/fluorescence conditions represented in fig. 4. There, two resolved UV-absorbing peaks at 17.34 and 17.7 min in fig. 4 corresponded to a zone of undifferentiated radioactivity that constituted about 34% of the total tritium recovered from that HPLC injection. In the LC/NMR analysis of neat bile, these corresponded to one broader, not so well-resolved peak at 24.8 min, as judged by the relative RT and the UV absorption profile (λmax, 250 nm). Similarly, the UV spectrum of the peak at 26.6 min in the LC/NMR analysis corresponded precisely to the UV absorption spectrum of the sharp UV-absorbing peak at 20 min in the HPLC/radioactivity analysis (λmax, 300 nm) shown in fig. 4; that peak constituted 21% of the total tritium recovered from that analysis.
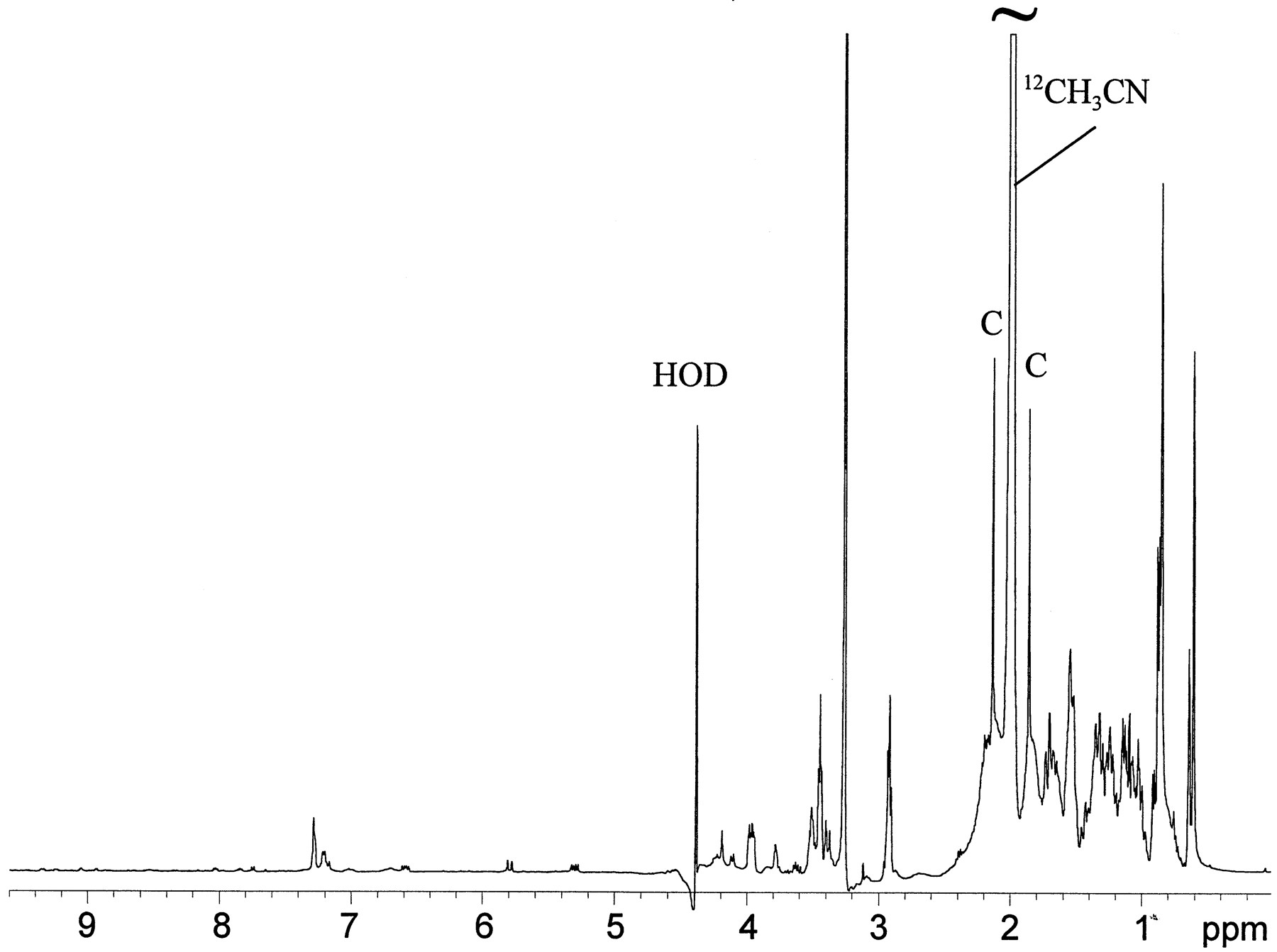
The full 1H NMR spectrum of the LC/NMR peak at 24.8 min (30,000 scans in 13.7 hr), is shown in fig.8. The residual solvent resonances were located at 4.4 and 2.0 ppm (HOD and the methyl resonance of acetonitrile, respectively). The sharp peaks (fig. 8, peaks C) located on both sides of the residual acetonitrile methyl resonance were the 13C satellite peaks of CH3CN. Because these peaks were present at levels corresponding to 0.55% of the integrated intensity of the12CH3CN resonance, they could serve as useful internal markers. Clearly, even though this was a spectrum of an isolated HPLC peak, the matrix contained within that peak volume was complex (fig. 8). Not only were the analytes of interest present, but also there was substantial chemical noise from components of the bile matrix (fig. 8, 2-4-ppm region). However, the1H NMR spectrum of the peak at 24.8 min contained two distinct sets of resonances clearly corresponding to metabolites of LY335979, which can be seen in the expansion of the aromatic region shown in fig. 9A. Note that LC/MS analysis of this material indicated a parent peak ofm/z 720 for both of the constituents, which corresponds formally to addition of one oxygen and a glucuronide moiety to LY335979. As table 3 shows, the chemical shifts, coupling patterns, and coupling constants allowed the structures of the two molecules to be deduced. Repeat analysis involving isolation of the leading edge of the peak in the LC/NMR microprobe clearly indicated only one set of resonances consistent with one set of the previously identified mixture of two molecules (fig.9B), thereby confirming the presence of two materials. For both molecules, the appearance of distinct sets of doublets at 5.28 and 5.32 ppm was immediately indicative of the anomeric proton of a glucuronic acid β-linked via oxygen substitution eitherortho and para to the ether linkage of the quinoline moiety, as shown in fig. 5. One glucuronide adduct, LY395599, is readily distinguishable from the other, LY395598, by the chemical shifts of the two sets of doublets in each compound.

LC/NMR stop-flow 1H spectrum corresponding to the peak with a RT of 24.8 min in rat bile.
LC/NMR analysis of the material corresponding to the peak with a RT of 17.3–17.7 min in the radiochromatographic analysis of bile (fig. 4) was carried out as described in Materials and Methods. Peaks C, 13C satellite peaks of CH3CN.

Expansion of the 5.0–9.5-ppm region of the stop-flow 1H spectrum shown in fig. 8, corresponding to the peak with a RT of 24.8 min in rate bile (A), and analysis of the leading edge of that peak in a subsequent HPLC experiment (B).
NMR data for LY395599 (IV), LY395598(V), and LY395597 (VI)
For the peak at 26.6 min analyzed in the stop-flow mode, 26,000 scans in 11.8 hr were sufficient to yield a 1H NMR spectrum, for which the resonances and coupling patterns are shown in table 3. This peak corresponded to the material shown by LC/MS to have two additional oxygen atoms plus a glucuronide moiety (m/z736), compared with LY335979. As was observed in the spectra described above, a doublet at 5.22 ppm is indicative of an anomeric proton of a glucuronic acid. Additionally, the triplet with a chemical shift at 7.6 ppm represents the aromatic proton at position 7, which coupling data indicate is flanked by two doublets at 6.85 and 7.53 ppm, representing the aromatic protons at positions 6 and 8, respectively. Substitution in the benzenoid portion of the quinoline ring was thus precluded. The observation that only five and not six unique proton resonances were found for the m/z 736 ion peak indicated that substitution had occurred in the pyridine ring of the quinoline, at either position 2, 3, or 4. The two remaining resonances at 6.68 and 8.29 ppm form an AB spin system consistent with those expected for an α-pyridone (a ketone ortho to the pyridine nitrogen) and so constitute adjacent vinylic protons located at positions 3 and 4, respectively, of the quinoline ring system. The O-β-linked glucuronide on the pyridine nitrogen of the quinoline ring shown in the cyclic lactam LY395597 in fig. 5 represents the structure of the bile metabolite entirely consistent with NMR and mass spectrometric data.
Discussion
Preliminary studies related to the metabolism of the P-glycoprotein inhibitor LY335979, which is being developed for potential reversal of the multidrug resistance seen in cancer chemotherapy (Dantzig et al., 1996; Starling et al., 1997), showed that there was rapid transformation of LY335979 by microsomes from rat, dog, monkey, and human liver. Although the approximate half-life values of 10 min in rat, dog, and monkey liver microsomal incubations and 20 min in human liver microsomal incubations, extrapolated from inspection of parent decay curves (fig.1), indicated rapid metabolism of LY335979, no major products with any appreciable retention in the reverse-phase chromatography systems used (fig. 2) were observed. Attempts were made to isolate the one prominent, although minor, observed metabolite (LY389551 in fig. 2), but these were not successful. It was possible that the primary metabolite(s) might be intrinsically unstable, subject to further metabolism, or both. LC/NMR was thus carried out to analyze the microsomal mixture directly. For this purpose, it was found that decomposition of the metabolites was avoided by direct lyophilization of the microsomal suspensions, followed by extraction of the solid residue with a 1:1 mixture of acetonitrile/D2O. In combination with LC/MS data obtained earlier, the LC/NMR analysis allowed unambiguous assignment of this in vitro metabolite as LY389551, corresponding to the N-oxide of LY335979. It should be noted that, although the other material identified in microsomal incubations, LY335995 (III in fig. 5), is a potential decomposition product of LY335979, it was a product observed only in conjunction with disappearance of the parent compound in incubations carried out with NADPH.
In the identification of microsomal metabolites described above, incubation of human liver microsomes relatively high in CYP3A catalytic activity (Wrighton and Stevens, 1992) led to much more rapid disappearance of LY335979 and a higher steady-state level of the metabolite during the course of the incubation than seen in incubations with human liver microsomes relatively low in CYP3A activity. An overlap of substrate specificity for CYP3A with that for P-glycoprotein has been noted (Wacher et al., 1995; Scheutz et al., 1996), which led us to carry out a preliminary study showing that LY335979 is a competitive inhibitor of CYP3A catalytic activity, with a Ki of 3.8 μM.3 Our results do not prove that CYP3A is the isozyme responsible for formation of LY389551 or the other metabolites resulting from transformations of the quinoline ring system. Nonetheless, these observations suggest that CYP3A is somehow involved in the metabolism of LY335979, and this will be studied as the compound is further developed.
Using the available radiolabeled material [3H]LY335979, it was possible to approximately define the disposition of the compound in bile-cannulated rats. Although exchange of the tritium label precluded quantitative recovery (table 1), it was possible to discover three major bile metabolites in the bile samples collected in 0–6 hr. As with the microsomal metabolite LY389551 (II in fig. 5), initial attempts to isolate these metabolites for NMR identification were not successful, with poor recovery of the material and appearance of various decomposition products (such as III in fig. 5, found only as a decomposition product from the handling of bile samples and not as a metabolite).
The isolation of bile metabolites is greatly complicated because of the inherent complexity of the matrix, which is related to the combined excretory and secretory properties of the fluid, resulting in numerous endogenous polar, nonpolar, and mixed interferences (Lindon et al., 1997; Gartland et al., 1989; Spraul et al., 1995; Mutlib et al., 1995). However, after location of radiopeaks in the bile using the HPLC conditions illustrated in fig. 4, three major areas of radioactivity were discerned. Attempts to better retain the solvent-front radioactivity using different chromatographic conditions or to directly analyze that material were not successful, but direct LC/NMR analysis of the bile using chromatographic conditions slightly different from those illustrated in fig. 4, in conjunction with LC/MS data, enabled us to identify the three major bile metabolites of LY335979 shown in fig. 5. Consistent with previous work on identification of sulfonylurea metabolites using LC/NMR,1 isolation of approximately 100 ng of material in the LC/NMR microprobe permitted1H NMR analysis.
The mass spectrometric evidence obtained earlier, suggesting that all metabolic transformations occurred in the quinoline portion of LY335979, made the subsequent LC/NMR analysis substantially easier. Chemical shifts for the protons associated with the quinoline moiety were easily discerned, as indicated in tables 2 and 3 and shown in figs. 3, 8, and 9. The resonances encompassing the numerous protons from the dibenzosuberane and piperazine portions of the molecule occurred at chemical shifts largely obscured by acetonitrile, the solvent used for HPLC (fig. 8). However, it should be noted that this can be substantially overcome by using per-deuterated acetonitrile in the LC/NMR analysis.
Whereas one metabolite identified in vitro (LY335995,III in fig. 5) is, formally, a cleavage product of LY335979, the other four metabolites (II andIV-VI in fig. 5) all involve transformations of the quinoline moiety of LY335979 and are consistent with what is known of the metabolism of that ring system. Transformations of the quinoline ring were first noted in studies of the metabolism of quinidine [the difficult early work has been nicely reviewed in the contribution byBrodie et al. (1951)], which has subsequently been studied thoroughly (Beedham et al., 1992; Bonora et al., 1977; and references cited therein). In addition to other quinoline-containing compounds such as amodiaquine (Jewell et al., 1995), the metabolism of quinoline itself is now known (Bott and Lingens, 1991; and references cited therein). In all of these examples, oxidation of the quinoline ring nitrogen occurs to yield anN-oxide (Beedham et al., 1992; Bonora et al., 1977; Jewell et al., 1995; Bott and Lingens, 1991), similar to the formation of II in this case. The bile metabolite VI probably represents further biotransformation of II, but the mechanism of its formation is not clear. In the cases discussed above, formation of an amide (2-quinolone) was observed, which could be the result of direct oxidation of the quinoline moiety or could involve rearrangement of theN-oxide (Yousif et al., 1982). Further oxidation of the N-oxide II or of an intermediate amide (not observed in these studies) would give VI (for example,Idowu et al., 1995).
Oxidation ortho or para to the ether linkage in the quinoline ring of LY335979, to give IV or V, respectively, is also logical. As with the analogous naphthalene ring of another well-studied example, propanolol, this might involve intermediate epoxide formation (Narimatsu et al., 1995; and references cited therein). As seen with all three bile metabolites, phase II metabolism to give glucuronide conjugates would then be expected (Green et al., 1994).
Consistent with the observations in microsomal incubations, profiling of the 0–6-hr rat urine samples showed significant polar radioactivity eluting at the solvent front of the reverse-phase chromatography systems used, but no parent compound. Subsequent incubations of [3H]LY335979 with microsomes showed that the material eluting at the solvent front was volatile, most probably3H2O (data not shown). In the urine samples, however, radioactivity in the samples collected over 0–6 hr was not volatile, although the samples collected at later times showed increasing volatility of the tritium. This pattern was also observed with the bile samples, as shown by the results in table 1. Our attempts to isolate the tritiated material eluting at the solvent front in both urine and bile were unsuccessful, as were attempts to analyze these materials directly by LC/NMR and LC/MS. Although further work needs to be carried out to elucidate these polar metabolites, our results suggest a possible explanation. Just as is known for propanolol (Narimatsu et al., 1995), these oxidative metabolites might be less stable than the parent compound (LY335979) to cleavage of the ether linkage. Noting that III is observed as an NADPH-dependent microsomal metabolite of LY335979, it could be speculated that various oxidized quinoline fragments could account for the unidentified polar radioactivity in bile and urine. Also, whereas exchange of tritium from compounds such as the oxidation product 5-[3H]hydroxyquinoline might be more substantial than that from the parent compound (possibly explaining the formation of 3H2O in microsomes), in vivo glucuronidation of these quinoline products could preclude exchange by conjugation of the free phenolic sites. Further studies of the metabolism of LY335979 are planned.
In this work we have shown that LC/NMR can be used to directly analyze metabolites in both liver microsome and bile matrices. Direct elucidation of the chemical structures of metabolites in their native matrices using LC/NMR, without laborious isolation and collection by preparative HPLC, represents a tremendous advantage for metabolism identification. Advances in solvent suppression techniques and LC/NMR microprobe performance now make it possible to routinely obtain1H, COSY, and TOCSY data on metabolites with molecular weights of about 500, in <2 hr for 1H spectra and overnight for COSY and TOCSY spectra when at least 1 μg of material is placed on column.
Acknowledgments
Thomas Lindsey, Michael Berna, Anthony Murphy, and Dr. Todd Gillespie generated mass spectral data for these metabolites. The preparation of radiolabeled LY335979, which was indispensable for carrying out the metabolism studies, was carried out at Amersham, patterned after procedures developed by Dr. Boris Czeskis. Marie Koenig carried out the rat surgery, allowing assessment of biliary excretion. We thank all of these helpful colleagues at Lilly Research Laboratories (Indianapolis, IN).
Footnotes
-
Send reprint requests to: William J. Ehlhardt, 0825 Drug Metabolism, Lilly Research Laboratories, Lilly Corporate Center, Indianapolis, IN 46285.
-
↵1 Maple SR, Woodland JM and Ehlhardt WJ. LC-NMR detection of the metabolites of the sulfonylurea oncolytic agent LY295501 in rat urine. Submitted for publication.
-
↵3 Wrighton SA, unpublished results.
- Abbreviations used are::
- WET
- water suppression enhanced through T1 effect
- CYP
- cytochrome P450
- COSY
- correlation spectroscopy
- TOCSY
- total correlation spectroscopy
- Received June 25, 1997.
- Accepted September 23, 1997.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}