


Abstract
Infection and inflammation impose a suppression in the expression and activity of several drug transporters and drug-metabolizing enzymes in liver. In the intestine, cytochrome P450 3A (CYP3A), P-glycoprotein (PGP/mdr1), and the multidrug resistance-associated protein 2 (MRP2) are important barriers to the absorption of many clinically important drugs; thus, the expression and activity of these proteins were examined in inflammation. Transport and metabolism were determined in jejunum segments isolated at 24 h from endotoxin-treated or control rats (n = 8) mounted in Ussing chambers. Transport and metabolism of 3H-digoxin, 5-carboxyfluorescein (5-CF), amiodarone (AM), and 7-benzyloxyquinoline (7-BQ) were measured for 90 min in the presence and absence of inhibitors. Reverse transcription-polymerase chain reaction was used to measure mRNA levels. As compared with controls, levels of mdr1a and mrp2 mRNA were significantly decreased by approximately 50% in the jejunum of LPS-treated rats. Corresponding reductions in the basolateral→apical efflux of digoxin, AM, and 5-CF were observed, resulting in significant increases in the apical→basolateral absorption of these compounds. Intestinal CYP3A mRNA levels and CYP3A-mediated metabolism of 7-BQ and AM were also decreased by approximately 50 to 70% (p < 0.05) in the LPS group. Mannitol permeability and lactate dehydrogenase release were not altered. These studies indicate that endotoxin-induced inflammation imposes a reduction in the intestinal expression and activity of PGP, mrp2, and CYP3A in rats, which elicits corresponding changes in the intestinal transport and metabolism of their substrates. Hence, infection and inflammatory diseases may impose variability in drug bioavailability through alterations in the intestinal expression and activity of drug transporters and metabolic enzymes.
Inflammation is a complex immunological response that is a component of many disease states, making it an important consideration in clinical therapeutics. An acute inflammatory reaction is initiated by a wide variety of pathological stimuli, including infection, tissue damage, trauma, or cellular stress resulting in the release of pro-inflammatory cytokines and modulation in the expression of many hepatic proteins. Numerous clinical reports indicate that drug biotransformation is compromised during infection and inflammation due to a down-regulation of cytochrome P450 caused by the elicited inflammatory response (Morgan, 1997; Renton, 2001; Slaviero et al., 2003). Inflammation is also known to alter the expression and activity of several drug efflux transporters (Hartmann et al., 2001, 2002; Goralski et al., 2003). The concomitant roles (i.e., removing xenobiotics from cells) and close cellular localization of metabolic enzymes and efflux transporters indicate that these proteins may function as a coordinate protective mechanism to limit systemic access of xenobiotics, likely contributing to the high interindividual variability that is observed for numerous drugs.
Cytochrome P450 3A is involved in the metabolic clearance of approximately 50% of drugs currently on the market and accounts for approximately 70% of total cytochrome P450 content in the small intestine (Guengerich, 1995; Wacher et al., 1998). The ATP-dependent drug efflux protein, P-glycoprotein (PGP1), encoded by the multidrug resistance gene (MDR1 in humans; mdr1a, mdr1b in rodents), is responsible for the active excretion of a wide variety of lipophilic cationic drugs from liver, kidney, and intestine. In contrast, the multidrug resistance-associated protein 2 (MRP2) is involved in the extrusion of lipophilic anions and their glutathione, glucuronide, and sulfate conjugates.
A variety of pharmaceutical and chemical agents, immune mediators, and disease states affect drug disposition by modulation of metabolism and/or transport. Expression of PGP, MRP2, and CYP3A are all reduced in animal livers during infection or inflammation (Morgan, 1997; Piquette-Miller et al., 1998; Tang et al., 2000; Payen et al., 2002). In vivo and in vitro studies indicate that interleukin-6 (IL-6) and other pro-inflammatory cytokines released during the inflammatory response are primarily involved in mediating this down-regulation (Sukhai et al., 2000; Hartmann et al., 2001; Lee and Piquette-Miller, 2001; Warren et al., 2001). Although the molecular mechanism of this phenomenon has yet to be elucidated, it is possible that these proteins share common regulatory pathways. Indeed, the pregnane X receptor (PXR), has been shown to regulate a network of drug-metabolizing and drug transporter genes in the liver and intestine, including CYP3A and MDR1 and MRP2 (Synold et al., 2001).
With respect to several acute phase protein markers, the intestine exhibits an acute phase response in a manner similar to that observed in the liver (Molmenti et al., 1993; Wang et al., 1998). However, the role of inflammation on the metabolic and transport proteins in the intestine has received little attention, despite the fact that the intestine is the first major barrier to xenobiotic absorption and that modulation of drug transport and/or metabolism likely impacts oral bioavailability. We conducted the present study in an animal model of inflammation, to ascertain the impact of experimentally induced acute inflammation (LPS-induced) on the expression and activity of PGP, MRP2, and CYP3A in intestinal tissue. Our results indicate that the intestines behave similar to the liver in the suppression of xenobiotic transport and metabolism during the inflammatory response.
Materials and Methods
Animals and Experimental Design. Male Sprague-Dawley rats (250–275 g) were purchased from Charles River Canada (Montreal, PQ, Canada), and studies were conducted in accordance with the guidelines of the Canadian Council on Animal Care. Rats were injected with 5 mg/kg i.p. endotoxin (LPS from Escherichia coli serotype O55:B5; Sigma-Aldrich, St. Louis, MO) dissolved in 0.5 ml of sterile saline, and controls received i.p. injections of 0.5 ml of sterile saline. Drinking water was provided but food was withheld after treatment to control for potential differences in food intake. Animals were sacrificed 24 h after injection of LPS or saline, a 20-cm segment of jejunum was excised, the lumen was rinsed with ice-cold Ringer's solution, and the intestine was opened along the mesenteric border. Sections of jejunum were immediately mounted in Ussing chambers (0.64-cm2 surface area) and allowed to equilibrate for 15 min with oxygenated Ringer's buffer. Enterocytes were harvested from the remaining tissue by scraping the luminal side and were frozen at –80°C for subsequent RNA isolation.
Reverse Transcription-Polymerase Chain Reaction (RT-PCR) Analysis of mRNA. Total RNA was extracted from mucosal scrapings of intestine segments using the Amersham QuickPrep Total RNA extraction kit (Amersham Biosciences Inc., Piscataway, NJ), and single-stranded cDNA was synthesized from 3 μg of RNA using the First Strand cDNA Synthesis Kit (MBI Fermentas, Flamborough, ON, Canada) according to manufacturer protocols. Serial dilutions (20- to 16,000-fold) of RT product were used to generate standard curves for PCR, and optimal amounts of template were determined from the linear portions of these curves (data not shown). Standard curves for RT-PCR were performed for each set of RNA samples analyzed. RT-PCR standard curves were highly reproducible. Selected RT-PCR results were also confirmed on Northern blots.
Amounts of RT product (cDNA template) used were: 25 ng (mdr1a, mrp2), 31.25 ng (PXR), 50 ng (mdr1b, IL-6), 62.5 ng (CYP3A), or 10 ng (GAPDH). The cDNA templates were amplified in the presence of 1.5 mM MgCl2, 200 μM deoxynucleoside-5′-triphosphate, and 50 pmol of forward and reverse primers in a total volume of 100 μl using a GeneAmp 2400 Thermocycler (PerkinElmer Life and Analytical Sciences, Woodbridge, ON, Canada). The reaction was initiated by addition of 2.5 U of Taq polymerase (MBI Fermentas), and amplification proceeded through 22 cycles for GAPDH, 25 cycles for PXR, 30 cycles for mdr1a, mrp2, and CYP3A, and 35 cycles for mdr1b. The PCR products were separated by electrophoresis on 2% agarose gels, stained with SYBR Gold nucleic acid stain (Molecular Probes, Eugene, OR), and visualized under ultraviolet light. Size of DNA bands was confirmed using Gene Ruler 100-bp DNA ladder (MBI Fermentas). Optical densities were normalized to GAPDH band intensities. PCR primers, obtained from the DNA Synthesis Centre (Hospital for Sick Children, Toronto, ON, Canada), are reported in Table 1.
PCR primer sequences
Functional Intestinal Ussing Chamber Studies. Procedures for the intestinal Ussing chamber studies were the same as those previously reported (Borchardt et al., 1996). Briefly, rat jejunum was opened along the mesenteric border, and 1-cm sections of jejunum were excised. Intestine was visually inspected prior to excision, and Peyer's patches were excluded from sections. Assembled diffusion chambers were placed in a 38°C heating block, connected to a 95% O2/5% CO2 airlift, and filled with 1.0 ml of 38°C Ringer's buffer (141 mM Na+, 5 mM K+, 1.2 mM Ca2+, 1.2 mM Mg2+, 122 mM Cl–, 25 mM HCO3–, 0.4 mM H2PO4–, 1.6 mM HPO42–), pH 7.4. Buffers of equal isotonicity containing 10 mM mannitol in the donor compartment and 8 mM glucose with 2 mM mannitol in the receiver compartment were used. The Ussing diffusion chambers and the airlift/heating block assembly were kindly provided by Dr. P. Swaan from The Ohio State University (Harvard Apparatus Inc., Holliston, MA).
Determination of PGP-Mediated Transport. The PGP transport studies were initiated by the addition of 3H-labeled digoxin and 10 μM unlabeled digoxin to either the apical or basolateral chamber to give a final concentration of 0.2 μCi/ml in the donor compartment. Mannitol (10 mM), traced with 0.2 μCi/ml 14C-labeled mannitol, was added to the donor side to serve as a permeability marker. For inhibition studies, 5 μM PSC-833 was added to the basolateral chamber prior to addition of digoxin. Duplicate samples of 100 μl were taken from the receiver chamber at t = 10, 20, 30, 45, 60, and 90 min for scintillation counting, and this volume was replaced with fresh buffer. Samples were taken from the donor chamber at the end of the transport study, and tissues were collected. Radioactivity was measured by liquid scintillation counting (Beckman LS5000TD; Beckman Coulter, Fullerton, CA) using a dual-label or single-label program, as appropriate.
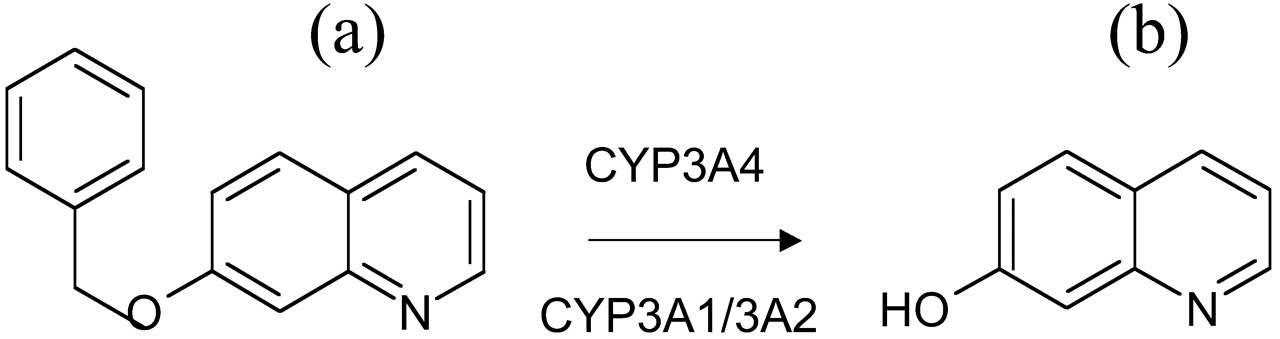
Determination of CYP3A-Mediated Metabolism. 7-Benzyloxyquinoline (7-BQ; BD Gentest, Woburn, MA) was used as a model substrate to assess CYP3A activity. The nonfluorescent 7-BQ is demethylated by rat CYP3A1/3A2 to the fluorescent metabolite, 7-hydroxyquinoline (7-HQ) (Fig. 1). A 250 μM concentration of 7-BQ (in Ringer's) was added to the apical side of each chamber, and a 300-μl sample was taken from the apical and basolateral sides at 30, 60, and 90 min. Fluorescence was then determined using a SpectraMAX Gemini XS plate reader (Molecular Devices, Sunnyvale, CA) with excitation and emission wavelengths of 360 and 520 nm, respectively. To assess the amount of metabolite remaining in the tissue, tissues from the chambers were digested with 150 μl of lysis buffer (Sigma-Aldrich) for 30 min and centrifuged at 250g for 10 min at 4°C. A 100-μl supernatant sample was collected and diluted with 50 μl of Ringer's buffer prior to measuring fluorescence.

Metabolism of 7-benzyloxyquinoline.
The metabolism of the nonfluorescent 7-benzyloxyquinoline (A) to the fluorescent 7-hydroxyquinoline (B) occurs through a demethylation by human CYP3A4 or rat CYP3A1/3A2.
Determination of Amiodarone (AM) Metabolism and Transport. The highly lipophilic PGP and CYP3A substrate, AM, was utilized to examine the combined influence of PGP and CYP3A on drug transport and metabolism (Yamreudeewong et al., 2003). AM (Sigma-Aldrich) was dissolved in methanol and diluted 100-fold with Ringer's buffer containing 8 mM glucose and 2 mM mannitol. AM (50 μg/ml) was added to the donor sides and duplicate 200-μl samples were taken from the receiver side at t = 30, 45, and 90 min. Samples obtained from donor and receiver sides and intestinal tissues were collected at the conclusion of the incubation period.
Tissue and buffer concentrations of AM and its major metabolite, desethylamiodarone (DEA), were analyzed by HPLC according to a validated assay (Jun and Brocks, 2001), with slight modification for extraction of drug and metabolite from gut tissue. DEA was a kind gift from Wyeth-Ayerst Research (Princeton, NJ). Intestinal tissue (100 μg) was homogenized in 0.4 ml of nonisotonic Sorenson phosphate buffer (pH 5.9) in the presence of 30 μl of internal standard solution (50 μg/ml ethopropazine HCl). After adding acetonitrile (300 μl), each tube was vortexed and centrifuged for 2 min at 2500g. The supernatant was transferred to new glass tubes, 3 ml of hexane was added, vortex-mixed for 30 s, and centrifuged, and the organic layer was transferred to clean tubes and evaporated to dryness in vacuo. The dried residue was reconstituted by adding 125 μl of mobile phase, and aliquots of 30 μl were injected into the HPLC. Standard curves generated from intestine yielded excellent linearity for both AM and DEA (r2 > 0.99).
Determination of MRP2-Mediated Transport. A previously described 5-CFDA/5-carboxyfluorescein (5-CF) efflux assay was used to study the functional activity of MRP (Lee and Piquette-Miller, 2001). In this assay, the nonfluorescent 5-CFDA passively and rapidly diffuses into cells, where it is converted to the fluorescent anion 5-CF by intracellular esterases. 5-CF is effluxed from cells by the MRP family of transporters and is not a substrate of PGP or the human organic anion transporter. 5-CFDA was dissolved in ethanol and diluted in Ringer's buffer to a final concentration of 50 μM (in 1% ethanol). Directional transport of the fluorescent anion, 5-CF, was monitored in both B→ A and A→ B directions, with 100-μl samples taken at t = 10, 20, 30, 45, 60, and 90 min, in the presence or absence of the MRP2 inhibitor, MK571 (100 μM). Fresh buffer was replaced at each sampling time point. Fluorescence of 5-CF was measured using a SpectraMAX Gemini XS plate reader (Molecular Devices) with excitation and emission wavelengths of 490 and 520 nm, respectively.
Assessment of Tissue Viability and Integrity. Mannitol diffusion, a paracellular permeability marker, was monitored between treatment groups for the experimental duration (90 min) to assess tissue integrity. Mannitol (10 mM), traced with 0.2 μCi/ml 14C-labeled mannitol, was added to the donor side and measured on the receiver side by liquid scintillation counting (Beckman LS5000TD; Beckman Coulter) at t = 10, 20, 30, 45, and 90 min.
The LDH in vitro toxicology assay kit (Sigma-Aldrich Canada, Oakville, ON, Canada) was used to measure membrane viability as a function of the amount of cytoplasmic LDH released into the medium. Following manufacturer directions, the Sigma LDH assay mixture (25 μl) was added to sample aliquots (50 μl) taken from the receiver chamber at each sampling time, mixtures were incubated at room temperature for 20 min, 10 μl of 1 M HCl was added, and absorbance was measured at 490 nm using an Ultrospec 2000 spectrophotometer (Amersham Biosciences UK, Ltd., Little Chalfont, Buckinghamshire, UK).
Morphological examination of intestinal tissue was achieved by immediately placing a 1-cm section of intestine in 10 to 15 ml of formalin (10% formaldehyde solution) fixative. This was followed by blocking with paraffin wax, sectioning to 6-μm thickness, and staining with eosin and hematoxylin.
Data Analysis and Statistical Analysis. Optical densities (O.D.) of bands obtained from agarose gels (PCR products) were quantitated using a Kodak DC120 camera and Kodak DS1D Scientific Imaging Software. Levels of PGP mRNA expression are reported as percentages of normalized values, as compared with control values. Normalized values were calculated as ratios: [(O.D.) mRNA/(O.D.) GAPDH mRNA]. The 90-min total area under the curve values for digoxin transport were calculated. Statistical analysis was performed using a Student's t test. Significance was assessed by a p value of 0.05 or less. Error bars in all graphs represent the standard error of the mean.
Results
Induction of Acute Inflammation in Rats. Although rats administered LPS displayed a pronounced formation of exudate around the eyes and nostrils and many developed diarrhea, there was no mortality in either the treated or the control groups. IL-6 mRNA expression, used as a marker for the presence of inflammation, was induced 2- to 6-fold throughout the small intestine 24 h after LPS treatment, with the greatest induction occurring in the jejunum (Fig. 2A).
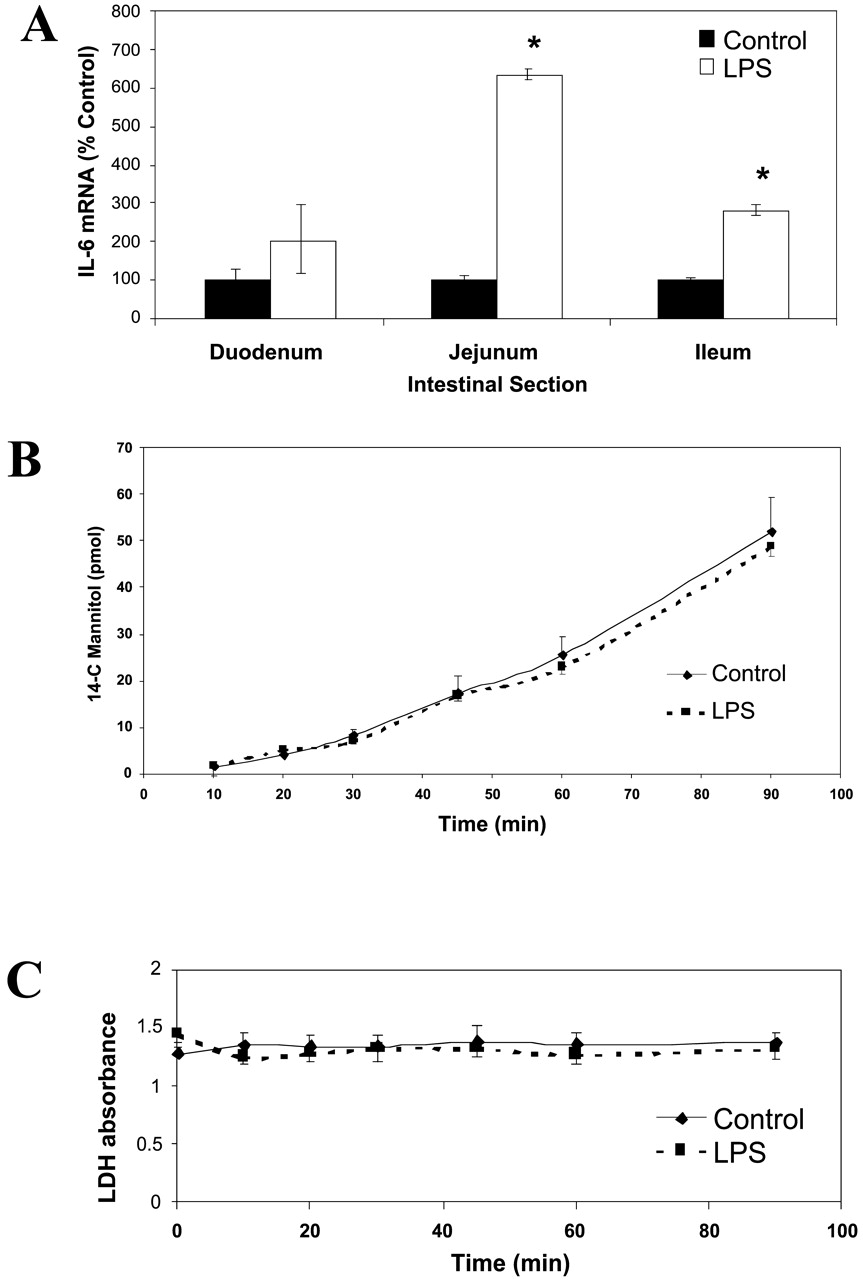
Tissue viability and integrity.
A, expression of IL-6 mRNA. Animals were treated with either saline or LPS (5 mg/kg), and different regions of the intestine (duodenum, jejunum, ileum, colon) were scraped at 24 h to obtain enterocytes. Semiquantitative RT-PCR was run on the isolated total RNA samples, as described under Materials and Methods. Values (mean ± S.E.M., n = 6) are reported as percentage of controls; raw intensities of the IL-6 genes were normalized to GAPDH values. ★, p < 0.05. B, mannitol permeability. Jejunum segments isolated at 24 h from control and LPS-treated rats were mounted in Ussing chambers. 14C-Mannitol (10 mM, 0.2 μCi/ml) was added to the basolateral side and B→ A permeability was measured over a 90-min interval. C, LDH release. Excised segments of jejunum isolated at 24 h from control and LPS-treated rats were mounted in Ussing chambers and buffer samples were obtained over a 90-min interval. LDH release was measured by UV absorbance as described under Materials and Methods.
Similar cell viability and membrane integrity were seen in intestinal segments obtained from LPS-treated and control rats. The directional transport of 14C-mannitol remained unchanged, and LPS treatment did not affect mannitol permeability in intestinal segments mounted on Ussing chambers (Fig. 2B). Levels of LDH did not significantly increase over the experimental duration and were similar between treatment groups (Fig. 2C). Slight changes in morphological characteristics were seen in histological sections of the LPS-treated animals, including loss of tissue color and enlarged spacing between adjacent villi. Nevertheless, similar cell viability and integrity were seen in treated and control samples.
Gene Expression. Levels of mdr1a mRNA were consistently and significantly reduced in each intestinal region by approximately 50% in LPS-treated animals (Fig. 3a). Levels of mdr1b mRNA also tended to be lower in the jejunum of LPS-treated animals; however, this change did not reach significance (Fig. 3, A and B). Expression of CYP3A was also suppressed in the jejunum of LPS-treated animals by 70% (29.6 ± 8.8% of controls, Fig. 3B). Likewise, Mrp2 mRNA expression was suppressed in response to LPS treatment (49.0 ± 4.7% of controls), whereas mrp3 levels were not significantly affected (Fig. 3B). PXR mRNA expression was also significantly reduced to 65.7 ± 6.7% of control levels in the jejunum of LPS-treated rats (Fig. 3B). Figure 3C depicts a representative image of the RT-PCR gels for each gene.

Expression of mRNA in intestinal segments (A) and jejunum (B) isolated from LPS-treated and control rats, with a representative image of RT-PCR gels for each gene (C).
Animals were treated with either saline or LPS (5 mg/kg), and different regions of the intestine (duodenum, jejunum, ileum, colon) were scraped at 24 h to obtain enterocytes. Semiquantitative RT-PCR was run on isolated RNA as described under Materials and Methods. Optical density of the mdr1a, mdr1b, mrp2, mrp3, CYP3A, and PXR PCR products were measured and normalized to GAPDH. Values (mean ± S.E.M., n = 6) are reported as percentage of controls (★, p < 0.05, ★★, p < 0.005).
3H-Digoxin Transport by PGP. At 24 h after LPS treatment, the AUC0–90 min B→ A 3H-Digoxin transport was reduced to 56 ± 6.3% of control values (Fig. 4A). Addition of the PGP-specific inhibitor, PSC-833, to the intestinal segments isolated from control rats reduced transport to 53 ± 7.9% of control levels, whereas addition of inhibitor to intestinal segments isolated from the LPS-treated rats did not further inhibit digoxin transport. The AUC0–90 min A→ B transport of digoxin, indicating net absorption, was increased from 100 ± 16% in controls to 143 ± 20% in LPS-treated animals. Due to high experimental variability, however, this did not reach statistical significance.
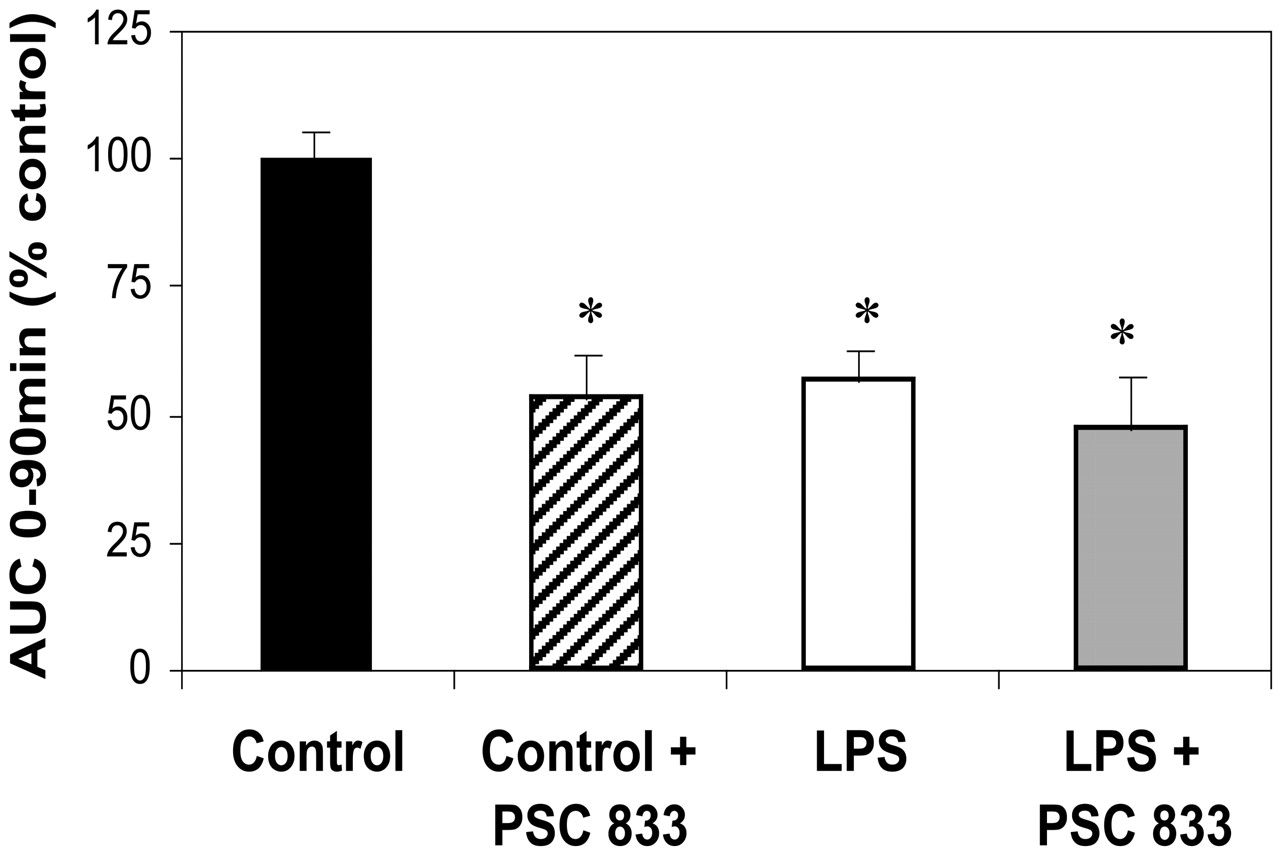
PGP-mediated efflux of 3H-digoxin in jejunum.
Jejunal segments isolated at 24 h from control or LPS-treated rats were mounted in Ussing chambers. Basolateral to apical transport (efflux) of 3H-digoxin (10 μM) was measured over 90 min, in the presence or absence of the PGP inhibitor, PSC-833 (5 μM). AUCs were compared, and values (mean ± S.E.M., n = 8 per group) are reported as percentage of controls, *, p < 0.05.
Benzyloxyquinoline Metabolism by CYP3A. As detected in media, intestinal CYP3A-mediated metabolism of 7-BQ to 7-HQ was significantly reduced in LPS-treated rats to 55%, 53%, and 52% of control values at 30, 60, and 90 min, respectively (Fig. 5). As compared with controls, significantly lower amounts of 7-HQ were also detected in the tissue lysates obtained from the treated animals (data not shown). Metabolism of 7-BQ to 7-HQ was only seen when intestinal segments were incubated with 7-BQ on the apical but not the basolateral side (data not shown), indicating apical localization of metabolic enzymes and poor diffusion of 7-BQ. Jejunum sections incubated with 7-HQ for periods of 30 to 120 min did not demonstrate significant changes or loss in total fluorescence, suggesting that further metabolism of 7-HQ does not occur. Furthermore, neither basolateral (B→ A) nor apical (A→ B) transport of 7-HBQ or 7-BHQ was altered by preincubation with the PGP-specific inhibitor, PSC-833 (data not shown). Hence, it is likely that neither 7-HQ nor 7-BQ is a substrate of PGP.

CYP3A-mediated metabolism of 7-BQ in jejunum.
Jejunal segments isolated at 24 h from control or LPS-treated rats were mounted in Ussing chambers, 7-BQ (250 μM) was added to the apical compartment, and samples were obtained at 30, 60, and 90 min. Concentrations of 7-HQ were measured in samples and standard curves by fluorescence detection (excitation 360 nm, emission 520 nm). Values (mean ± S.E.M., n = 3) are expressed as concentrations of 7-HQ.
Amiodarone Transport and Metabolism. Consistent with previous studies reporting interaction of AM with PGP (Katoh et al., 2001), the B→A transport (10% of total drug added) of AM was significantly greater than its A→B transport (3% of total drug added) after 90 min of incubation, suggesting PGP-mediated efflux. Transport in jejunum segments obtained from the LPS-treated animals displayed a 50% reduction in B→A efflux and an increase of A→B absorption to 153% of controls (p < 0.05). HPLC analysis of tissue lysates obtained at 30 and 90 min after incubation indicated that the majority of AM and its major metabolite, DEA, remained within the intestinal tissues (data not shown).
Since levels of the CYP3A metabolite, DEA, were below detection limits in the majority of buffer samples obtained from the donor and receiver chambers, we examined the impact of LPS treatment on the metabolism of AM based on data obtained in tissue lysates. Concentration ratios of DEA/AM were 50% lower (p < 0.01) in tissue lysates obtained from LPS-treated rats (0.26 ± 0.09) as compared with those obtained from control rats (0.51 ± 0.07) after 90 min. A 50% reduction in the concentration ratio of DEA/AM (p < 0.05) was also seen after 30-min incubations.
5-Carboxyfluorescein Transport by Mrp2. MRP activity, as measured by the AUC0–90 min B→ A 5-CF transport, was reduced to 31.7 ± 3.9% of control values in LPS-treated animals at 24 h (Fig. 6A). Adding the MRP-specific inhibitor, MK571, to intestinal segments isolated from control rats reduced transport by 41.8 ± 7.3%. In contrast, addition of inhibitor to intestinal segments isolated from the LPS-treated rats did not further inhibit 5-CF transport. The AUC0–90 min A→ B transport of 5-CF, indicating net absorption, was significantly increased from 100 ± 11% in controls to 173 ± 23% in LPS-treated rats (p < 0.05).

MRP-mediated efflux of 5-CF in jejunum.
Jejunal segments isolated at 24 h from control or LPS-treated rats were mounted in Ussing chambers. Basolateral to apical transport (efflux) of 5-CF (50 μM) was measured over 90 min, in the presence or absence of the MRP inhibitor MK571 (100 μM). Concentrations of 7-HQ were measured in samples and standard curves by fluorescence detection (excitation 490 nm, emission 520 nm). AUCs were compared, and values (mean ± S.E.M., n = 6 per group) are reported as percentage of controls, ★★, p < 0.005.
Discussion
Previous studies have demonstrated that inflammation reduces the hepatic expression and activity of the drug efflux transporters mdr1 and mrp2 (Piquette-Miller et al., 1998; Tang et al., 2000). This suppression is mediated primarily by the pro-inflammatory cytokines, particularly IL-6 (Sukhai et al., 2000, 2001; Hartmann et al., 2001, 2002). Whether inflammation-mediated changes in mdr1 or mrp2 expression occur in epithelial tissues such as the intestine is not known. However, there is increasing evidence that the intestinal mucosa responds to endotoxin in a manner similar to the well characterized hepatic acute phase response (Molmenti et al., 1993; Wang et al., 1998). Endotoxin is known to stimulate the production of IL-6 in liver (Billiar et al., 1992). Indeed, we observed an increased expression of IL-6 mRNA in the intestinal segments of LPS-treated rats that is consistent with that reported by Wang et al. (1998).
Overall, we found that LPS-induced inflammation imposed a down-regulation in the intestinal mRNA expression of the mdr1 and mrp2 transporters, similar to that which has been previously reported in liver (Goralski et al., 2003; Piquette-Miller et al., 1998; Tang et al., 2000). Although levels of mdr1a mRNA were reduced in all intestinal regions of LPS-treated rats, we found that mdr1b expression was low and not significantly altered. A low and often undetectable expression of mdr1b in the intestine has been reported by other laboratories (Salphati and Benet, 1998; Smit et al., 1998). Investigations in knockout mice have clearly shown that mdr1a is the major determinant of PGP-mediated drug efflux within the intestine (Stephens et al., 2002).
Consistent with the changes in mRNA, we found that LPS treatments imposed significant reductions in PGP- and MRP2-mediated transport in the intestine, whereas the permeability of mannitol was unaffected. As compared with controls, significant reductions in the basolateral to apical (B→ A) efflux of model substrates of PGP (digoxin) and MRP (5-CF) were seen in intestinal segments isolated from the LPS-treated group. In these animals, the residual B→ A transport could not be further reduced by addition of specific inhibitors such as MK571 or PSC-833 and likely reflect passive diffusion. Hence, it is likely that PGP- and MRP2-mediated efflux of digoxin and 5-CF may be fully suppressed in the LPS-treated rats. Because mRNA levels of PGP and MRP2 were not completely repressed, this may indicate that other post-translational factors may also be involved. Of interest was that the elevated apical to basolateral (A→ B) digoxin and 5-CF flux, reflective of substrate absorption, suggests that a net increase in bioavailability of PGP and MRP2 substrates is likely to occur in vivo during an inflammatory response.
Our expression and activity data of PGP/mdr1a and MRP2 are in agreement, leading us to believe that the reductions in both PGP and MRP2 activities are the result of a regulatory mechanism of acute inflammation. Furthermore, these studies indicate that statistically significant alterations in intestinal drug absorption can be predicted as a result of altered drug transporter levels. Conversely, it could be argued that mucosal damage and enhanced cell sloughing could result from LPS. However, since tissue viability (as measured by LDH release) and mannitol permeability were not significantly different between LPS-treated and control animals, it is unlikely that alterations in membrane integrity are responsible for the observed changes in expression and activity of PGP and MRP2. Likewise, it is important to consider the potential contribution of other transporters and metabolic enzymes. Although 5-CF efflux is reflective of total MRP activity, levels of mrp1 and mrp3 were not significantly affected and mRNA expression of other MRP isoforms was not detectable, indicating that changes in 5-CF transport likely reflected changes in mrp2 activity. Although it has been reported that the PGP substrate digoxin is metabolized in rat liver (Salphati and Benet, 1999), digoxin is not metabolized in intestinal tissue (Sababi et al., 2001). Furthermore, although the organic anion transporter OATP2 plays a role in the cellular uptake of digoxin in the liver, the OATP2 isoform is not expressed in the intestine (Cattori et al., 2001). Studies in mdr1a(–/–) mice have confirmed that transport by PGP is the sole route of intestinal efflux for digoxin (Stephens et al., 2002).
Our data demonstrated that LPS-induced inflammation also imposes significant reductions in the intestinal mRNA expression of CYP3A (Fig. 3B). Likewise, corresponding decreases in the CYP3A-mediated intestinal metabolism of 7-BQ to 7-HQ were seen in the LPS-treated rats. Metabolic studies in rat and human liver microsomes have demonstrated that metabolism of 7-BQ to 7-HQ (a highly fluorescent metabolite) is very specific for CYP3A (Renwick et al., 2001; Stresser et al., 2002). In liver, 7-HQ is further metabolized to its glucuronide conjugate by rat UDP-glucuronosyltransferase; the glucuronide metabolites retain fluorescence. Although delineation of the individual roles of PGP and CYP3A in inflammatory-mediated changes in drug disposition is important, it is particularly critical to ascertain the impact of their coregulation during an inflammatory response. Consequently, using the dual PGP/CYP3A substrate, AM, we assessed how inflammation might impact its transport and metabolism and, hence, its net bioavailability/absorption. We found that inflammation imposed reductions in both the CYP3A-mediated metabolism of AM to DEA and the PGP-mediated B→ A efflux of AM, overall resulting in a net increase in the A→ B absorption of AM in the LPS-treated rats. A similar extent of increased A→ B absorption was seen for both AM and digoxin, suggesting that inflammation-induced alterations in the expression and activity of both CYP3A and mdr1 do not have a cumulative impact on the oral bioavailability/absorption of their substrates. It is likely that for most dual substrates, either mdr1-mediated efflux or CYP3A-mediated metabolism serves as the rate-limiting barrier to drug absorption.
Major findings from these studies indicate significant and concurrent reductions in the expression and activity of PGP/mdr1a, mrp2, and CYP3A within intestinal tissue during acute inflammation. Hence, a coordinate system that regulates multiple drug transporters and drug-metabolic genes likely exists in response to inflammatory stimuli. Recently, activation of the PXR nuclear receptor has been reported to induce expression of CYP3A (Kliewer et al., 1998), MDR1 (Geick et al., 2001), and MRP2 (Kast et al., 2002). This has been seen in both liver and intestine (Xie et al., 2000; Staudinger et al., 2001). Furthermore, negative regulation of PXR has been reported to occur via an IL-6-mediated mechanism in human hepatocytes (Pascussi et al., 2000). To date, this has not been examined in vivo or in intestinal tissues. Hence our results, demonstrating a significant reduction in PXR mRNA levels in LPS-treated animals in conjunction with suppression of CYP3A, mdr1, and mrp2, suggest potential involvement or coregulation of PXR during inflammation. Further studies examining the involvement of PXR in the basal expression and negative regulation of these genes are currently being performed in PXR knockout and wild-type animal models of inflammation (Jekerle et al., 2003).
Intestinal efflux transporters and metabolic enzymes also contribute to drug clearance, including secretion of drugs into bile and direct exsorption of drugs into the intestinal lumen. Hence inflammatory stimuli are likely to impose changes in the bioavailability and clearance of numerous drugs that are substrates of PGP, MRP2, or CYP3A, thus increasing the possibility of adverse drug reactions or therapeutic failure.
Our findings indicate that the intestine is affected during an inflammatory response. Alterations in the drug transport or metabolism occurring within the intestine, as well as the liver, should therefore be considered when predicting drug disposition during inflammation. Increased and variable drug absorption is likely to occur in inflammation, and proper precautions must be taken when determining dose and regimen of drug therapy. Understanding this phenomenon may aid in better predicting therapeutic efficacy or toxicity, and raises the importance of acknowledging the presence of drug-disease interactions.
Acknowledgments
Technical assistance was kindly provided by Jing Wang.
Footnotes
-
↵1 Abbreviations used are: PGP, P-glycoprotein; mdr1, multidrug resistance gene; mrp2, multidrug resistance-associated protein 2; CYP3A cytochrome P450 3A; IL, interleukin; PXR, pregnane X receptor; LPS, lipopolysaccharide; RT-PCR, reverse transcription-polymerase chain reaction; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; PSC-833, valspodar; OATP, organic anion transporting polypeptide; 7-BQ, 7-benzyloxyquinoline; 7-HQ, 7-hydroxyquinoline; AM, amiodarone; DEA, desethylamiodarone; HPLC, high performance liquid chromatography; 5-CF, 5-carboxyfluorescein; 5-CFDA, 5-carboxyfluorescein diacetate; A→B apical to basolateral; B→A, basolateral to apical; MK571, 3-[[3-[2-(7-chloroquinolin-2-yl)vinyl]phenyl]-(2-dimethylcarbamoylethylsulfanyl)methylsulfanyl] propionic acid; LDH, lactate dehydrogenase; O.D., optical density; AUC, area under the curve.
-
Financial support was provided by an operating grant obtained from the Canadian Institute of Health Research (CIHR). Dr. Piquette-Miller is a recipient of the Rx&D Health Research Foundation-CIHR Research Career Award.
- Received June 16, 2003.
- Accepted September 2, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}