


Abstract
Prediction of drug–drug interactions due to cytochrome P450 isoform 3A4 (CYP3A4) overexpression is important because this CYP isoform is involved in the metabolism of about 30% of clinically used drugs from almost all therapeutic categories. Therefore, it is mandatory to attempt to predict the potential of a new compound to induce CYP3A4. Among several in vitro–in vivo extrapolation methods recently proposed in the literature, an approach using a scaling factor, called a d factor, for a given hepatocyte batch to provide extrapolation between in vitro induction data and clinical outcome has been adopted by leading health authorities. We challenged the relevance of the calibration factor determined using a set of 15 well-known clinical CYP3A4 inducers or the potent CYP3A4 inducer rifampicin only. These investigations were conducted using six batches of human hepatocytes and an established HepaRG cell line. Our findings show that use of a calibration factor is preferable for clinical predictions, as shown previously by other investigators. Moreover, the present results also suggest that the accuracy of prediction through calculation of this factor is sufficient when rifampicin is considered alone, and the use of a larger set of fully characterized CYP3A4 clinical inducers is not required. For the established HepaRG cell line, the findings obtained in three experiments using a single batch of cells show a good prediction accuracy with or without the d factor. Additional investigations with different batches of HepaRG cell lines are needed to confirm these results.
Introduction
Over the last 15 years, drug–drug interactions (DDI) have become one of the emerging topics in clinical drug development process (Boulenc and Barberan, 2011). In the late 1990s health authorities issued dedicated guidelines, which have been recently updated, related to the detection and consequences of DDIs (CDER 2011; CHMP 2012). The cytochrome P450 3A subfamily enzymes play a major role in the metabolism of about 30% of clinically used drugs from almost all therapeutic categories (Zanger and Schwab, 2013). Therefore, CYP3A4 isoform induction has been of particular interest, with several attempts to predict clinical consequences from in vitro results (Fahmi et al., 2008a,b; Almond et al., 2009; Fahmi and Ripp, 2010; Einolf et al., 2014).
In the majority of reported cases, the induction mechanism is due to activation of key transcription factors. Kliewer et al. (1998) first identified an orphan nuclear receptor, pregnane X receptor (PXR), that transcriptionally activates the CYP3A gene by interacting with the PXR response elements in the genes. Even if the final physiologically relevant effect is the enzyme activity, the mRNA increase is the most intrinsic metric for investigating CYP induction. Indeed, the mRNA expression data are in general more sensitive compared with enzymatic activity for detecting induction in human hepatocytes, and give more information if the drug is also a P450 inhibitor (Fahmi et al., 2010).
Predictive mathematical models incorporating either induction alone or induction in combination with inhibition mechanisms have been applied by a number of authors (Fahmi et al., 2008b; Shou et al., 2008; Fahmi and Ripp, 2010; Kirby et al., 2011; Templeton et al. 2011). Dynamic models based on an inducer concentration-time profile to account for the change in enzyme expression have also been proposed (Almond et al., 2009; Fahmi et al., 2009).
Some years ago, a calibration factor approach was proposed, which used a set of clinically well-known CYP3A4 inducers in a mathematical static model (Fahmi et al., 2008a). In this method, a d factor was determined for each in vitro human hepatocyte batch used in the assay. The d parameter represented an empirical calibration factor for the purpose of in vitro to in vivo induction scaling, and its value was estimated through correlation and minimization of predicted and observed area under the curve (AUC) ratios for the set of known inducers. The method initially proposed by Fahmi et al. (2008a) has been adopted by the U.S. Food and Drug Administration in its DDI guidance (CDER 2011).
Human hepatocyte donors used in in vitro induction models show a substantial interbatch variability due to considerable differences in the quality and viability of the isolated hepatocytes as well as interdonor variability in their basal cytochrome P450 levels that consequently leads to variation in their respective levels of induction by an inducer compound (Shou et al., 2008). Such interindividual variability is not uncommon and imposes a degree of uncertainty in predicting the clinical consequences of induction. Therefore, alternatives to the use of primary human hepatocyte cultures as models have been investigated.
Among these alternatives, the human HepaRG cell line is one of the most suitable human hepatic cell lines due to the retention of key liver functionality (Kanebratt and Andersson, 2008; Turpeinen et al., 2009; Templeton et al. 2011). This model is considered useful for the evaluation of DDIs as most of the common CYP isoform activities have been measured in this cell line and shown to be both selectively inhibited and induced by prototypical CYP-selective inhibitors and inducers at comparable levels to those of primary cultures of human hepatocytes (Turpeinen et al., 2009). Recently, in vitro HepaRG CYP3A4 induction data were used to predict a large number of DDIs. The investigators demonstrated similar predictive accuracy using the HepaRG cell line compared with the primary hepatocyte culture model (Grime et al., 2010).
In the current work, d factor values have been determined, for each of the six cryopreserved human hepatocyte batches as well as for the HepaRG cell line through the comparative use of both total and unbound plasma clinical Cmax values for 15 well-known inducers. Three approaches have been evaluated to establish optimal accuracy: 1) d not calculated (i.e., d = 1); 2) d value determination based on predicted and observed effects of rifampicin only; 3) d value estimated through minimization of the distance between predicted and observed AUC ratios for the set of 15 known inducers. To reach our conclusion in terms of the recommended method, we also took into account time and resources needed for each of them.
Materials and Methods
Compounds.
Carbamazepine, nifedipine, phenobarbital, phenytoin, pioglitazone, pleconaril, rifampicin, rifapentine, rosiglitazone, troglitazone, and aprepitant were purchased from Sigma-Aldrich (St. Louis, MO). Four proprietary compounds, obtained from Sanofi Research and Development, called SARA, SARB, SARC, and SARD were also selected based on the availability of their clinical and in vitro induction results.
Chemicals.
Dimethylsulfoxide (DMSO) and the cytotoxic references diclofenac, 4-hydroxytamoxifen, and menadione were purchased from Sigma-Aldrich (St. Louis, MO). All other chemicals and reagents used were obtained from usual commercial sources, and were of the highest commercially available grade.
Cell Media for Cryopreserved Human Hepatocytes.
The plating medium was composed of Ham’s F-12 and Williams’ E medium (50/50, v/v), both purchased from GIBCO/BRL (Bethesda, MD), supplemented with 10% decomplemented fetal calf serum (GIBCO, Paisley, United Kingdom), 10 mg/l insulin, 0.8 mg/l glucagon (Sigma-Aldrich, St. Louis, MO), 100 IU penicillin G, and 100 µg/ml streptomycin (GIBCO).
Culture medium was also composed of Ham’s F-12 and Williams’ E medium (50/50, v/v) that was devoid of serum but supplemented with 3.6 g/l HEPES, 4 mg/l ethanolamine, 10 mg/l transferring, 1.4 mg/l linoleic acid-albumin, 252 mg/l d-glucose, 44 mg/l sodium pyruvate, 50 mg/l ascorbic acid, 104 mg/l arginine, and 0.7 g/l l-glutamine (all purchased from Sigma-Aldrich).
Cell Media for HepaRG Cells.
The plating medium was composed of Williams’ E medium with Glutamax (GIBCO/BRL) and supplemented with HepaRG Thaw, Seed, and General Purpose Supplement (BIOPREDIC International, Rennes, France). For culture, Williams’ E medium with Glutamax was supplemented with HepaRG Serum-free Induction Supplement (BIOPREDIC International). Cells were seeded onto 48-well collagen I–coated plates purchased from BD Biosciences (Bedford, MA).
Human Hepatocytes and HepaRG Cells.
Six batches of plateable cryopreserved human hepatocytes were used. They were obtained from GIBCO/Life Technologies (Carlsbad, CA) for CD-Hu4237 and CD-Hu8084, from BD Gentest (Woburn, MA) for BD-281 and BD-295, from Bioreclamation (Baltimore, MD) for IVT-IBG, and from In Vitro ADMET Laboratories (Columbia, MA) for HH1024. One single batch of cryopreserved HepaRG cells (1247818) was obtained from GIBCO/Life Technologies.
Thawing Procedures for Cryopreserved Human Hepatocytes and HepaRG Cells.
Briefly, the vials containing the cryopreserved cells were removed from liquid nitrogen storage, thawed in a 37°C water bath (75–90 seconds), and then quickly poured into prewarmed seeding medium following the vendor’s protocol. Experiments with the same batch of HepaRG cells were performed using three independent thawings to investigate the reproducibility of the effects of the panel of inducers (interstudy variability).
Human Hepatocyte Cell Culture and Treatment.
Before seeding, cell viability was estimated using the Trypan blue dye exclusion test. The human hepatocyte cell density was adjusted to 0.8 × 106 viable cells per ml of medium. The hepatocytes were then seeded onto collagen I–coated 48-well plates at 0.16 × 106 viable cells per well in 200 μl of medium. Plating efficiency was evaluated around 4 hours after seeding for the hepatocytes and then daily over the 2 additional days of culture in the presence of CYP inducers. Cells were cultured in a 37°C thermostatically controlled incubator with 5% CO2 and 95% relative humidity.
After the cell attachment period, the plating medium was removed, and the hepatocytes were treated daily for 2 consecutive days (approximately 48 hours), with 100 µl per well of fresh culture medium containing either the vehicle control (DMSO) or the investigated compounds at eight preoptimized concentrations (based on compounds’ cytotoxicity, solubility, and EC50 [concentration resulting in half-maximal induction] found in literature) ranging from 0.01 to 30 µM for rifampicin, rifapentine, aprepitant, and troglitazone, from 0.01 to 60 µM for SARD, from 0.03 to 100 µM for pioglitazone, SARB, and SARA, from 0.1 to 300 µM for nifedipine, phenytoin, rosiglitazone, SARC, and pleconaril, from 0.3 to 1000 µM for carbamazepine, and from 1 to 3000 µM for phenobarbital. For each batch of hepatocytes, two separate wells for each concentration of a test compound were prepared. The final solvent (DMSO) concentration in the incubation medium ranged from 0.1% to 1% (depending on compound solubility). For incubations in the absence of compound (vehicle control), DMSO, as vehicle, was added, to incubation medium to provide the same final solvent concentration range of 0.1% to 1% as that used for the test compounds. Cell donor information is summarized in Table 1. Basal enzymatic activities for each human hepatocyte batch are summarized and compared with historical data in Table 2.
Cell donor information
Metabolic capacity assessment of human hepatocyte preparations
HepaRG Cell Culture and Treatment.
Before seeding, cell viability was estimated using the Trypan blue dye exclusion test. The HepaRG cell density was adjusted to 1.2 × 106 viable cells/ml of medium. The HepaRG cells were then seeded onto collagen I–coated 48-well plates at 0.36 × 106 viable cells per well in 300 µl of medium. Plating efficiency was evaluated around 6 hours after seeding and daily over the 2 additional days of culture in the presence of CYP inducers. Cells were cultured in a 37°C thermostatically controlled incubator with 5% CO2 and 95% relative humidity. After a cell attachment period of 3 days, the plating medium was removed, and the HepaRG cells were treated daily for 2 consecutive days under the same conditions as described earlier for human hepatocytes. Basal enzymatic activities are summarized in Table 3.
Metabolic capacity assessment of HepaRG cells
Assessment of Test Compounds Cytotoxicity.
Microscopical examination of hepatocyte morphology was used to evaluate cytotoxicity during incubation of each test compound. Moreover, to quantify the potential cytotoxic effects of the compounds in each experiment, a cytotoxicity assessment was also performed in parallel to the induction experiments. Specifically, test compound cytotoxicity was evaluated using an in vitro toxicity kit that measured ATP levels in the cells after a 48-hour exposure period to the test compound (CellTiter-Glo; Promega, Madison, WI).
Assessment of Test Compounds Stability.
The concentration, in medium, of each compound was measured at three different time points (0, 2, and 24 hours, in duplicate) during the last day of their respective incubation period to determine the exposure of human hepatocytes to the incubated compounds. Liquid chromatography with tandem mass spectrometry (LC-MS/MS) analyses were performed using an Acquity UPLC System I-Class, equipped with a Waters Acquity UPLC BEH C18 column (2.1 mm i.d. × 100 mm length, 1.7 µm particle size) coupled to a Xevo TQS mass spectrometer (all from Waters, Milford, MA).
The percentage of remaining compound at 2 or 24 hours was calculated as follows:

Induction Assay.
Quantification of CYP3A4 mRNA induction was performed using the Quantigene Plex 2.0 technology. The reagents used in the assay, including bDNA molecules (preamplifier, amplifier, label probe, and streptavidin-conjugated R-phycoerythrin [SAPE]), were obtained from the QuantiGene Plex 2.0 assay kit (Affymetrix, Santa Clara, CA). After a 48-hour incubation period with the test compounds, the cell culture medium was removed, and the cells were lysed with 100 µl of diluted lysis mixture buffer to release the RNA. Specific mRNA transcripts for CYP3A4 and β2-microglobulin (β2M, the housekeeping gene) were captured on their respective beads through a specific probe interaction during an overnight hybridization. Unbound materials were washed from the beads (complexed with probe set and mRNA) using a Hydroflex magnetic plate washer (Tecan, Männedorf, Switzerland). The signal was amplified by a sequential hybridization of DNA probes (2.0 preamplifier, 2.0 amplifier and biotinylated labeled probe, respectively) for 1 hour at 50°C. Three washes were performed after each hybridization step. After a final wash, SAPE was added, and the beads were incubated for 30 minutes at room temperature. The beads were then washed to remove unbound SAPE, and the samples were analyzed on a Luminex 200 system (Luminex, Austin, TX) or a Bio-Plex 200 system (Bio-Rad Laboratories, Hercules, CA). The levels of SAPE fluorescence were proportional to the amounts of mRNA transcripts captured by the beads.
Two duplicate assays (n = 2) were performed for all the described experimental samples. All multiplex data were derived from measuring median reporter fluorescence from 50 beads per gene per well assayed, and were presented as median fluorescence intensity. All data were corrected for background signals determined in the absence of target mRNAs. The gene of interest and the housekeeping gene (β2-microglobulin, β2M) levels (median fluorescence intensity) were both determined in two different tubes of the same sample, and the mean of the two values was determined. Thereafter, the measured amount of the gene of interest was normalized to the levels of the housekeeping gene in the same sample.
All results were expressed as the expression level of the investigated gene in the treated-hepatocytes, relative to control conditions (calibrator).The calibrator used was “untreated hepatocytes” (i.e., hepatocytes treated over the same period of time with DMSO alone). The fold-induction for each investigated compound relative to the level of vehicle control mRNA expression was calculated as follows: Fold-induction was calculated for each concentration of test compound and for each individual hepatocyte preparation. The maximal fold-induction (Emax) and the concentration resulting in half-maximal induction (EC50) of CYP3A4 for each compound were determined after fitting of the fold-induction values to a sigmoidal curve, using biost@t-speed (internal software).
Fold-induction was calculated for each concentration of test compound and for each individual hepatocyte preparation. The maximal fold-induction (Emax) and the concentration resulting in half-maximal induction (EC50) of CYP3A4 for each compound were determined after fitting of the fold-induction values to a sigmoidal curve, using biost@t-speed (internal software).
In Vitro–In Vivo Prediction of CYP Induction: Determination of the d Value.
The d parameter in eq. 1 represents an empirical calibration factor for the purpose of in vitro–in vivo induction scaling. A unique value was determined for each human hepatocyte batch and for each HepaRG experiment. This value was determined through comparison of the predicted and observed exposure ratio [AUCi/AUC, i.e., clinically observed ratio of area under the curve plasma concentration/time of the relevant administered CYP3A4 probe substrate, in the presence (AUCi) and absence of the inducer compound (AUC)] for different CYP3A4 probe substrates such as midazolam, nifedipine, or simvastatin. This equation only considers the potential induction effect of a compound on CYP3A4 expressed in the liver. The plasma concentration, Cmax, of the perpetrator (inducer compound) in the corresponding clinical trials was either expressed as its unbound concentration, with Fup incorporated into the equation or total concentration (without Fup in the equation). When unbound Cmax was used, the in vitro EC50 was also corrected for the unbound fraction of test compound in the hepatocyte assay (i.e., Fuhep), which was calculated using a quantitative structure-activity relationship (QSAR) model as previously described elsewhere (Kilford et al., 2008). Fup and Fuhep values for each test compound are reported in Table 4. (1)where fm is the fraction of the substrate probe drug metabolized by CYP3A4 (e.g., 0.91 for midazolam), [Inducer] is the unbound or total plasma concentration of the perpetrator inducer (i.e., Cmax), and d factor, EC50, and Emax are as previously defined.
(1)where fm is the fraction of the substrate probe drug metabolized by CYP3A4 (e.g., 0.91 for midazolam), [Inducer] is the unbound or total plasma concentration of the perpetrator inducer (i.e., Cmax), and d factor, EC50, and Emax are as previously defined.
Clinical trials used in the analyses
Total of 23 trials and 18 victim CYP3A4 substrate/perpetrator inducer compound pairs for the set of 15 test inducer compounds. Fup is the fraction unbound in plasma and Fuhep is the fraction unbound in the in vitro hepatocyte model of each of the test inducer compounds.
Data Set and Analysis.
Three approaches were evaluated: 1) d not calculated (d = 1); 2) d value determination based on the predicted (with eq. 1) and observed effects of rifampicin alone; and 3) d value estimated through minimization of the distance between the predicted (calculated with eq. 1) and observed AUC ratios for the set of the 15 well-known inducers, using the Brent’s method in “optim” function of R software version 3.0 (www.r-project.org).
The retained distances were GMFE (geometric mean fold error), RMSE (root mean squared error), RMSLE (root mean squared logarithmic error), and MAE (mean absolute error), which are defined as the following:




In each case, the scaling factor d was estimated per batch using all perpetrator compounds except one to simulate real conditions (when inducer potency of a new chemical entity is investigated). The AUC ratio of the remaining perpetrator was then predicted using the optimal d. This approach avoids an optimistic estimation of the error, as estimation of d and prediction of the AUC ratio were not performed on the same set of data (see Fig. 1).

Schematic presentation of the statistical investigations workflow when all inducers (23 clinical trials in total, called method 3 in Materials and Methods) are used, with methods comparison.
The predictions were classified as true positives or true negatives with respect to the potential induction effect of the test compound if both the predicted and observed AUC ratios were ≤0.8-fold (20% decrease in AUC) or >0.8-fold, respectively. Predictions were classified as false positives or false negatives with respect to the potential induction effect if the observed AUC ratios were not predicted appropriately within the 0.8-fold cutoff criteria.
To compare the relative predictability of the different methods, the positive predictive error (PPE) and the negative predictive error (NPE) values were calculated using the following equations:
 The PPE is defined as the proportion of in vitro studies that predicted a risk but for which no clinical DDI was observed. The NPE is the proportion of studies that were predicted as providing no risk of induction but actually demonstrated a DDI risk. The lowest PPE, NPE, GMFE, and RMSE values obtained indicated the best prediction outcomes.
The PPE is defined as the proportion of in vitro studies that predicted a risk but for which no clinical DDI was observed. The NPE is the proportion of studies that were predicted as providing no risk of induction but actually demonstrated a DDI risk. The lowest PPE, NPE, GMFE, and RMSE values obtained indicated the best prediction outcomes.
Methods were compared through use of the predicted to observed AUC ratios for the victim substrate probe by considering the number of predictions within a 2-fold error (0.5 ≤ predicted/observed AUC ratio ≤ 2.0). Among the methods employed to investigate the four minimization processes, only the best one, based on quality criteria described earlier, was selected for this last comparison. Statistical investigations and method comparison are summarized in Fig. 1.
Results
Viability and Morphology of the Cultured Hepatocytes.
Hepatocyte cultures were evaluated daily by phase-contrast microscopy, and were considered to exhibit normal hepatocyte morphology for initiating experiments when the confluence ranged between 80% and 100%.
Daily morphologic observations indicated that human hepatocytes treated with vehicle (0.1% to 1% DMSO) exhibited normal hepatocyte morphology. These observations, supported by the ATP content measurements performed on the last day of incubation (after 48 hours), showed that most of the test compounds did not exhibit any cytotoxic effects when used at concentrations up to 1000 µM. Cytotoxic concentrations of test drugs for each hepatocyte preparation and HepaRG experiment are summarized in the Supplemental Data.
Stability of Compounds.
The remaining concentration of each parent test compound in culture medium was determined by LC-MS/MS at various time points (0, 2, and 24 hours, in duplicate) during the second day of incubation to check that cells were well exposed to test compound. These analyses demonstrated that human hepatocytes and HepaRG cells were well exposed to unchanged drug for the majority of the tested compounds, at least at the highest noncytotoxic concentrations. For the lowest concentrations of nifedipine, pioglitazone, rosiglitazone, troglitazone, SARA, and SARB (i.e., the worst case), hepatocytes were well exposed after 2 hours of incubation while at least 80% of each respective parent compound had been metabolized at the last 24-hour sampling time point for at least one batch. Compounds concentrations and percentages remaining at 24 hours are presented in the Supplemental Data.
Effect of Test Compound Treatment on the CYP3A4 mRNA Expression in Human Hepatocytes and HepaRG Cells.
The clinical trials (n = 23) used in this analysis are referenced in Table 4. The 15 compounds were each tested in three to six different batches of cryopreserved human hepatocytes and three thawings of HepaRG cells, using CYP3A4 mRNA fold-induction as an end-point measurement. Dose–response data were fitted using a sigmoidal Emax model, and EC50 and Emax values were determined (Tables 5 and 6, Fig. 3). Representative dose–response curves, obtained with rifampicin in the different human hepatocyte preparations and HepaRG cells, are illustrated in Fig. 2. Only dose–response curves that fitted the following acceptance criteria were used. The dose-response curve had at least six data points. Though more than six concentrations were typically tested, some dose–response curves were bell-shaped due to compound cytotoxicity, solubility limitations, or other factors, and in these cases, data points at higher concentrations were excluded for EC50 and Emax calculations. A sigmoidal model fitted the dose-response data with less than 20% EC50 coefficient of variation. The responses of the human hepatocytes and HepaRG cells in the presence of the positive control (i.e., rifampicin) were within the normal ranges reported in the literature: CYP3A4 gene expression was potently increased, with mean Emax at 57-fold ± 44 and 17-fold ± 0.33, and mean EC50 at 1.0 ± 0.91 µM and 2.4 ± 0.77 µM, respectively.
Summary of in vitro induction parameters from test compound concentration/fold change in CYP3A4 mRNA curves obtained with the six human cryopreserved hepatocyte batches
Summary of in vitro induction parameters from test compound concentration/fold change in CYP3A4 mRNA curves obtained with HepaRG cells (three experiments)
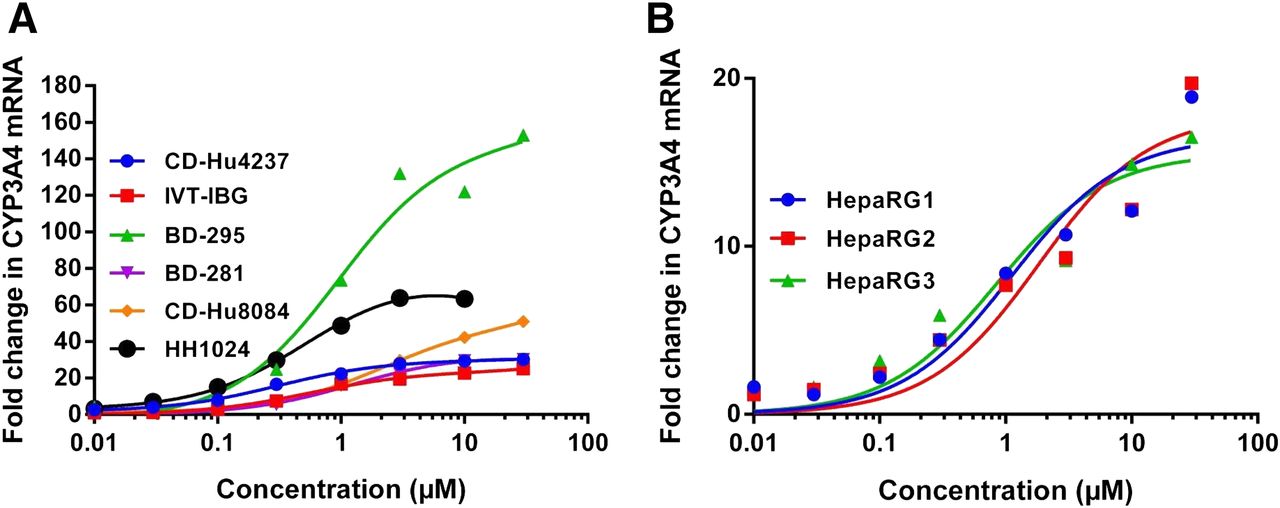
Representative concentration–response curves for rifampicin obtained with biost@t-speed software. Shown are (A) six cryopreserved hepatocyte batches and (B) results for three independent thawing periods used for single batch of HepaRG cells.

In vitro induction parameters EC50 (A, B) and Emax (C, D) obtained with six human cryopreserved hepatocyte batches (left) and HepaRG cells (three independent thawing periods) (right), derived from the test compound concentration–fold change in CYP3A4 mRNA curves.
The 15 tested compounds (references and proprietary sanofi compounds) induced CYP3A4 gene expression in all the human hepatocyte donors, with mean EC50 values ranging from 1.0 ± 0.91 µM for rifampicin to 310 ± 92 µM for phenobarbital, and mean Emax values ranging from 7.0 ± 4.8-fold for pleconaril to 57 ± 44-fold for rifampicin.
The tested compounds also induced CYP3A4 gene expression in HepaRG cells, with mean EC50 values ranging from 1.2 ± 0.57 µM for rifapentine to 325 ± 17 µM for phenobarbital, and mean Emax values ranging from 2.5 ± 0.41-fold for troglitazone to 17 ± 0.33-fold for rifampicin.
Determination of the d Value for the Human Hepatocyte Batches.
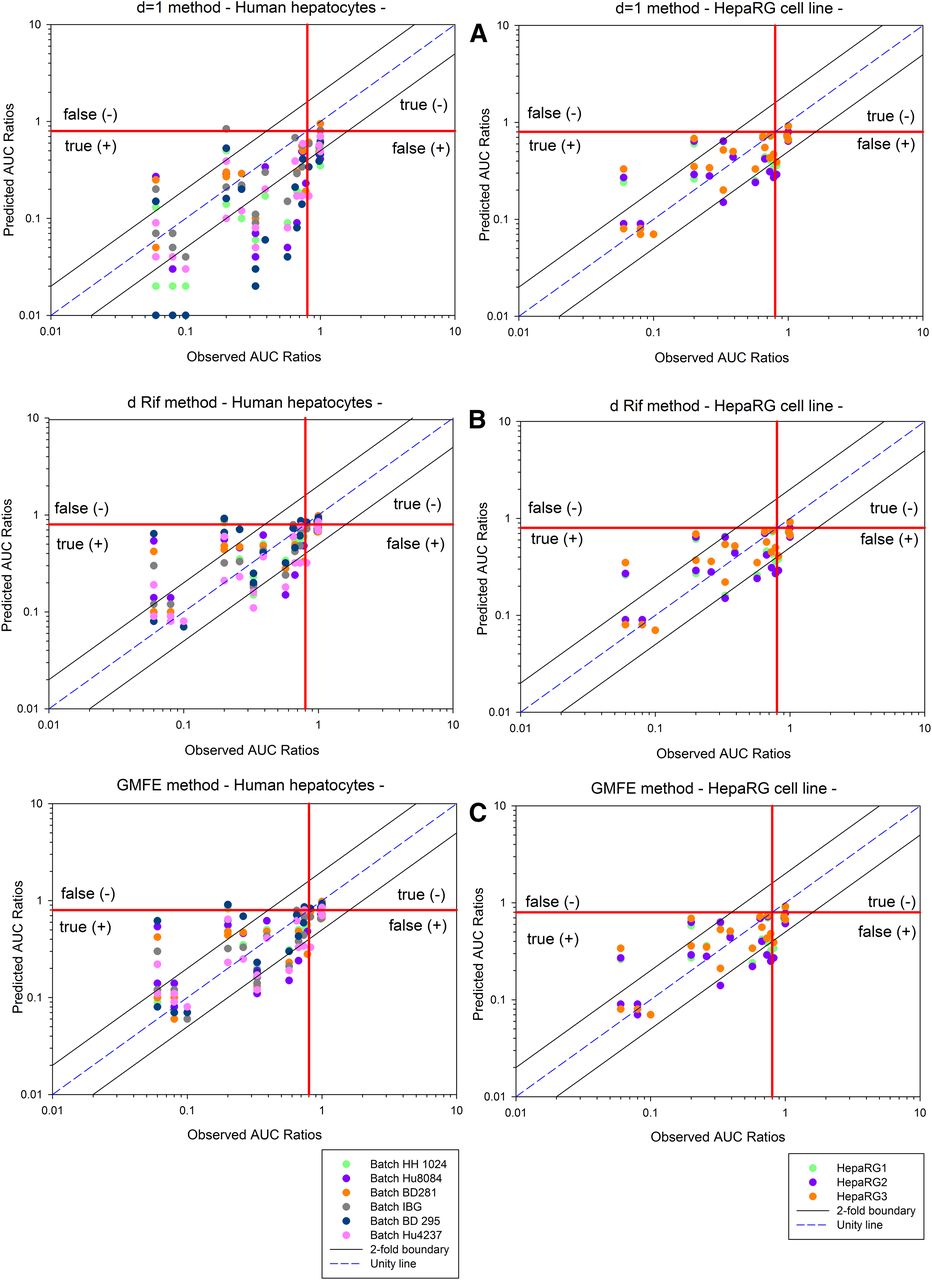
For each human hepatocyte batch, the scaling parameter d was determined using the three approaches described in the Materials and Methods section: 1) d value = 1; 2) d value determination based on predicted and observed effects of rifampicin alone; and 3) d value estimated through minimization of the distance between predicted and observed AUC ratios for the set of 15 well-known inducers with four retained distances (GMFE, RMSE, RMSLE, and MAE). The three methods were evaluated using different values for the hepatic inducer plasma concentration: unbound (with Fup) or total (without Fup) Cmax. To compare the relative predictability of the different methods, GMFE and RMSE were calculated as quality prediction criteria. Considering the overall total six batches, predicted versus observed AUC ratios, in each test condition, are presented in Fig. 4. As shown in Table 7, similar results were obtained with all four methods of minimization, GMFE, RMSE, RMSLE, or MAE. Therefore, only GMFE was selected for comparison with the other methods in Fig. 4. As shown in this figure and in Table 7, the NPE percentage is 20%–100% higher, whatever the method evaluated, when unbound concentrations (EC50 and Cmax) are incorporated, but it has a tendency to lead to higher values of PPE (Table 7). Allocating the scaling parameter d to a value of 1 provides the lowest accuracy in comparison with using a calculated scaling value [GMFE = 2.5, 46% within 2-fold (Table 7)]. Moreover, there was a trend for more-biased predictions toward overprediction of induction when d = 1, with the higher PPE value (16%) (Table 7 and Fig. 4). Hence, the determination of a d factor calculated for each hepatocyte batch is the method of choice.

Comparison of predicted and observed AUC ratios for each batch of human hepatocyte cultures and each independent thawing period of the HepaRG batch. Predicted AUC ratios are calculated using three different methods: (A) d equal unity, (B) d determined with rifampicin alone as calibrator, and (C) d factor obtained through GMFE error function for human hepatocytes in primary culture (left) or HepaRG cell line (right). The dotted lines represent the 2-fold error (i.e., 2-fold boundary), and the solid line represents the line of unity.
Summary of the DDI predictions of tested approaches for induction-based DDI of 129 predicted AUC ratios
Six cryopreserved human hepatocyte batches, 20 to 22 trials per batch. One trial is one observed AUC ratio compared with the corresponding predicted value. See Materials and Methods for calculation of PPE, NPE, RMSE, and GMFE.
The method using the full set of 15 compounds to calculate d values did not increase the quality criteria compared with the method using rifampicin alone. As shown in Table 7, the quality criteria RMSE, GMFE, NPE and PPE were similar, with both methods reflecting no improvement in the prediction when the set of compounds are used, when total Cmax is considered. In addition, comparison of both methods indicates that the d values are equivalent for a given tested human hepatocyte batch, despite having clearly different d values (range: 0.10–0.56) in the two methods for each hepatocyte batch reflecting the interbatch variability response (Table 9).
Determination of the d Value for the HepaRG Cell Line.
For each thawing of one batch of HepaRG cell line, the scaling parameter (i.e., d) was determined using the three approaches outlined earlier with the inducer plasma Cmax concentration expressed as either unbound (with Fup) or total (without Fup). As described earlier for human hepatocyte preparations, the relative predictability of each approach was compared by calculating the GMFE, RMSE, NPE, and PPE as quality prediction criteria.
Taking the three experiments together, the predicted versus observed AUC ratios in each condition are presented in Fig. 4. Similarly to the human hepatocyte preparations, the use of the d factor calculated without Fup was the best method to avoid underestimation, and similar results were obtained with all methods of minimization, GMFE, RMSE, RMSLE, or MAE (Table 8). Therefore, only GMFE was selected for comparison with the other methods in Fig. 4. As shown in this figure and in Table 8, the NPE percentage is 0 in most of cases, when total Cmax concentrations are incorporated, whatever the test method applied.
Summary of the DDI predictions of tested approaches for induction-based DDI of 69 predicted AUC ratios
Three experiments with one batch of HepaRG cell line, 23 trials per experiment. One trial is one observed AUC ratio compared with the corresponding predicted value. See Materials and Methods for calculation of PPE, NPE, RMSE, and GMFE.
Values for d determined by two methods and total Cmax inducer concentrations: 1) d factor obtained through GMFE error function and 2) d determined with rifampicin alone as calibrator for six human hepatocyte preparations in primary culture
The d factor value calculated with GMFE method is the average of the d values determined for each batch (see Materials and Methods and Figure 1 for details).
As observed with the human hepatocyte preparations, the third approach, which used the set of 15 compounds to calculate the d values, did not increase the quality criteria (RMSE, GMFE, PPE, and NPE) compared with the second approach that used rifampicin alone. Allocation of the scaling parameter d to a value of 1 provides the same level of accuracy as that observed for a calculated scaling value [GMFE = 1.6, 77% within 2-fold (Table 8)]. Moreover, using the d value determination based on the predicted and observed effects of rifampicin alone and incorporating its total Cmax inducer concentration, the d values were close to 1 [range: 0.92 to 0.99 (Table 10)], indicating that calculation of a d value is unnecessary when a test compound is incubated with HepaRG cells.
Values for d determined by two methods and total Cmax inducer concentrations: 1) d factor obtained through GMFE error function and 2) d determined with rifampicin alone as calibrator for three experiments with the HepaRG cell line
The d factor value calculated with GMFE method is the average of the d values determined for each experiment (see Materials and Methods and Figure 1 for details).
To summarize the results obtained with the HepaRG cell line (Fig. 4), the preferred approach for both optimal prediction of potential induction and ease of use is the first method incorporating a d factor value equal to 1 (i.e., no d factor) and total Cmax compound concentration (without Fu).
Discussion
In the current study, six different batches of human cryopreserved hepatocytes and one batch of HepaRG cells, analyzed after three independent thawing periods, were treated with 15 compounds at concentrations ranging from 0.01 to 3000 µM. For each investigated batch, an empirical calibration factor for the purpose of in vitro–in vivo scaling, called the d factor, was determined (eq. 1). Gene expression, as a measure of the in vitro induction potential of these compounds, was evaluated at concentrations up to their respective maximal noncytotoxic concentration. The metabolic stability of the test compounds in human hepatocytes and HepaRG cells was also investigated, as recommended by regulatory guidelines.
Quantification of test compounds by LC-MS/MS during day 2, the final day of incubation, demonstrated that human hepatocytes and HepaRG cells were well exposed to unchanged compound at least for the highest concentrations. However, 6 of 15 compounds exhibited a non-negligible metabolism after 24 hours incubation at the lowest concentrations for at least one batch. This suggests the effective concentration for induction process was less than those considered to derive EC50 and Emax. There is no obvious way to address this problem because induction mechanism and metabolic depletion of parent compound occur simultaneously. To address this potential issue, some authors have proposed considering the time weighted average concentration but have showed no improvement regarding quality criteria (Zhang et al., 2014).
The Emax values determined in vitro were found to be highly variable from one batch of human hepatocytes to another, and limited interexperimental variability was demonstrated for the HepaRG cells (n = 3) with both rifampicin alone (Fig. 2) and for all test compounds (Fig. 3). This variability in Emax values between different donors reflects the different magnitude of response in the donors and justifies the use of the d factor. Variability in hepatocyte plating efficiency and cell-viability during the 2-day duration of the experiment may also contribute to this observed variability. Typically, there was relatively little interindividual variability in the EC50 values in our study (Fig. 3). Of note, comparison of EC50 values for human hepatocytes and HepaRG show that the EC50 is similar for all of the test inducers (Fig. 3, Table 5, 6).
For each human hepatocyte batch and HepaRG cell line, the scaling parameter d was determined using the three approaches described in the Materials and Methods section using the unbound or total hepatic inducer plasma concentration.
For human hepatocyte batches, the use of in vitro and in vivo unbound concentrations tended to increase false negatives, even though in terms of 2-fold error the methods considering unbound concentration were slightly more accurate (Table 7). However, the use of the d factor calculated without Fup (i.e., Cmax total used) was the best method to avoid underestimation of clinical outcome (the lowest NPE percentage) (Table 7). This statement was specifically true with HepaRG in vitro model, which also showed a better accuracy in terms of 2-fold error (Table 8).
Despite these findings showing that unbound input concentrations (both in vitro and in vivo) yielded more accurate predictions of clinical induction magnitude, we currently recommend using the total concentration values when predicting positive/negative outcomes in clinical induction studies as this will not yield false negatives. This is consistent with the previous claims of others (Einolf et al. 2014). However it must be acknowledged that our dataset had only four clinical noninducers; thus, to thoroughly test free versus total concentrations as the relevant input values for prediction, a larger dataset that is enriched with drugs that show an induction response in vitro but not in vivo, as well as being highly protein bound (e.g., fup < 0.1), would be needed.
For experiments with human hepatocyte batches, comparison of the three methods (i.e., d equal to unity, d determined with rifampicin alone, and d determined with the set of compounds) showed that incorporation of the d factor provided the best prediction. Omitting the calibration factor led to a clear overestimation of the clinical effects of the test inducer. Moreover, our data provided no strong rational to suggest that a more accurate prediction was obtained when the set of 15 known test inducers were evaluated. Indeed, the d factor determined with rifampicin alone appeared to be sufficiently robust because it provided similar predictive accuracy to that obtained with the full set of test compounds (see Table 7 and Fig. 4).
In conclusion, the current investigation with the human hepatocyte in vitro model has demonstrated that the optimal determination of the calibration factor d, in terms of both the predictive accuracy of clinical outcome and level of resources required, is the method using the known CYP3A4 inducer rifampicin alone. Because rifampicin is classically used as a prototypical positive control inducer in each in vitro experiment, this option provides a method to incorporate the d calibrator in each experiment to reduce the impact of interexperimental variability for any human hepatocyte batch. In addition, rifampicin is metabolically stable in vitro (Supplemental Data) and thereby provides robust EC50 and Emax determination.
The HepaRG cell line has been previously proposed as an in vitro model to investigate induction processes (Grime et al., 2010). In our experiments with this cell line, using the set of 15 known inducers resulted in in vitro Emax values that tended to be lower than those obtained when human hepatocyte batches were used as the in vitro model (Fig. 3). Because HepaRG cell line exhibits a higher basal activity of CYP3A4 compared with the various cryopreserved human hepatocyte batches (Table 2, 3), the lower Emax values obtained in the HepaRG cell line may be partly explained by a lower induction potential. In view of the low Emax values (Fig. 3 and Table 8), the calibration factor did not appear to improve the accuracy of the prediction obtained with HepaRG cells in contrast to the results obtained with the batches of human hepatocytes.
As previously stated in the literature, since similar predictive accuracy has been obtained with HepaRG cell line and human hepatocytes, data from both in vitro models can be used interchangeably within the same laboratory to predict in vivo clinical outcome for CYP3A4 induction (McGinnity et al., 2009). In terms of the accuracy in prediction of clinical outcome as shown by 2-fold error (%), our data also suggest comparable reliability of the two models (Tables 7 and 8).
To sum up, our findings show that application of the calibration factor d to in vitro values obtained from experiments to evaluate potential inducers in human hepatocytes is preferable for prediction of clinical outcome. However they also suggest that the calculation of this factor d does not require the use of a complete set of 15 known CYP3A4 inducers. The use of rifampicin alone, which is already systematically incorporated as a prototypical positive CYP3A4 control in each in vitro experiment, should be sufficient to predict clinical outcome. When the HepaRG established cell line is used as the in vitro model, the findings obtained in three experiments, using a single batch of cells, show a good prediction accuracy with or without d factor. However, this finding is derived from values obtained with one single batch of HepaRG cells (three independent thawing periods). Additional studies using different batches of HepaRG cells will be required to provide definitive data that could confirm these results.
Acknowledgments
The authors thank Dr. Magalie Pardon, Sophie Vivier, Dr. François Donat, Dr. Terence Appelqvist, Dr. Roger Botham, and Dr. Jean-Marie Martinez for reviewing the manuscript and for their helpful suggestions.
Authorship Contributions
Participated in research design: Vermet, Klieber, Fabre, Boulenc.
Conducted experiments: Vermet, Raoust, Ngo.
Contributed new reagents or analytic tools: Vermet, Ngo.
Performed data analysis: Vermet, Esserméant, Boulenc.
Wrote or contributed to the writing of the manuscript: Vermet, Esserméant, Klieber, Fabre, Boulenc.
Footnotes
- Received May 26, 2015.
- Accepted October 13, 2015.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AUC
- area under the curve
- DMSO
- dimethylsulfoxide
- DDI
- drug–drug interaction
- Emax
- maximal fold-induction
- EC50
- concentration resulting in half-maximal induction
- fm
- fraction metabolized
- Fuhep
- fraction unbound in hepatocytes
- Fup
- fraction unbound in plasma
- GMFE
- geometric mean fold error
- LC-MS/MS
- liquid chromatography with tandem mass spectrometry
- MAE
- mean absolute error
- NPE
- negative predictive error
- PPE
- positive predictive error
- PXR
- pregnane X receptor
- RMSE
- root mean squared error
- RMSLE
- root mean squared logarithmic error
- SAPE
- streptavidin-conjugated R-phycoerythrin
- Copyright © 2015 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}