


Abstract
These studies were designed to characterize the disposition and metabolism of atomoxetine hydrochloride [(−)-N-methyl-γ-(2-methylphenoxy)benzenepropanamine hydrochloride; formerly know as tomoxetine hydrochloride] in Fischer 344 rats and beagle dogs. Atomoxetine was well absorbed from the gastrointestinal tract and cleared primarily by metabolism with the majority of its metabolites being excreted into the urine, 66% of the total dose in the rat and 48% in the dog. Fecal excretion, 32% of the total dose in the rat and 42% in the dog, appears to be due to biliary elimination and not due to unabsorbed dose. Nearly the entire dose was excreted within 24 h in both species. In the rat, low oral bioavailability was observed (F = 4%) compared with the high oral bioavailability in dog (F = 74%). These differences appear to be almost purely mediated by the efficient first-pass hepatic clearance of atomoxetine in rat. The biotransformation of atomoxetine was similar in the rat and dog, undergoing aromatic ring hydroxylation, benzylic oxidation (rat only), and N-demethylation. The primary oxidative metabolite of atomoxetine was 4-hydroxyatomoxetine, which was subsequently conjugated forming O-glucuronide and O-sulfate (dog only) metabolites. Although subtle differences were observed in the excretion and biotransformation of atomoxetine in rats and dogs, the primary difference observed between these species was the extent of first-pass metabolism and the degree of systemic exposure to atomoxetine and its metabolites.
Atomoxetine hydrochloride (Fig. 1) (LY139603; formerly known as tomoxetine hydrochloride) is known chemically as (−)-N-methyl-γ-(2-methylphenoxy)benzenepropanamine hydrochloride. Atomoxetine is a potent inhibitor of the presynaptic norepinephrine transporter with minimal affinity for other monoamine transporters or receptors (Wong et al., 1982; Gehlert et al., 1993). This compound has been developed as a therapeutic agent for the treatment of attention deficit/hyperactivity disorder in children, adolescents, and adults.

Mean plasma concentrations of atomoxetine and radioactivity following a single intravenous bolus (A) or oral gavage dose (B) of 14C-atomoxetine to rats.
Error bars represent standard errors of the mean.
Atomoxetine is predominantly metabolized by CYP2D6 (Ring et al., 2002), an important source of intersubject variability in metabolism for a number of drug substances (Wolf and Smith, 1999). Because the metabolism of atomoxetine is influenced by the polymorphic expression of CYP2D6 (Sauer et al., 2003), the pharmacokinetics (clearances) of atomoxetine have a bimodal distribution with two distinct populations (Farid et al., 1985).
Rats and dogs do not express CYP2D6 and, in general, do not show polymorphic metabolism of CYP2D6 substrates. Although the human pharmacokinetics (Farid et al., 1985) and metabolism (Sauer et al., 2003) of atomoxetine have been previously characterized, little is known about the fate of atomoxetine in either rats or dogs. The objectives of these studies were to determine the plasma pharmacokinetics of atomoxetine following either oral or intravenous administration, as well as to characterize its metabolism and elimination in the Fischer 344 rat and beagle dog.
Materials and Methods
Reference Compounds and Other Materials.
The following compounds were synthesized at Lilly Research Laboratories: atomoxetine hydrochloride, [LY139603; (−)-N-methyl-γ-(2-methylphenoxy)benzenepropanamine hydrochloride]; 3-14C-atomoxetine hydrochloride (radiochemical purity, 99%; specific activity, 22.34 μCi/mg),2H7-atomoxetine,N-desmethylatomoxetine hydrochloride [N-γ-(2-methylphenoxy)benzenepropanamine hydrochloride], 3-14C-N-desmethylatomoxetine oxalate (radiochemical purity, 99%; specific activity, 66.7 μCi/mg), 4-hydroxyatomoxetine oxalate [N-methyl-γ-(2-methyl-4-hydroxyphenoxy) benzenepropanamine oxalate], 3-14C-4-hydroxyatomoxetine oxalate (radiochemical purity, 99.4%; specific activity, 55.8 μCi/mg), 2-hydroxymethylatomoxetine hydrochloride [N-methyl-γ-(2-hydroxymethylphenoxy)benzenepropanamine hydrochloride], 2-carboxyatomoxetine hydrochloride [(+)-N-methyl-γ-(2- carboxyphenoxy)benzenepropanamine hydrochloride], 4-hydroxy-N-desmethylatomoxetine hydrochloride [N-methyl-γ-(2-methyl-4-hydroxyphenoxy)benzenepropanamine hydrochloride], 2,4-dihydroxyatomoxetine hydrochloride [N-methyl-γ-(2-hydroxymethyl-4-hydroxyphenoxy)benzenepropanamine hydrochloride], and 4-hydroxy-2-carboxyatomoxetine hydrochloride [N-methyl-γ-(2-carboxy-4-hydroxyphenoxy)benzenepropanamine hydrochloride].
Sulfatase (Aerobacter aerogenes) and β-glucuronidase type HP-2 (Helix pomatia) were purchased from Sigma-Aldrich (St. Louis, MO). HPLC1 grade ammonium acetate and acetonitrile were purchased from Fisher Scientific (Fair Lawn, NJ). Ultima-Gold XR, Permafluor V, and Ultima-Flo M were purchased from PerkinElmer Life Sciences (Boston, MA).
Animal experiments.
All animal experiments were conducted according to protocols approved by the Eli Lilly Animal Care and Use Committee. The dosing solution used for all animal studies was prepared by dissolving atomoxetine hydrochloride and 14C-atomoxetine hydrochloride in sterile deionized water. All doses are expressed as free base concentrations of atomoxetine.
Fischer 344 rat.
Male Fischer 344 rats (8 to 10 weeks old/200 to 250 g) were obtained from Harlan Sprague-Dawley (Indianapolis, IN) and acclimated for at least 3 days before use. Food and water were supplied ad libitum at all times throughout the experiment. For the radiolabeled excretion study, three rats were administered a single oral (gavage) dose of14C-atomoxetine (50 mg/kg; 50 μCi/kg). Urine was collected at intervals of 0 to 6, 6 to 12, 12 to 18, 18 to 24, and 24 to 48 h. Fecal and cage washing samples were collected at 24-h intervals for up to 48 h.
For the radiolabeled pharmacokinetic study, 78 rats were divided into two groups. Each animal was administered either a single oral (gavage) dose of 14C-atomoxetine (50 mg/kg; 50 μCi/kg) or a single intravenous dose of 14C-atomoxetine (5 mg/kg; 50 μCi/kg). Blood was collected for the oral portion at 0.167, 0.25, 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 24, and 48 h and for the intravenous portion at 0.083, 0.167, 0.25, 0.5, 0.75, 1, 2, 4, 6, 8, 12, 24, and 48 h. Three rats were sampled for each time point. Plasma was obtained by centrifugation and stored at −70°C until analysis. Twelve additional rats were dosed orally with14C-atomoxetine (50 mg/kg; 100 μCi/kg) for metabolite identification in plasma. Blood was collected and plasma was pooled from three rats at 0.5, 2, 8, and 24 h.
To determine the role of biliary excretion in the rat, three cannulated rats where administered a single oral (gavage) dose of atomoxetine (50 mg/kg; 50 μCi/kg). Urine and bile were collected at intervals of 0 to 8, 8 to 16, 16 to 24, and 24 to 48 h. Fecal and cage washing samples were collected at 24-h intervals for up to 48 h.
Beagle dog.
Three female beagle dogs were obtained from Marshal Farms (North Rose, NY). For the radiolabeled excretion and pharmacokinetic study, three dogs (ages 2 to 4 years; weight 13.3–14.5 kg) were placed in individual stainless steel metabolism cages. Animals were fasted overnight, before and 2 h after drug administration. Adult dogs were given a single intravenous dose of atomoxetine (2 mg/kg; 5 μCi/kg). Following a 2-week washout, the same dogs were given a single oral dose of atomoxetine (2 mg/kg; 5 μCi/kg). Urine was collected from all dogs at 0 to 6, 6 to 12, 12 to 24, 24 to 48, 48 to 72, and 72 to 96 h intervals. Fecal and cage washing samples were collected every 24 h for 96 h. Blood was drawn at 0.083, 0.167, 0.25, 0.5, 1, 2, 4, 8, 12, and 24 h. Aliquots were withdrawn for determination of radioactivity, and the remainder was centrifuged to obtain plasma. For identification of plasma metabolites, three dogs were dosed orally with atomoxetine (2 mg/kg; 10 μCi/kg), blood was collected, and plasma was pooled at 0.5, 2, 8, and 24 h.
In vitro incubations.
Subcellular fractions from naive Fischer 344 rat and beagle dog livers were prepared by differential centrifugation (van der Hoeven and Coon, 1974). Microsomes from rat and dog livers (2 mg of protein/ml) were incubated with 14C-atomoxetine (50 μM) at 37°C. Incubations were conducted in a 100 mM sodium phosphate buffer (pH 7.4) in the presence of an NADPH-regenerating system (2 mM NADPH, 10 units of glucose-6-phosphate dehydrogenase, and 20 mM glucose 6-phosphate). After 1 h of incubation, proteins were precipitated with acetonitrile (0.5 ml), and the mixture was centrifuged. The resulting supernatant was removed and diluted with water before HPLC or LC/MS analysis.
Bioanalytical Analysis of Atomoxetine and Its Metabolites.
A validated atmospheric pressure chemical ionization LC/MS/MS assay was used to quantify atomoxetine, N-desmethylatomoxetine, and 4-hydroxyatomoxetine in heparinized plasma. Briefly, the method consisted of solid phase extraction (Varian SDB-XC cartridge) of analytes and the internal standard (2H7-atomoxetine) from 0.5 ml of plasma. The cartridges were washed with 1 ml of methanol/water (15:85 v/v) and eluted with 0.75 ml of 0.1% trifluoroacetic acid in acetonitrile. The samples were dried under nitrogen in a Zymark Turbo-Vap at approximately 45°C. The samples were reconstituted in 100 μl of acetonitrile and then mixed with an additional 25 μl of water. The instrument used was a PE Sciex API III MS/MS with a Shimadzu LC-10 HPLC system. The plasma extracts were analyzed on a Brownlee Spheri-5 ODS column (4.6 × 100 mm, 5-μm particle size) with a mobile phase consisting of 5 mM ammonium acetate, 0.2% formic acid, and 0.03% trifluoroacetic acid in acetonitrile/water (85:15, v/v). The flow rate was maintained at 1.0 ml/min. Analysis was carried out in positive atmospheric pressure chemical ionization (heated nebulized interface) mode using selected ion monitoring. The transitions fromm/z 256 → 44 (atomoxetine),m/z 272 → 44 (4-hydroxyatomoxetine),m/z 242 → 30 (N-desmethylatomoxetine), and m/z 262 → 44 (internal standard) were monitored. The limits of quantitation of the assay were from 0.25 to 25 ng/ml for atomoxetine, and from 1 to 100 ng/ml for both 4-hydroxyatomoxetine andN-desmethylatomoxetine. Plasma samples above the quantitative limit were diluted with plasma and reanalyzed.
Plasma Pharmacokinetic Analysis.
Noncompartmental analysis was used to determine the pharmacokinetics of atomoxetine, N-desmethylatomoxetine, 4-hydroxyatomoxetine, and radioactivity. Maximal plasma concentration (Cmax) and time of maximal plasma concentration (Tmax) were assessed by visual inspection. The terminal elimination half-life was calculated using the relationship 0.693/k, where k is the elimination rate constant. The AUC0-t was calculated up to the last measurable time point t by the trapezoidal rule. The AUC0-t was extrapolated to infinite time using k. From the intravenous plasma concentration data, clearance, volume of distribution, and the absolute oral bioavailability of atomoxetine were determined.
In Vitro Plasma Protein Binding.
Plasma (0.5 ml) was spiked with 14C-atomoxetine (75 to 1500 ng/ml),14C-N-desmethylatomoxetine (150 to 3000 ng/ml), or 14C-4-hydroxyatomoxetine (15 to 1500 ng/ml) and incubated for 1 h at 37°C. Following ultracentrifugation [100,000 rpm (approximately 430,000g, 37°C, 4 h], the amount of radioactivity in the supernatant was determined by liquid scintillation counting (LSC). The fraction of atomoxetine, N-desmethylatomoxetine, or 4-hydroxyatomoxetine bound to protein was calculated from the radioactivity concentrations in the spiked sample and the supernatant.
Analysis of Radioactivity.
The radioactivity in bile, plasma, urine, and cage washings were determined by LSC after the addition of scintillation cocktail (Ultima-Gold XR). Fecal samples were prepared for radioactivity determination by making a 1:1 water/feces homogenate. Aliquots of whole blood and fecal homogenates were pipetted into sample oxidizer combustion cones, weighed, air-dried, and combusted in a sample oxidizer. The blood and fecal samples prepared in this manner were then counted after addition of Permafluor V.
Metabolite Identification.
Aliquots of urine samples from individual animals were centrifuged before analysis by HPLC and LC/MS. Bile samples were diluted with an equal volume of 50 mM ammonium acetate and centrifuged before analysis. Aliquots of plasma (1 ml) from individual animals were pooled (3 ml, total volume) and extracted with 6 ml of acetonitrile. The samples were centrifuged, and the supernatant was evaporated to dryness in a 50°C water bath under a stream of nitrogen. Plasma extracts were reconstituted in 300 μl of 0.2% formic acid in water and centrifuged before analysis.
HPLC conditions.
A Shimadzu HPLC system consisted of two model LC-10AD pumps, a SIL-10A autosampler, a DGU-3A degasser, a CTO-10A column heater, and a model SCL-10A controller with a Berthold model LB 507A radiodetector equipped with either a 150-μl yttrium solid cell or a 500-μl liquid cell were utilized. For profiling plasma and urine metabolites, samples were separated on a Zorbax Eclipse XDB-C18 (5 μm particle size, 4.6 × 150 mm) column using a gradient of 0.05 M ammonium acetate and acetonitrile. The initial solvent composition was 90:10 0.05 M ammonium acetate/acetonitrile and increased linearly until a solvent composition of 40:60 0.05 M ammonium acetate/acetonitrile was attained in 30 min. The column was maintained at 30°C, and the flow rate was 1.0 ml/min. The concentration of each metabolite in plasma and urine was estimated by injecting an aliquot of an extract into the column, collecting the peak of interest as it eluted from the column and measuring the amount of radioactivity in the sample by LSC. The total radioactivity in the injected sample was estimated by passing an aliquot through the HPLC autoinjector and collecting the whole sample before the sample passed through the column. Ultima-Gold scintillation cocktail (15 ml) was added to each sample, and the level of radioactivity determined.
LC/MS and LC/MS/MS analysis.
Microsomal preparations, bile, urine, and plasma extracts were analyzed for metabolites and parent compound by LC/MS and LC/MS/MS on either a Finnigan TSQ-7000 or LCQ mass spectrometer in positive ion electrospray mode. LC/MS was performed by injecting aliquots of extracts onto a Zorbax Eclipse XDB-C18 column coupled to the mass spectrometer via a splitting tee. Chromatographic conditions were the same as described above for HPLC profiling except that the concentration of the ammonium acetate was reduced to 0.025 M. The column effluent was split such that the flow rate to the ion source was approximately 400 μl/min, the remaining being diverted to a radiochemical detector. Full scan analysis was programmed to scan from m/z 200 to 800 every second. The capillary heater was set to 225°C, and the spray voltage was 5 kV. For MS/MS experiments the relative collision energy was set at 35%.
Hydrolysis of conjugates.
Glucuronide and sulfate conjugates (approximately 1 μg) isolated from urine were hydrolyzed to their corresponding deconjugated metabolites. Enzymatic hydrolysis was accomplished by incubating the isolated conjugates in 0.2 M sodium acetate buffer, pH 5.0, with either β-glucuronidase (containing 100,500 units/ml β-glucuronidase and 4,000 units/ml sulfatase) or sulfatase (17 units/ml) at 37°C for up to 16 h. Incubations were also conducted in the absence of β-glucuronidase or sulfatase and in the presence of β-saccharolactone (0.03 M).
Results
Identification of in Vitro Metabolites.
Atomoxetine was extensively metabolized by Fischer 344 rat (4.6% atomoxetine remaining after 1 h) and beagle dog (39.7% atomoxetine remaining after 1 h) hepatic microsomes. The major metabolite produced by each of the species evaluated was identified as 4-hydroxyatomoxetine (m/z 272). Collision-induced dissociation (CID) analysis of the m/z 272 resulted in fragment ions at m/z 148 and 44, resulting from protonated C6H5(CH=CHCH2)NHCH3(N-methyl-3-phenenyl-propylamine) and CH2NHCH3, respectively (Table 1). This metabolite coeluted with synthetic 4-hydroxyatomoxetine and had an identical CID spectrum. Furthermore, the metabolite was isolated from the microsomal preparations by HPLC for further structural characterization. The1H and 13C NMR data obtained for the metabolite and the synthetic standard were nearly identical (Table 2). Slight differences in the chemical shifts of protons and carbons near the secondary amine were attributed to different salt forms (oxalate for the standard and acetate for the metabolite).
Retention time, molecular ions, and characteristic product ions of atomoxetine and its metabolites
Comparison of NMR data obtained for authentic standard of 4-hydroxyatomoxetine oxalate and the major metabolite in microsomal preparations

In both dog and rat microsomal preparations,N-desmethylatomoxetine (m/z 242), 2-hydroxymethylatomoxetine (m/z 272), and 4-hydroxy-N-desmethylatomoxetine (m/z258) were also formed. These metabolites had identical retention times and CID spectra as synthetic standards.
Excretion of Radioactivity.
At 48 h after the administration of a single oral dose (50 mg/kg) of 14C-atomoxetine, the recovery of radioactivity in excreta was greater than 95% (Table3) in the rat. The majority of radioactivity was excreted in the first 24 h. Approximately 66 and 29% of the radioactivity was found in urine and feces, respectively, and approximately 2% of the administered dose was recovered in the carcasses at 48 h. In bile duct cannulated rats, the mean total recovery of radioactivity was 99%. Approximately 49% of the dose was recovered in bile, 35% in urine, 2.5% in feces, and 13% in the carcass.
Excretion of radioactivity following a single oral dose of14C-atomoxetine administered to Fischer 344 rats and bile duct cannulated Fischer 344 rats
In the dog, approximately 91% of the total radioactive dose was recovered by 96 h after the administration of a single oral dose (2 mg/kg) of 14C-atomoxetine (Table4). More than 77% of the administered dose was excreted during the first 48 h of collection. As observed in the rat, renal excretion was the primary route of radiocarbon elimination.
Excretion of radioactivity following a single oral dose of14C-atomoxetine administered to beagle dogs
Plasma Pharmacokinetic Evaluation.
Fischer 344 rat
Noncompartmental pharmacokinetic parameters of atomoxetine,N-desmethylatomoxetine, 4-hydroxyatomoxetine, and radioactivity in plasma are shown in Table5 for the Fischer 344 rat. Following intravenous administration, the apparent distribution and elimination half-lives of atomoxetine were 0.09 and 1.4 h, respectively. Although the elimination half-life was rapid, a possible longer residual phase was observed from 12 to 24 h (Fig.1). Little or no radioactivity was partitioned into red blood cells. Total radioactivity in the plasma declined more slowly than atomoxetine, and had a half-life of 7 h. Atomoxetine accounted for approximately 30% of the total14C-equivalent exposure (AUC). Plasma concentrations of 4-hydroxyatomoxetine andN-desmethylatomoxetine, the two major in vitro metabolites, were negligible and their AUC represented less than 1% of the total14C-equivalent exposure (AUC) in rat. The large amount of uncharacterized radioactivity suggested that atomoxetine forms other metabolites, or 4-hydroxyatomoxetine andN-desmethylatomoxetine underwent further oxidation and/or conjugation. Clearance and volume of distribution of atomoxetine were 36.4 ml/min/kg and 4.31 l/kg, respectively.
Pharmacokinetic parameters of atomoxetine, N-desmethylatomoxetine, 4-hydroxyatomoxetine, and 14C-equivalents in Fischer 344 rats following the administration of a single intravenous bolus (5 mg/kg; 50 μCi/kg) or oral gavage (50 mg/kg; 50 μCi/kg) dose of14C-atomoxetine as a hydrochloride salt
The plasma concentration versus time plot of atomoxetine,N-desmethylatomoxetine, 4-hydroxyatomoxetine, and radioactivity following a single oral dose is shown in Fig. 1. TheCmax of atomoxetine was 0.17 μg/ml and occurred at 2 h after the dose, whereas the correspondingCmax value for radioactivity was 2.06 μg-equivalent/ml and was achieved at 0.5 h. Atomoxetine AUC only accounted for 2% of the total 14C-equivalent exposure (AUC), indicating that atomoxetine underwent extensive hepatic first-pass metabolism in the rat. Only minor amounts of 4-hydroxyatomoxetine and N-desmethylatomoxetine were present in plasma. Interestingly, the half-life of 4-hydroxyatomoxetine appeared to be prolonged compared with the other analytes evaluated. The absolute bioavailability of atomoxetine in the rat was only 4.0%.
Beagle dog.
The mean terminal plasma half-life of atomoxetine after a single intravenous dose of atomoxetine to dogs was 3.4 h (Table6). Total radioactivity declined with a mean apparent half-life of 8.2 h. The plasma concentration versus time plot of atomoxetine and radioactivity is shown in Fig.2. Atomoxetine,N-desmethylatomoxetine, and 4-hydroxyatomoxetine accounted for approximately 34, 24 and 2% of the total14C-equivalent AUC, respectively. Systemic clearance and volume of distribution of atomoxetine were 7.4 ml/min/kg and 1.4 l/kg, respectively.
Mean pharmacokinetic parameters of atomoxetine, N-desmethylatomoxetine, 4-hydroxyatomoxetine, and 14C-equivalents in beagle dogs following administration of a single 2 mg/kg (5 μCi/kg) intravenous or oral dose of 14C-atomoxetine as a hydrochloride salt

Mean plasma concentrations of atomoxetine and radioactivity following a single intravenous bolus (A) or oral dose (B) of 14C-atomoxetine to three dogs.
Error bars represent standard errors of the mean.
Following a single oral dose, the meanCmax and AUC for atomoxetine were 0.73 μg/ml and 4.3 μg·h/ml. The corresponding values for plasma radioactivity were 1.4 μg-equivalent/ml and 15.1 μg-equivalent·h/ml. Plasma concentrations versus time plot of atomoxetine, N-desmethylatomoxetine, 4-hydroxyatomoxetine, and radioactivity are shown in Fig. 2. AtCmax, atomoxetine accounted for approximately 28% of the radioactivity, compared with a value of 39% after an intravenous dose. The absolute oral bioavailability of atomoxetine was 74% in the dog.
In Vitro Plasma Protein Binding Evaluation.
Atomoxetine was highly bound to protein in dog plasma with mean binding of approximately 97%, while it was slightly less bound in rat plasma (89%). N-Desmethylatomoxetine (98% in dog and 90% in rat) binding was similar to atomoxetine, while the binding of 4-hydroxyatomoxetine (60% in dog and 55% in rat) to plasma protein was substantially less than atomoxetine. Binding was concentration-independent over the ranges tested in both species.
Identification of in Vivo Metabolites.
Fischer 344 rat
Approximately 59% of the administered dose was eliminated within the first 48 h in the urine. A representative HPLC radiochromatogram of pooled urine collected over 48 h following oral administration of 14C-atomoxetine is shown in Fig.3. The predominant urinary metabolite exhibited an MH+ ion at m/z448. The product ion spectrum of m/z 448 produced characteristic fragment ions at m/z 272 (protonated atomoxetine + O), m/z 148 (N-methyl-3-phenenyl-propylamine), andm/z 44 (CH2NHCH3) (Table 1). β-Glucuronidase (H. pomatia) hydrolysis of this conjugated metabolite resulted in 4-hydroxyatomoxetine, confirming the major metabolite in urine as 4-hydroxyatomoxetine-O-glucuronide. A secondary urinary metabolite exhibited an MH+ ion atm/z 434, which suggested that the metabolite was the glucuronide conjugate of 4-hydroxy-N-desmethylatomoxetine. The product ion spectrum of m/z 434 produced a fragment ion atm/z 258, which is likely due to loss of dehydroglucuronic acid from the conjugate. β-Glucuronidase hydrolysis of the conjugate resulted in 4-hydroxy-N-desmethylatomoxetine, confirming this metabolite in urine as 4-hydroxy-N-desmethylatomoxetine-O-glucuronide. Other metabolites identified in rat urine included hydroxy-2-carboxyatomoxetine-O-glucuronide (m/z 478), dihydroxyatomoxetine-O-glucuronide (m/z 464), 4-hydroxy-N-desmethylatomoxetine, and 4-hydroxyatomoxetine. No appreciable amount of unchanged parent drug was present in urine. The relative amount of each metabolite in urine was determined by radiochemical detection and is presented in Table7.
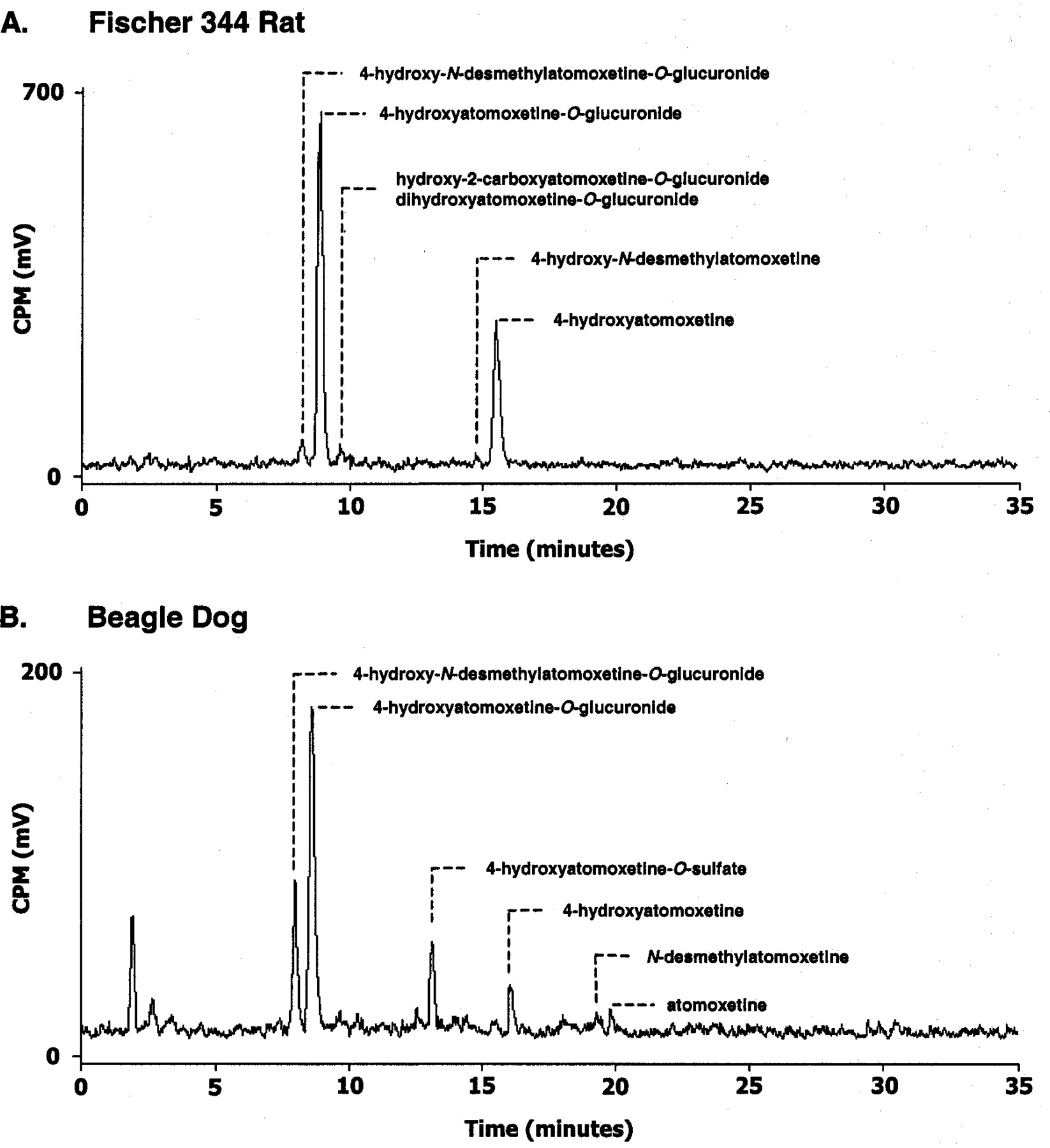
Representative HPLC radiochromatogram of urine collected from Fischer 344 rat (0–48 h) (A) and beagle dog (0–96 h) (B) administered a single oral dose of14C-atomoxetine.
Estimated amounts of atomoxetine and its metabolites in urine following a single oral dose of 14C-atomoxetine to Fischer 344 rats and beagle dogs
In plasma from orally dosed rats, atomoxetine, 4-hydroxy-N-desmethylatomoxetine-O-glucuronide, 4-hydroxyatomoxetine-O-glucuronide, hydroxy-2-carboxyatomoxetine-O-glucuronide, dihydroxyatomoxetine-O-glucuronide, 2-carboxyatomoxetine (m/z 286), and 4-hydroxyatomoxetine were detected. The relative amount of each metabolite in 2-h plasma is shown in Table 8. The major circulating species in plasma was 4-hydroxyatomoxetine-O-glucuronide at all time points evaluated (0.5, 2, 8, and 24 h), and this metabolite accounted for approximately 70% of the total plasma radioactivity at each time point evaluated.
Estimated amounts of atomoxetine and its metabolites in plasma following a single oral dose of 14C-atomoxetine to Fischer 344 rats and beagle dogs
Beagle dog.
Urine samples from the first 96 h after dosing were used for metabolite identification. A representative HPLC radiochromatogram of pooled urine collected over 96 h following oral administration of14C-atomoxetine is shown in Fig. 3. The major metabolite was identified in a similar manner as in rat urine, as 4-hydroxyatomoxetine-O-glucuronide. The other prominent metabolite present in urine was unique to dog. The electrospray MS of this metabolite gave an apparent protonated molecular ion ofm/z 352, and the CID product ion spectrum of them/z 352 ion showed characteristic ions atm/z 148 and 44 along with a smaller ion atm/z 272 (Table 1). The LC/MS/MS fragmentation suggested that this metabolite was a sulfate conjugate of a hydroxylated species. Sulfatase hydrolysis on the conjugate resulted in 4-hydroxyatomoxetine, confirming this metabolite in urine as 4-hydroxyatomoxetine-O-sulfate. In addition, 4-hydroxy-N-desmethylatomoxetine-O-glucuornide and 4-hydroxyatomoxetine, N-desmethylatomoxetine, and unchanged atomoxetine were also present in urine. The amount of urinary radioactivity accounted for by each metabolite and atomoxetine was estimated and is shown in Table 7.
Following an oral administration, only atomoxetine was detected in 0.5-h plasma sample. In the 2- and 8-h plasma samples, atomoxetine, 4-hydroxyatomoxetine-O-glucuronide, 4-hydroxy-N-desmethylatomoxetine-O-glucuronide, 4-hydroxyatomoxetine, and N-desmethylatomoxetine were present. The 4-hydroxyatomoxetine-O-glucuornide accounted for approximately 24% of the total radioactivity in the 2-h plasma sample (Table 8).
Discussion
Atomoxetine was well absorbed following oral administration to rats and dogs; however, profound differences in the absolute oral bioavailability of atomoxetine were observed between these species. Low oral bioavailability was observed in rats (approximately 4%), whereas dogs possessed relatively high oral bioavailability (approximately 74%). The poor oral bioavailability in the rat appears to be mediated by the efficient first-pass clearance of atomoxetine, resulting in poor systemic exposure to atomoxetine but high exposure to its metabolites. In the dog, atomoxetine does not undergo extensive first-pass metabolism, and exposure to circulating metabolites was relatively low compared with atomoxetine exposure.
In vitro metabolism studies using hepatic microsomal fractions from rat and dog livers produced similar oxidative metabolites. The metabolic pathways determined in hepatic microsomal preparations were confirmed by in vivo experiments. In both species, the primary route of systemic elimination for atomoxetine was metabolism with the majority of its metabolites being excreted into the urine. The primary oxidative (phase I) metabolite of atomoxetine was identified as 4-hydroxyatomoxetine, which was subsequently conjugated to form the major ultimate metabolite, 4-hydroxyatomoxetine-O-glucuronide, in each of the species evaluated. Likewise, the primary circulating plasma metabolite was 4-hydroxyatomoxetine-O-glucuronide. Based on the structures of its identified metabolites, three phase I metabolic pathways predominate the biotransformation of atomoxetine in the rat and dog: aromatic ring hydroxylation, benzylic hydroxylation, andN-demethylation. Although each of these metabolic pathways is distinct, aromatic ring hydroxylation appears to be the penultimate step for the further biotransformation of each of these metabolites. Conjugation by O-glucuronidation and O-sulfation of the hydroxylated metabolites was the only phase II metabolic pathway to participate in the further biotransformation of the oxidized atomoxetine metabolites. No metabolites were observed that indicated the formation of reactive intermediates or electrophilic species, i.e., dihydrodiol-derived metabolites or glutathione-conjugated metabolites.
The metabolism of atomoxetine was similar for both species; however, there were some species differences in the biotransformation of atomoxetine (Fig. 4). Specifically, the benzylic oxidation pathway was absent in dog, and the sulfate conjugation of the 4-hydroxyatomoxetine was only observed in dog. With regard to the 4-hydroxyatomoxetine-O-sulfate, there are a number of compounds that demonstrate a similar species specificity forO-sulfate conjugate formation in dogs compared with rodents. For example, prenalterol (Hoffman et al., 1982) and denopamine (Furuuchi et al., 1985) demonstrate a similar degree of species specificity in O-sulfation over O-glucuronidation in the dog compared with the rat. However, many other compounds do not elicit this behavior and it is unclear what structural requirements dictate this metabolic behavior. It is interesting to note that prenalterol, denopamine and 4-hydroxyatomoxetine each containpara-hydroxylated aromatic moieties which undergo eitherO-sulfation (dog only) or O-glucuronidation (dog and rodent).

Metabolic pathways of atomoxetine in rats and dogs.
The compounds in brackets are intermediates that were not observed in urine or plasma. For both dihydroxyatomoxetine-O-glucuronide and hydroxy carboxyatomoxetine-O-glucuronide, multiple structural isomers were observed and specific hydroxylation sites are not shown.
The major route of excretion of atomoxetine-derived radioequivalents was via the urine, while fecal excretion was a secondary route in both the rat and dog. Very little atomoxetine was excreted intact; indicating that direct renal or biliary elimination of atomoxetine is a minor route of systemic clearance. Fecal excretion of radioactivity was clearly due to biliary elimination and not due to unabsorbed dose. In the dog and rat, the same degree of fecal excretion of atomoxetine-derived radioequivalents was observed following both intravenous (data not shown) and oral administration of14C-atomoxetine. Furthermore, the amount of radioactivity eliminated into the bile of cannulated rats (49% of the total dose) exceeded the amount of the dose excreted in the feces of intact rats (29% of the total dose). The primary metabolite eliminated in the bile was 4-hydroxyatomoxetine-O-glucuronide (greater than 90% of the radioactivity eliminated in the bile). Thus, it appears that 4-hydroxyatomoxetine-O-glucuronide undergoes enterohepatic cycling resulting in deconjugation and reabsorption of 4-hydroxyatomoxetine. This conclusion is further supported by the prolonged plasma half-life of 4-hydroxyatomoxetine in the rat following oral administration. However, this prolonged half-life was not observed following intravenous administration, probably due to the low plasma concentrations of 4-hydroxyatomoxetine following this route of administration.
Recently, the metabolism and excretion of atomoxetine in humans was characterized in a clinical study (Sauer et al., 2003). A comparison of the metabolic profile and excretion of atomoxetine in rats, dogs, and humans show that the overall metabolism and disposition was similar among these species. As observed in the rat and dog, atomoxetine was well absorbed from the gastrointestinal tract in humans and cleared primarily by metabolism with the majority of the dose being excreted into the urine. Furthermore, the primary oxidative metabolite of atomoxetine was 4-hydroxyatomoxetine, which was subsequently conjugated forming 4-hydroxyatomoxetine-O-glucuronide.
In humans, atomoxetine is predominantly metabolized by CYP2D6 (Ring et al., 2002); and, therefore, its pharmacokinetics are influenced by the polymorphic expression of this enzyme (Farid et al., 1985; Sauer et al., 2003). However, regardless of CYP2D6 phenotype, atomoxetine undergoes the same routes of biotransformation as observed in the rat and dog: aromatic ring-hydroxylation, benzylic oxidation, andN-demethylation. Although subtle differences are observed among each of the species evaluated, the overall disposition of atomoxetine was similar in the rat, dog, and human.
Acknowledgments
We thank the following scientists for their assistance in these experiments: Heidi Weiss for technical expertise and input, as well as Marie Koenig and Carma Maples for conducting the live phase of these studies. Special thanks to J. Matthew Clemens for scientific contributions during the early in vivo metabolite identification studies. Finally, we thank Dean Clodfelter for efforts in the analyses of the bulk C-14-labeled materials.
Footnotes
- Abbreviations used are::
- HPLC
- high pressure liquid chromatography
- LC/MS
- liquid chromatography-mass spectrometry
- AUC
- area under the time verssus plasma concentration curve
- LSC
- liquid scintillation counting
- CID
- collision-induced dissociation
- LC/MS/MS
- combined liquid chromatography-tandem mass spectrometry
- CL
- clearance
- Cmax
- maximal plasma concentration
- Tmax
- time to maximal plasma concentration
- Vss
- volume of distribution
- F
- absolute oral bioavailability
- Received June 13, 2002.
- Accepted October 7, 2002.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}