


Abstract
Induction of cytochrome P450 (P450) can impact the efficacy and safety of drug molecules upon multiple dosing with coadministered drugs. This strategy is focused on CYP3A since the majority of clinically relevant cases of P450 induction are related to these enzymes. However, the in vitro evaluation of induction is applicable to other P450 enzymes; however, the in vivo relevance cannot be assessed because the scarcity of relevant clinical data. In the preclinical phase, compounds are screened using pregnane X receptor reporter gene assay, and if necessary structure-activity relationships (SAR) are developed. When projects progress toward the clinical phase, induction studies in a hepatocyte-derived model using HepaRG cells will generate enough robust data to assess the compound’s induction liability in vivo. The sensitive CYP3A biomarker 4β-hydroxycholesterol is built into the early clinical phase I studies for all candidates since rare cases of in vivo induction have been found without any induction alerts from the currently used in vitro methods. Using this model, the AstraZeneca induction strategy integrates in vitro assays and in vivo studies to make a comprehensive assessment of the induction potential of new chemical entities. Convincing data that support the validity of both the in vitro models and the use of the biomarker can be found in the scientific literature. However, regulatory authorities recommend the use of primary human hepatocytes and do not advise the use of sensitive biomarkers. Therefore, primary human hepatocytes and midazolam studies will be conducted during the clinical program as required for regulatory submission.
Introduction
Induction of drug metabolizing enzymes (DMEs) is an important mechanism for clinical drug-drug interactions (DDIs). Induction of DMEs occurs after multiple dosing of a drug (perpetrator) and manifests in increased clearance of another drug (victim) or their own clearance (autoinduction). These DME changes occur mainly via nuclear receptor–meditated increases in gene transcription. AstraZeneca (AZ) has developed strategies that assess the potential for induction throughout preclinical (all project phases up to first time in human) and clinical (all clinical project phases including first time in human) testing (Fig. 1). The different elements of the strategy are detailed in Fig. 1. Although this strategy has been developed to investigate CYP1A, CYP2B6, and CYP3A induction, the major focus is on CYP3A because of its importance in drug clearance and the fact that the most induction DDIs are related to CYP3A.

Schematic detailing AZ induction strategy from preclinical to clinical assessment.
Briefly, at early stages of a project many compounds are synthesized to focus on answering specific questions and developing structure-activity relationships (SARs) to understand interactions with the target and key off-target receptors or enzymes. The number of compounds produced at this stage requires assays to be high throughput. Induction is one of the key off-target effects to understand at this stage. To allow the generation of induction SARs, nuclear receptor–linked reporter gene assays are used. These assays are relatively high throughput and give an indication not only of the compound’s binding to the receptor but also the subsequent activity of the target gene triggered as a result of this binding.
As projects progress toward the clinical phase, the reporter gene data are supplemented and ultimately superseded by induction studies in a hepatocyte-derived cell model, i.e., HepaRG (Kanebratt and Andersson, 2008b). These data support the selection of a compound as a clinical candidate. The end point of this assay is measurement of mRNA levels that provide a sensitive indication of induction. At this point, HepaRG data are used for internal decision making because regulatory authorities consider data from this model only as complementary to primary human hepatocytes to predict induction liability in vivo.
Following this, the understanding of the compound continues to build either by specific DDI studies (particularly important if the compound induces CYP1A or CYP2B6) or via the impact on its own pharmacokinetics (PK) or those of co-medications. If the compound is flagged not to be an inducer in the HepaRG assays, this is confirmed by evaluating 4β-hydroxycholesterol—a sensitive biomarker for CYP3A early in the clinical phase I multiple ascending dose finding study. Furthermore, an in vitro study in human hepatocytes is eventually conducted to ensure that we have a full understanding of the lack of induction potential of the compound and to abide by the current regulatory guidance.
If the compound is identified as an inducer in the HepaRG assays this is investigated early in the clinical program by evaluating 4β-hydroxycholesterol to assess the clinical relevance. Currently, regulatory authorities do not recommend submission of 4β-hydroxycholesterol data as a sensitive marker for CYP3A induction. Therefore, for clinical induction assessment this strategy includes an additional study with midazolam, a recommended sensitive CYP3A substrate, to provide suitable data for regulatory submission.
Nuclear Receptor Assays
A number of different nuclear receptors have been shown to be involved in the regulation of cytochrome P450 (P450) enzymes, including the pregnane X receptor (PXR), constitutive androstane receptor (CAR), aryl hydrocarbon receptor, etc. (Chai et al., 2013). The strategy described here focuses on CYP3A and PXR, which is the major transcriptional controller involved (Chai et al., 2013). It should be noted that PXR is responsible for the transcriptional regulation of other genes besides CYP3A, such as MDR1; however, these will not be considered here. Furthermore, although it has been shown that there is a degree of crosstalk between PXR and CAR in the induction of CYP3A (Chai et al., 2013), the CAR receptor is not the main focus of the strategy. Briefly, the mechanism of action of PXR involves the binding of drug in the cytosol; translocation to the nucleus; recruitment of the transcriptional partner, retinoid X receptor; and binding to the xenobiotic response element in the promoter region of the CYP3A gene (Chai et al., 2013).
The ability of a compound to interact with PXR and trigger the CYP3A induction can be assessed through ligand binding studies. Such studies involve the displacement of a bound ligand or transactivation reporter gene assays, where the CYP3A promoter region drives the transcription of a detection protein such as luciferase. These methods are higher throughput in nature and can be used to screen large numbers of molecules for their ability to induce CYP3A. The link between PXR activation and induction in cell systems such as HepaRG has been previously established (McGinnity et al., 2009). Sinz et al. (2006) clearly indicated that the results from PXR reporter gene assay could also be used to evaluate the potential of 170 xenobiotics to induce CYP3A in vivo when correlating the data with known in vivo drug interaction profiles.
One of the advantages of these higher throughput methods is that they can be applied in the early phases of drug discovery projects to provide SAR information on binding to PXR. These data, combined with the x-ray crystal structure of PXR, can be used in drug design to understand the SAR and thus remove potential CYP3A induction liability (Handa et al., 2015). This is quicker and more cost effective than using cell lines such as HepaRG or human hepatocytes for these types of SAR studies.
HepaRG Induction Assay
HepaRG is a human hepatoma cell line that differentiates toward hepatocyte-like cells. In the differentiated cell the mRNAs encoding DMEs attain levels of expression close to those found in freshly isolated primary human hepatocytes. The HepaRG cells are metabolically competent and also express liver hepatocyte drug transporters (Kanebratt and Andersson, 2008a; Anthérieu et al., 2010; Lübberstedt et al., 2011; Zanelli et al., 2012). It has also been shown by several laboratories that HepaRG responds to prototypical inducers such as omeprazole, phenobarbital, and rifampicin at both mRNA and enzyme activity levels (Aninat et al., 2006; Kanebratt and Andersson, 2008b; Kaneko et al., 2009; McGinnity et al., 2009).
Close concordance has been observed between P450 induction in HepaRG cells and primary human hepatocytes. Previously, excellent concordance had been demonstrated between CYP3A mRNA maximum observed induction (Emax) and EC50 and efficiency ratios (Emax/EC50) for 10 prototypical CYP3A inducers and noninducers in HepaRG cells and human hepatocytes (McGinnity et al., 2009). A further 23 AZ compounds with unknown induction potential and a variety of therapeutic targets and molecular structures have since been studied in both HepaRG cells and human hepatocytes (two or three donors per compound) and support the conclusion that HepaRG cells and primary human hepatocytes respond correspondingly to new chemical series (see the Supplemental Material) . A recent study by Vermet et al. (2016) further supports this conclusion with a set of 15 well-known clinical CYP3A4 inducers comparing HepaRG with six batches of primary human hepatocytes (Vermet et al., 2016).
Since the ultimate goal of in vitro studies is to provide accurate prediction of enzyme induction in vivo, the HepaRG data were correlated to in vivo induction caused by model compounds in Kanebratt and Andersson (2008b) and Grime et al. (2010). Both studies indicated that the results from HepaRG cells can be used to judge the induction potential of compounds in vivo using various static modeling equations. Based on this, it can be concluded that HepaRG cells provide an excellent surrogate for predicting the CYP3A induction potential by test compounds and candidate drugs in vivo. The induction response in HepaRG cells is generally similar to the response in primary hepatocytes; therefore, similar criteria for classifying compounds as inducers or noninducers can be used for the two cell models. However, if the target pharmacology of a compound is involved directly or indirectly in mRNA regulation or P450 protein synthesis this may also result in a compound being labeled as an inducer or repressor of P450 enzymes. Since the HepaRG cell line is hepatocarcinoma derived, these effects may be more marked for oncology targets. HepaRG cells provide several advantages, such as being a reliable source of cells and having a consistent response to inducers, which can be used to predict levels and variability. The predictions of CYP3A induction results using data from HepaRG cells fall well within the range of known interindividual variability of induction in vivo. The use of HepaRG cells will also be a powerful tool to build up a consistent historical database of P450-inducing compounds.
The HepaRG assay is used during lead generation/lead optimization before candidate drug selection to test the compound for P450 induction potential. Compounds are incubated at six different concentrations over 48 hours with the HepaRG cells. The mRNA expression of CYP1A2, CYP2B6, and CYP3A4 is measured, dose-response curves are plotted, and the F2 (concentration of the compound that elicits a 2-fold increase over the base line), Emax, and EC50 (when possible) values are reported. Levels of induction are put into context with predicted clinically relevant concentrations, and where appropriate modeled using static and dynamic equations. All modeling approaches are used to generate a relative risk assessment for induction within a drug project and the significance of these is considered on an individual project basis, taking into account the therapy area and likely patient population. Various static equations are used depending on the in vitro parameters available and to establish the worst-case in vivo induction prediction for a compound. These include in-house correlation approaches based on the F2 value and relative induction score values (Kanebratt and Andersson, 2008b; Grime et al., 2010). Additionally, the Food and Drug Administration (FDA) published R3 relative induction score equation is used with a d value of 1 as the worst-case prediction (http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf). The use of correlation approaches and basic static models has been shown to be as (or more) predictive than mechanistic static models to identify inducers (Einolf et al., 2014). However, for a limited number of compounds static modeling is followed by dynamic modeling using the Simcyp software platform (Simcyp, Sheffield, UK).
Human Hepatocytes Induction Analysis
Cultured human hepatocytes are primary human liver cells and as such are considered the closest in vitro representation of the in vivo situation. For this reason they are recommended by the United States (http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf) and European (http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf) regulatory authorities for the in vitro assessment of P450 induction . However, the use of human hepatocytes is restricted by the relatively low availability of suitable platable and inducible cryopreserved cells. This makes the in vitro testing for P450 induction of all potential drug candidates in hepatocytes impractical. However, of greater concern with primary hepatocytes is the massive de-arrangement of transcriptome, proteome, and functionality during the de-differentiation process when cells are cultured (Ulvestad et al., 2012; Bell et al., 2016). Lauschke et al. (2016) presented striking results on the de-differentiation process of primary hepatocytes in two-dimensional cultures. The study indicated that the de-differentiation process is orchestrated by a surge of noncoding RNA, mainly miRNA during the time cells are attaching to the plate. Such de-differentiation is not seen in three-dimensional spheroid cultures, which may constitute a more in vivo–like phenotype of the liver cell. The impact of this de-arrangement for P450 induction has not been fully explored. In particular, the induction via CAR—which is not via direct activation by inducers such as phenobarbital but includes the plasma membrane epidermal growth factor receptor and several cytoplasmic factors—raises concerns as to how all of these are maintained in hepatocyte cultures (Kobayashi et al., 2015).
Since primary human hepatocytes are recommended by the FDA and European Medicines Agency (EMA) to determine P450 induction, such studies are initiated for compounds that have shown absence of CYP3A induction in the clinical phase I study through monitoring 4β-hydroxycholesterol levels in order to confirm the results in HepaRG cells. For compounds that increase 4β-hydroxycholesterol levels by >25%, and thus indicate CYP3A induction, the clinical DDI studies supersede the preclinical induction studies. At this stage, hepatocytes may occasionally have another role in the risk assessment of complex DDIs. Examples include the assessment of compounds known to be both inducers and time-dependent inhibitors of the same enzyme, compounds with significant cytotoxicity in the HepaRG cell line, and for compounds known to have pharmacological effects on mRNA regulation or protein formation. These studies are currently recommended by the regulatory authorities to confirm or refute P450 induction. Based on prior data from the HepaRG cell line these studies can be individually designed to correctly position the test compound concentration range, maximize data generation, and answer the specific question posed for that compound. Additionally, where compounds require confirmation of a lack of P450 induction late in the development process, it is prudent to use human hepatocytes at this point. For all compounds studied with cultured human hepatocytes it is recommended to measure induction of both P450 mRNA and enzyme activity to provide a more complete risk assessment, especially when assessing known time-dependent inhibitors or potent inducers, where induction of activity is more likely to reach Emax than for mRNA.
4β-Hydroxycholesterol Clinical Biomarker Assessment
The propensity of a drug to alter human cytochrome CYP3A activity through induction has generally been assessed by administration of exogenous probe drugs such as midazolam that are selective for the enzyme. Endogenous biomarkers selective for CYP3A have been identified such as urinary 6β-hydroxycortisol/cortisol and plasma 4β-hydroxycholesterol. Although not approved as a biomarker by the regulatory agencies (Ma et al., 2013), 4β-hydroxycholesterol is one of the most promising biomarkers and is increasingly used. This biomarker now forms an integral part of the AZ induction strategy.
This potential use as a biomarker of induction was first identified by Bodin et al. (2001). These researchers identified that the 4β-hydroxycholesterol concentrations in patients taking antiepileptic drugs associated with CYP3A induction had elevated plasma concentrations of 4β-hydroxycholesterol compared with other antiepileptic drugs. Treatment of patients with efavirenz, stated as a medium inducer by the FDA, resulted in a more than 2-fold increase in plasma concentrations of plasma 4β-hydroxycholesterol (Josephson et al., 2008). The study by Bodin et al. (2001) also showed that the weak inducer ursodeoxychilic acid increased 4β-hydroxycholesterol concentrations by 45% in noncomplicated gall stone patients.
4β-Hydroxycholesterol concentrations in subjects have been shown to be very stable at steady state (CV between 4.8% and 13.2% over 3 months) (Diczfalusy et al., 2009) and has a long half-life of 17 days. It is sensitive to induction of both CYP3A4 and CYP3A5. These attributes make 4β-hydroxycholesterol suitable as a marker for CYP3A induction but not for single dose studies investigating CYP3A inhibition (Leil et al., 2014). Gut CYP3A is likely to have minimal impact on the circulating concentrations of 4β-hydroxycholesterol, and hence the large dynamic range observed with midazolam because significant first-pass metabolism will not be observed. Recently, Gjestad et al. (2016) claimed that changes in gut CYP3A actually will result in changes in 4β-hydroxycholesterol levels, and thus both gut and liver CYP3A will contribute to the plasma 4β-hydroxycholesterol levels. However, CYP3A content in the gut compared with liver content is low, and thus would be a minor contributor to the total plasma levels. 4β-Hydroxycholesterol in plasma has historically been analyzed with a liquid chromatography–tandem mass spectrometry method (van de Merbel et al., 2011); however, more recently a more sensitive method based on related methodologies has been introduced (Goodenough et al., 2011).
At AZ the use of 4β-hydroxycholesterol is built into all multiple ascending dose studies during phase I, irrespective of the in vitro data generated during the preclinical research and development phase. This was in response to the clinical induction seen with AZD1208, which is described in more detail subsequently. Single samples are collected before the first dose and at steady state at the end of the treatment period, which should preferably last for a minimum of 14 days to ensure identification of weak inducers. The time course induction study by Diczfalusy et al. (2009) clearly showed that a low dose of rifampicin (20 mg), mimicking weak induction of CYP3A, resulted in a significant increase in 4β-hydroxycholesterol levels at 14 days. Samples are collected at all dose levels. Initially, samples from the highest dose are analyzed, and any showing >25% change in the 4β-hydroxycholesterol levels leads to analysis of samples from all dose levels to define a dose-response relationship. A simple ratio of post-treatment against pretreatment concentrations is reported as part of the clinical study report. The data are also added to the in vitro understanding to integrate all induction knowledge to determine a dose-response relationship for induction and likely impact on subsequent drug development plans.
A number of studies have been conducted on the dose-response relationship of 4β-hydroxycholesterol induction. The plasma concentrations of 4β-hydroxycholesterol have been measured after 2 weeks of treatment with rifampicin at 20, 100, and 500 mg/day (Kanebratt et al., 2008) or 10, 20, and 100 mg/day (Björkhem-Bergman et al., 2013, 2014). The different doses were given to mimic weak, moderate, and strong induction of CYP3A as described in the FDA and EMA guidance on DDIs. Significant induction of 4β-hydroxycholesterol concentrations (Fig. 2) or the 4β-hydroxycholesterol/cholesterol ratio has been shown for all doses. The induction ratio showed significant (1.5-fold) induction at low-dose rifampicin (20 mg) and gives confidence that this biomarker can also capture weak induction of CYP3A. The change of 4β-hydroxycholesterol concentrations or (when alterations in cholesterol metabolism are suspected) the 4β-hydroxycholesterol/cholesterol ratio after rifampicin treatment is in concordance with changes to other CYP3A markers such as the quinine metabolic ratio (Kanebratt et al., 2008), cortisol ratio (Mårde Arrhén et al., 2013), and midazolam area under the curve (AUC) (Björkhem-Bergman et al., 2013). The advantage of 4β-hydroxycholesterol compared with these other markers is that no other drug is given and only one blood sample before and after treatment is needed. There is no need for lengthy urine collections (as is the case for the cortisol ratio) or several blood samples (as is the case for the midazolam AUC).
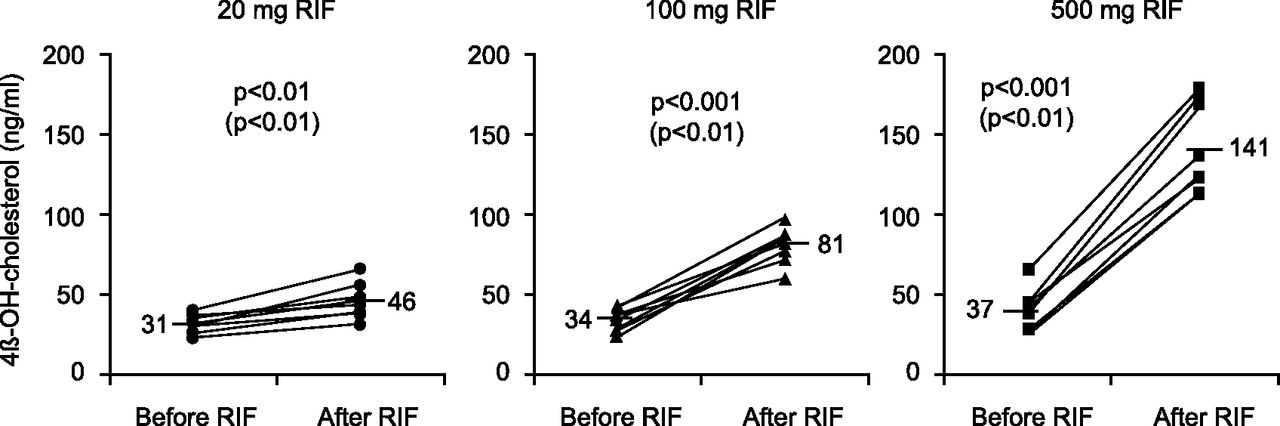
Plasma concentrations of 4β-hydroxycholesterol before and after 2 weeks of treatment with daily doses of 20, 100, and 500 mg rifampicin (Kanebratt et al., 2008, figure published with permission from the publisher).
If 4β-hydroxycholesterol levels are not changed in the multiple ascending dose study the lack of induction potential in vivo will be investigated in primary human hepatocytes to comply with regulatory recommendations. Only if a signal of induction of CYP3A is seen (>25% change), as indicated by 4β-hydroxycholesterol levels measured in the multiple ascending dose studies, will an in vivo clinical study using midazolam as a CYP3A probe substrate be conducted to comply with regulatory requirements. The in vivo study is done to verify the potential for induction of CYP3A by the AZ compound.
Clinical Midazolam Induction Study
Midazolam is the regulatory recommended sensitive probe substrate to evaluate CYP3A induction in the in vivo situation. Midazolam is almost exclusively metabolized by CYP3A in the gut and in the liver to 1-hydroxymidazolam and 4-hydroxymidazolam (Venkatakrishnan et al., 2001), making it a suitable probe substrate. The standard AZ in vivo clinical study design looking at the perpetrator potential for CYP3A induction is a fixed-sequence two-way crossover study with one treatment consisting of a single oral dose of midazolam (7.5 mg) and the second treatment being predosing with the AZ compound up to steady state or for at least 14 days at the maximum therapeutic dose, followed by a single oral dose of midazolam. The dosing of the AZ compound will continue for the entire PK sampling period for midazolam.
Case Study: AZD1208
Here, one rare AZ case study is presented in which all of the in vitro models tested negative but the in vivo results showed potent induction of CYP3A. AZD1208 tested negative in HepaRG cells and primary human hepatocytes but showed a positive induction potential in vivo. AZD1208 (Fig. 3) is an orally available, small molecule Pan proviral integration site for the Moloney murine leukemia virus kinase inhibitor intended for treating patients with acute myeloid leukemia (Keeton et al., 2014). In vitro studies in human microsomes and recombinant systems suggested that the phase 1 metabolism of AZ1208 is primarily mediated via CYP3A.

Structure of AZD1208.
Preclinical characterization of AZD1208 in the HepaRG assay suggested no potential for P450 induction, as evidenced by lack of mRNA or activity increase. In human cryopreserved hepatocytes, up to a concentration of 30 µM, there was no increase in mRNA or activity after 48-hour incubation (Table 1). For the methodology and full data see Supplemental Table 1. The exposure in preclinical species (rat and dog) increased with time and duration of treatment, consistent with their single dose PK data.
Effect of AZD1208 on CYP3A4 mRNA in HepaRG and human hepatocytes
In first time trials with humans, two studies were conducted in parallel. One study focused on understanding the PK, safety, and tolerability in acute myeloid leukemia patients, while the other study (conducted in Asian countries) focused on the same objectives in solid tumor patients. Both studies started evaluation at 120 mg, and doses up to 800 or 900 mg/day of AZD1208 as a capsule formulation were further explored (Cortes et al., 2016).
The PK data after a single dose in both studies were similar and generally increased dose proportionally up to 540 mg and less than dose proportionally above 540 mg. There was greater variability at doses above 540 mg, consistent with variable absorption due to solubility and dissolution limits of AZD1208, which led to less than a dose-proportional increase with dose. The PK data in solid tumor patients were better described due to prolonged exposure characterization after single doses of AZD1208. The half-life could not be fully characterized in all patients even with prolonged sample collection, and in subjects where this could be characterized it was ∼37 hours (18–74 hours). This is likely an underestimate of half-life since this could not be fully characterized in many subjects where the elimination profile was not fully defined.
After multiple doses, exposures generally decreased with increasing doses and duration of dosing. Based on the prolonged elimination profile and the half-life after a single dose, accumulation of AZD1208 was expected after multiple dosing. However, the exposure of AZD1208 decreased with time, dose, and duration, indicative of time-dependent PK. To understand the reason for this apparent autoinduction several approaches were evaluated, including determination of 4β-hydroxycholesterol levels at predose and at day 15 of the solid tumor study following doses of 700 and 800 mg. The results from this evaluation are shown in Fig. 4, which shows significant increases in 4β-hydroxycholesterol levels after repeated dosing of AZD1208—achieving average steady-state plasma concentrations of 1238 and 1521 ng/ml for the 700 and 800 mg doses, respectively. The results clearly indicate that AZD1208 induces CYP3A activity, and since AZD1208 is also metabolized by CYP3A this explain the decreased exposure with repeated dosing, which is consistent with increased CYP3A-mediated clearance.
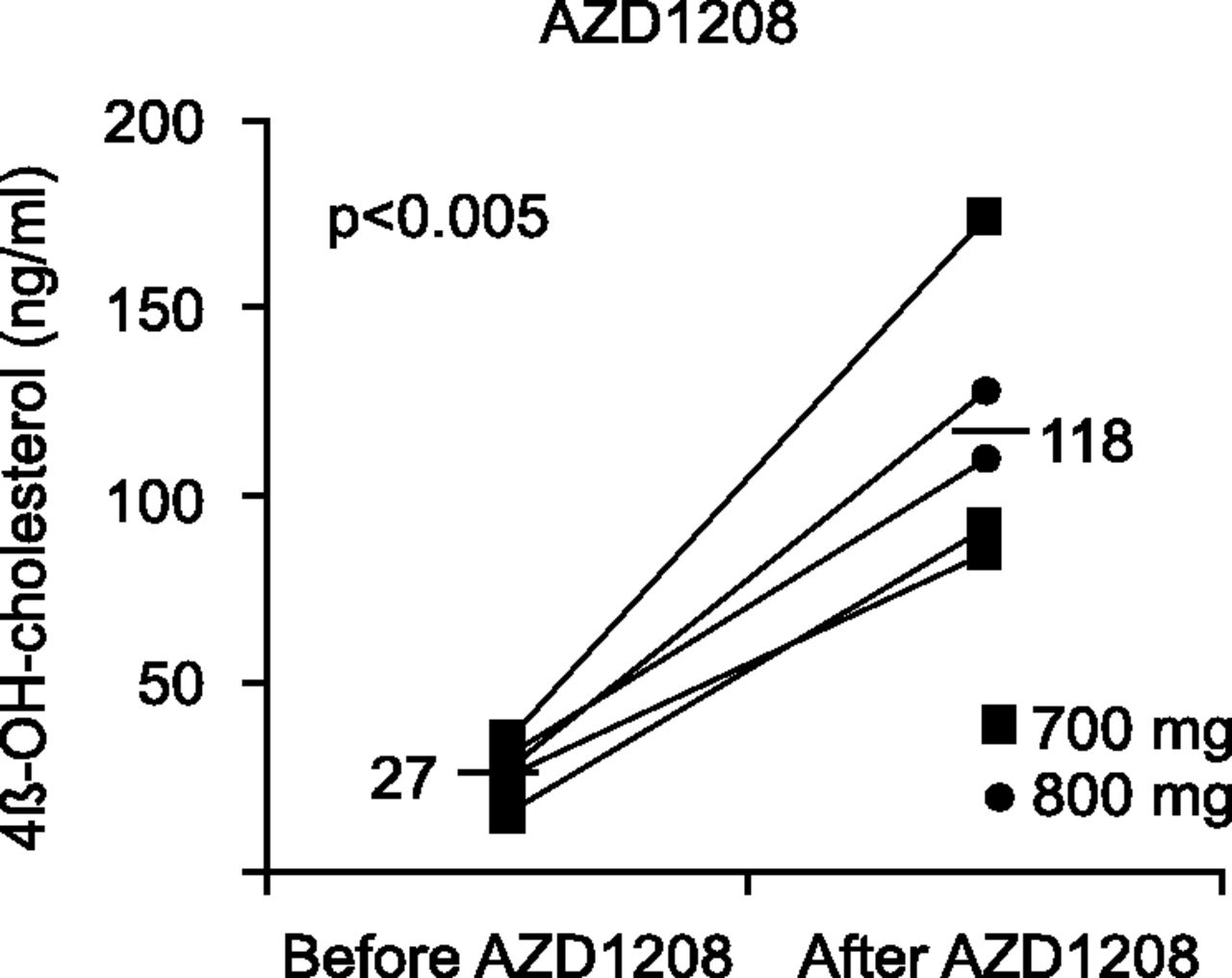
Plasma concentrations of 4β-hydroxycholesterol before and after 2 weeks of treatment with daily doses of 700 or 800 mg AZD1208.
Although the evaluation of 4β-hydroxycholesterol could be performed only on a limited number of subjects at the higher doses (due to administrative clinical conduct reasons), the results could be extrapolated to other lower doses where similar autoinduction effects could be observed. Further work is ongoing in our laboratories to understand the mechanism of this increased CYP3A activity and the reason for the lack of evidence from HepaRG and human cryopreserved hepatocyte induction studies. Our initial analysis indicates that the mechanism of this CYP3A induction appears to be unique and different from traditional induction mechanisms and several alternative approaches are being evaluated to understand this further. Importantly, this mechanism(s) of induction is not present in currently used in vitro systems; hence, these models would not identify other substances with a similar mechanism of induction.
Discussion
The AZ induction strategy integrates in vitro assays and in vivo studies to make a comprehensive assessment of the induction potential of a new chemical entity and also incorporates regulatory recommendations to comply with regulatory guidance documents. The development of a multiple assay P450 induction strategy enables induction to be assessed throughout the drug development process with the ability to match the assay to the throughput and depth of data required. Additionally, the assessment of induction of CYP1A, CYP2B6, and CYP3A in the HepaRG assay for selected compounds during lead optimization ensures coverage of all the major nuclear receptors involved in P450 induction and can direct further investigations. The higher relative availability of HepaRG compared with inducible human hepatocytes allows a fuller assessment of more compounds earlier in the drug development process. This enables six concentrations to be investigated as standard for every compound, thus ensuring a clear concentration-response evaluation and that clinically relevant concentrations are assessed through compound development. The availability of large batches of HepaRG cells has also enabled models to be generated that predict expected changes in midazolam AUC (CYP3A probe drug) from in vitro parameters such as F2 or EC50 with Emax.
Additionally, our strategy replaces three human hepatocyte donors with a single HepaRG experiment. Due to the correlation between HepaRG and hepatocytes, and in combination with predictive models, this allows compounds to be classified as inducers or noninducers and the next steps in the induction strategy decision tree to be selected. The argument that induction should be investigated in human hepatocytes from at least three donors to reflect interindividual variability in human response still needs to be confirmed. There are several factors that could impact the responsiveness of a batch of hepatocytes to induction such as the quality of preparation, disease state, and ongoing drug treatment at the time of liver resection. It should also be emphasized that the P450 enzyme expression and activities in primary hepatocyte cultures rapidly decline with time. The induction potential in a primary culture of hepatocytes is thus measured over a period when the P450 enzyme expression is decreasing or has already fallen to a very low basal level, which may confound the results. The studies by Grime et al. (2010) and Vermet et al. (2016) clearly showed that primary hepatocytes and HepaRG cells are equally effective in predicting the induction response within the variability observed in vivo by known CYP3A inducers. Therefore, HepaRG cells should be a good replacement for primary hepatocytes in drug discovery in order to evaluate potential CYP3A inducers in vivo.
Our strategy as presented focuses on CYP3A induction due to its importance in drug clearance. However, with the utilization of the aryl hydrocarbon receptor and CAR reporter gene assays in the initial stages the strategy can be extended to investigate CYP1A and CYP2B6 induction. Additionally, analysis of internal compounds assessed in HepaRG has shown the incidence of specific CYP2B6 inducers to be less than 1%, thus confirming the relative lack of importance of induction via CAR rather than PXR activation. While HepaRG cells have been shown to be induced by prototypical inducers of both CYP1A and 2B6 (Aninat et al., 2006; Kanebratt and Andersson, 2008b; McGinnity et al., 2009; Anthérieu et al., 2010) the correlation of the induction response with in vivo studies has not been established for these isoforms because of few compounds being identified as in vivo inducers. Therefore, the strategy currently lacks predictive power for specific inducers of CYP1A2 and 2B6. As requested by the regulatory authorities our P450 induction strategy utilizes mRNA as the sole endpoint. Induction of mRNA is not in a one-to-one relation with induction of enzyme activity; therefore, any predictions of induction risk through in vivo AUC or Cmax changes will always represent a worst-case scenario.
The FDA and EMA guidance documents refer to fresh or cryopreserved hepatocytes as the gold standard for evaluation of enzyme induction in vitro, and information from other in vitro systems such as reporter gene assays or HepaRG cells are considered complementary only. This is despite a number of convincing publications that PXR reporter gene assays and HepaRG cells are useful models to predict P450 induction in vivo, especially CYP3A induction (which is the major concern). CYP3A is induced by many drugs and the predictive value of in vitro results has therefore been possible to evaluate. However, the scientific evidence is now very convincing that primary hepatocytes do not offer any advantages over PXR reporter gene assays or HepaRG cells. On the contrary, the quality and stability of primary hepatocytes in culture as discussed previously may lead to confounding results in induction response evaluation.
The FDA and EMA guidance documents provide little information on the use of biomarkers for investigating possible interactions by new chemical entities. However, several recent publications have presented the use of biomarkers in the field of drug interactions. For instance, published data have convincingly demonstrated that induction of CYP3A can be estimated by the biomarker 4β-hydroxycholesterol when compared with the sensitive CYP3A probe drug midazolam. The rifampicin dose-response study (Kanebratt et al., 2008) and the case study (AZD1208) provide good evidence that 4β-hydroxycholesterol is a sensitive biomarker for CYP3A inducers. The AZD1208 case study is a challenging scenario where neither HepaRG nor primary human hepatocytes indicated possible CYP3A induction but in vivo clearly indicated induction of CYP3A, as shown by both nonlinear kinetics and an increase in 4β-hydroxycholesterol levels. As a result of this example, the strategy recommends inclusion of the measurement of 4β-hydroxycholesterol in all multiple ascending dose studies irrespective of in vitro results.
There are both ethical and practical advantages to using sensitive and selective biomarkers. The use of reliable biomarkers would eliminate the need to expose subjects to an unnecessary drug and is also easy to incorporate into clinical protocols. Samples for measuring a biomarker could be included in any clinical study when exposing subjects or patients to the study drug in a multiple dosing study, preferably for a minimum of 14 days. In summary, screening compounds in the preclinical phases using PXR and/or HepaRG cells generate enough robust data to assess the compounds inducing liability in vivo. Rare cases such as AZD1208 have been seen where no currently used in vitro method predicted CYP3A induction in vivo. Therefore, all compounds are recommended to include the sensitive CYP3A 4β-hydroxycholesterol biomarker in the multiple ascending dose study to investigate CYP3A induction liability early in the clinical program. 4β-Hydroxycholesterol is as sensitive as midazolam in assessing CYP3A induction and could replace exposure to an unnecessary drug such as midazolam to study CYP3A induction in vivo.
Since the introduction of this strategy, there has been one identified example of a positive 4β-hydroxycholesterol response from a cohort of more than 10 compounds assessed. This example was previously identified by in vitro assays detailed in the strategy as having an induction risk. There have been no further instances of compounds with mechanisms similar to AZD1208 within this cohort.
Authorship Contributions
Participated in research design: Rollison, Johansson, Kanebratt, Lambert, Vishwanathan, Andersson.
Performed data analysis: Jones, Rollison, Johansson, Kanebratt, Lambert, Vishwanathan, Andersson.
Wrote or contributed to the writing of the manuscript: Jones, Rollison, Johansson, Kanebratt, Lambert, Vishwanathan, Andersson.
Footnotes
- Received June 9, 2016.
- Accepted October 19, 2016.
↵
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.

Abbreviations
- AUC
- area under the curve
- AZ
- AstraZeneca
- CAR
- constitutive androstane receptor
- DDI
- drug-drug interaction
- DME
- drug metabolizing enzyme
- EMA
- European Medicines Agency
- FDA
- Food and Drug Administration
- P450
- cytochrome P450
- PK
- pharmacokinetics
- PXR
- pregnane X receptor
- SAR
- structure-activity relationship
- Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}