


Abstract
Seratrodast (ABT-001, AA-2414) undergoes cytochrome P450 (CYP)-dependent metabolism to a major (5-methylhydroxy seratrodast; 5-HOS) and a minor 4′-hydroxy seratrodast metabolite in human liver microsomes. The mean apparent Km andVmax for the formation of 5-HOS were 15.5 μM and 589.0 pmol 5-HOS formed/mg protein/min, respectively. Chemical inhibition using isoform-selective CYP inhibitors, correlation of 5-HOS formation with several isoform-specific CYP activities in a panel of liver microsomes, metabolism by microsomes derived from CYP cDNA-expressed B-lymphoblastoid cells, and immunoinhibition by isoform-specific anti-CYP antibodies indicated that 5-HOS formation is catalyzed by CYP3A and CYP2C9/10, with a minor contribution from CYP2C8 and CYP2C19. At clinically relevant concentrations, seratrodast was found to inhibit tolbutamide methylhydroxylation (IC50 = 60 μM), (S)-mephenytoin 4′-hydroxylation (IC50 = 50 μM), and coumarin 7-hydroxylation (IC50 = 95 μM), indicating the potential for significant clinical interactions. The inducers of CYP3A and/or CYP2C9 (e.g. rifampicin and phenytoin) are likely to alter the disposition of seratrodast.
Seratrodast (ABT-001; AA-2414) (fig. 1) is a potent thromboxane A2 inhibitor that was synthesized at Takeda Chemical Industries Ltd., Osaka, Japan. Seratrodast has been recently approved for the treatment of asthma in Japan. Both phase I and II metabolism are involved in the elimination of seratrodast in animal models. Seratrodast is metabolized to a major (5-HOS)1 and minor (4′-HOS) metabolite by human liver microsomes (fig. 1).
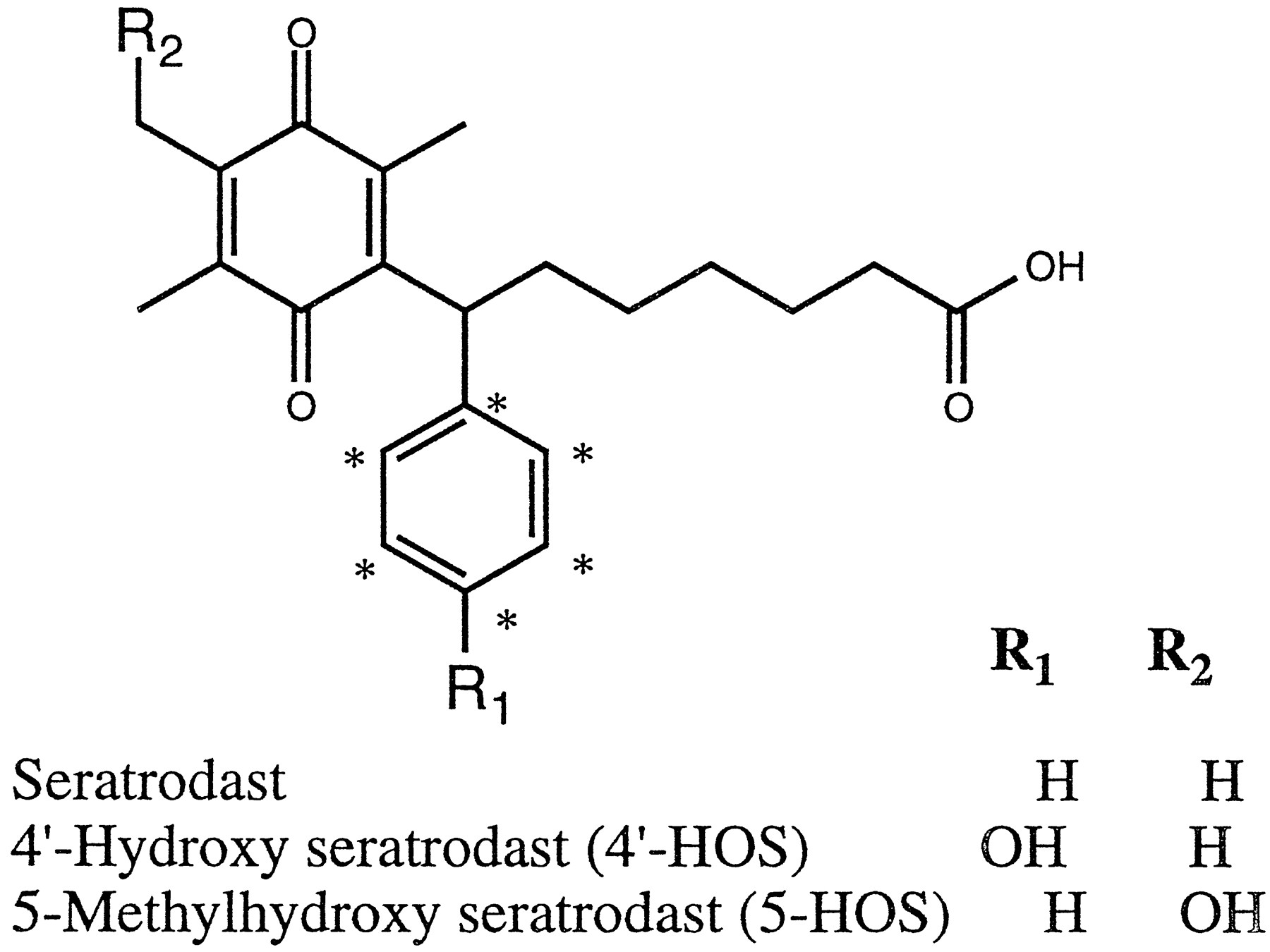
Structures of seratrodast and its human liver microsomal metabolites.
* Carbon-14 label.
For the treatment of asthma, seratrodast may be coadministered with several other therapeutic agents, such as theophylline and albuterol. This could potentially lead to interactions between seratrodast and coadministered drugs that are metabolized by the same enzymes that metabolize seratrodast. The purpose of this study was to identify the enzyme systems involved in the oxidative metabolism of seratrodast, with the intention of predicting the potential drug interactions.
Materials and Methods
α-Naphthoflavone, 8-methoxypsoralen, quinidine, 4-methylpyrazole, troleandomycin, tolbutamide, retinol, nifedipine, glucose 6-phosphate, glucose-6-phosphate dehydrogenase, β-NADP+, and MTT were purchased from Sigma Chemical Co. (St. Louis, MO). Ketoconazole and sulfaphenazole were purchased from Research Biochemicals International (Natick, MA). (S)-Mephenytoin was obtained from Salford Ultrafine Chemicals (Manchester, UK). Human B-lymphoblastoid-derived CYP microsomes and antihuman CYP2A6 monoclonal antibodies were purchased from Gentest Corp. (Woburn, MA). Rabbit antihuman-CYP3A4 and antihuman-CYP2C9 antibodies were purchased from Dr. Jerome Lasker (Mt. Sinai Medical Center, NY). Seratrodast was uniformly labeled with carbon-14 in the phenyl ring by Takeda Chemical Industries Ltd. (fig.1). For microsomal incubations (0.5 ml final volume), 2.5 μl of the appropriate seratrodast stock solution was added to achieve the desired final concentration, and each incubation contained 0.1 μCi of [14C]seratrodast (specific activity: 97.6 μCi/mg; >98% pure). Metabolite standards 4′-HOS and 5-HOS were obtained from Takeda Chemical Industries Ltd.
Microsomes.
Human livers were obtained from the International Institute for the Advancement of Medicine (Exton, PA). Microsomes were prepared by differential centrifugation as described previously (1). Microsomal protein concentration was determined using a bicinchoninic acid assay kit procedure (Pierce Chemical, Rockford, IL), with bovine serum albumin as the standard. Total CYP was determined by means of ferrous carbon monoxide complex formation (2). The following isoform-specific CYP-mediated activities of the microsomal bank were characterized (1): 7-ethoxyresorufin O-deethylase (CYP1A2) (3), coumarin 7-hydroxylase (CYP2A6) (4), [1,2-3H2]tolbutamide methylhydroxylase (CYP2C9) (5), S(+)-mephenytoin 4′-hydroxylase (CYP2C19) (6), [O-methyl-14C]dextromethorphanO-demethylase (CYP2D6) (7),N,N-dimethylnitrosamine N-demethylase (CYP2E1) (8), and erythromycin N-demethylase (CYP3A) (8).
Incubation.
Because seratrodast is a photosensitive compound, appropriate measures were taken to minimize exposure to light during experimentation. Individual incubations (final volume: 0.5 ml) were conducted in 4-ml borosilicate amber vials and consisted of 1 mg microsomal protein/ml in 100 mM phosphate buffer (pH 7.4), with final concentrations of 5 mM magnesium chloride, 5 mM glucose 6-phosphate, 1 mM β-NADP+, and 1 unit of glucose-6-phosphate dehydrogenase/ml. The drug, buffer, and microsomes were mixed and kept at 37°C for 5 min, and the reaction was started by adding the NADPH-generating system. The reaction was stopped by adding 0.5 ml of acetonitrile and vortexing. After centrifuging for 10 min at 3000 rpm, the supernatant was evaporated under nitrogen, and the residue was reconstituted in 200 μl of mobile phase. Samples were kept refrigerated until analysis. Just before the radio-HPLC analysis, an aliquot of the sample (130 μl) was mixed with 10 μl of MTT (10 mg/ml in methanol) to oxidize 2H-seratrodast and its metabolites to their respective quinone forms.
Radio-HPLC Method.
Separations were achieved at ambient temperature on a Beckman Ultrasphere 5 μm 4.6 × 250 mm C18 column. A step gradient of 30–45% acetonitrile in buffer (50 mM ammonium acetate, pH adjusted to 3.5 with trifluoroacetic acid) for 45 min, followed by 45–80% acetonitrile in buffer for >5 min, then followed by 80% acetonitrile in buffer for 7 min was used as column eluent at a flow rate of 1 ml/min. The HPLC retention times of reference standards of seratrodast, 4′-HOS, and 5-HOS were 54.4, 36.9, and 35.3 min, respectively.
Metabolism by B-Lymphoblastoid Microsomes.
Incubations with B-lymphoblastoid microsomes were conducted essentially as described above with 25 μM seratrodast and 3 mg of microsomal protein/ml, with an incubation time of 120 min. In the case of CYP2A6 microsomes, 50 mM Tris buffer (pH 7.5) was used instead of phosphate buffer.
Kinetics of Metabolite Formation.
The concentration range for studying the kinetics of seratrodast metabolism by human liver microsomes was 1–100 μM, with an incubation period of 10 min at 37°C. Due to lack of solubility, higher concentrations could not be used. The incubation period for kinetic experiments with B-lymphoblastoid microsomes was 120 min at 37°C. The manufacturer has shown that the reactions catalyzed by these microsomes are linear up to 120 min. Protein concentrations in incubations with B-lymphoblastoid microsomes were equivalent to 20 pmol CYP2C8, 60 pmol CYP2C9-arg, 100 pmol CYP2C9-cys, 25 pmol CYP2C19, and 130 pmol CYP3A4. Kinetic parameters were calculated by the weighted Lineweaver-Burk method using EnzymeKinetics version 1.3 software (Trinity Software, Campton, NH).
Correlation Analysis.
Correlation of the formation of 5-HOS with CYP isoform-specific activities in the microsomes prepared from a panel of human livers (N = 10) was studied at a concentration of 25 μM seratrodast (0.2 μCi/ml). Statistical analysis was performed using Instat 2.01 (GraphPad, San Diego, CA). Two-tailed Student’st test for paired data was performed to calculatep values.
Chemical Inhibition.
In chemical inhibition experiments, methanolic stock solutions of inhibitors were added just before the addition of a drug. An equivalent quantity of methanol was added for control incubations. Concentration of seratrodast in all chemical inhibition experiments was 20 μM. In the case of the mechanism-based inhibitors, troleandomycin and tienilic acid, the mixture of microsomes, inhibitor, and the NADPH-generating system was preincubated for 10 min at 37°C before the addition of seratrodast. The p value was calculated using one samplet test (Instat 2.01, GraphPad).
Immunoinhibition by Anti-CYP Antibodies.
Microsomes (FGL852, equivalent to 50 pmol CYP), IgG, and phosphate buffer were combined and kept for 3 min at 37°C, followed by 15 min at room temperature. The drug (20 μM final concentration) was then added, and the mixture was kept at 37°C for 5 min. The reaction was started by adding an NADPH-generating system, and incubation was conducted at 37°C for 15 min. Sample work-up and analysis were as described previously. The ratio of preimmune IgG and anti-CYP IgG was varied to achieve a range of anti-CYP IgG/CYP ratios while keeping the total amount of IgG added constant.
Other Assays.
[O-methyl-14C]DextromethorphanO-demethylase (10 μM) (7), 7-ethoxyresorufinO-deethylase (0.2 μM) (3), [1,2-3H2]tolbutamide methylhydroxylase (100 μM) (5), coumarin 7-hydroxylase (0.5 μM) (4),p-nitrophenol hydroxylase (30 μM) (9),S-mephenytoin 4′-hydroxylase (80 μM) (6), and terfenadine hydroxylase (10 μM) (10) activities were assayed by literature methods using HGD057 microsomes.
Results
Metabolite Formation.
Seratrodast was metabolized by human liver microsomes to a major (5-HOS) and a minor (4′-HOS) metabolite in an NADPH-dependent manner (fig. 1). In incubates, seratrodast and its metabolites existed primarily in their respective hydroquinone forms. Hence, the samples were oxidized with MTT before HPLC analysis to convert them completely to their respective quinone forms (11). The rate of formation of the major metabolite was linear for up to 10 min of incubation and a protein concentration of 1 mg/ml. Hence, 1 mg microsomal protein/ml and a 10-min incubation period were used for all experiments, except where specifically mentioned otherwise. Due to the low rate of formation, accurate data could not be obtained with 4′-HOS within the linear range of 5-HOS formation. Thus, this study is mostly devoted to the formation of 5-HOS. In a panel of liver microsomes (N = 10), the rate of formation of 5-HOS was found to vary 9.7-fold (330 ± 272 pmol 5-HOS formed/mg protein/min; range: 106–1033 pmol 5-HOS formed/mg protein/min) (fig. 2).

Variability in the rate of 5-HOS formation by a panel of human liver microsomes (N = 10).
Incubations were conducted for 10 min, with 1 mg/ml of protein, 25 μM seratrodast, and an NADPH-generating system. Values are the means of two determinations. Numbers in parentheses are the actual rates for the respective liver microsomes.
Metabolism of Seratrodast by Microsomes Derived form CYP cDNA-Expressed Lymphoblastoid Cells.
CYP1A2, CYP2A6, CYP2D6, and CYP2E1 B-lymphoblastoid microsomes did not metabolize seratrodast (table 1). CYP2B6, CYP2C8, CYP2C9-arg, CYP2C9-cys, CYP2C19, and CYP3A4 microsomes formed 5-HOS, whereas CYP2C8, CYP2C9-arg, CYP2C9-cys, and CYP2C19 microsomes formed 4′-HOS as well. The results of this study indicated the intrinsic capability of several isoforms of CYP to mediate the formation of 5-HOS and 4′-HOS.
Metabolism of seratrodast by specific CYP c-DNA transfected human B-lymphoblastoid cell microsomes
Kinetics of Seratrodast Metabolism.
The formation of 5-HOS followed monophasic Michaelis-Menten kinetics. In the concentration range tested, the mean apparentKm and Vmax values for the formation of 5-HOS were 15.5 ± 3.1 μM (range: 11.8–18.3 μM) and 589.0 ± 357.0 pmol of 5-HOS formed/mg protein/min (range: 177.6–948.1 pmol of 5-HOS formed/mg protein/min), respectively (table 2). The mean CLint =Vmax/Km was 36.9 ± 21.0 μl/mg protein/min (range: 15.1–57.7 μl/mg protein/min). Among B-lymphoblastoid microsomes, CYP2C9-arg exhibited the highest capacity to form 5-HOS, with a Km of 16.5 μM, aVmax of 410.6 pmol of 5-HOS formed/nmol CYP/min, and a CLint of 24.9 μl/nmol CYP/min, with CYP2C8 and CYP2C19 having substantial activity. In comparison, CYP3A4 was found to have a very low capacity (CLint = 0.9 μl/nmol CYP/min) to form 5-HOS. CYP2C9-arg, possibly the predominant CYP2C9 allelic variant in human liver (12), has several-fold higher capacity to form 5-HOS than another allelic variant, CYP2C9-cys (table 2), as has been observed with tolbutamide methylhydroxylation (13). All three isoforms of CYP2C were found to be capable of forming the minor metabolite 4′-HOS (table 1). Kinetic parameters obtained with all three CYP2C isoforms are approximately similar (table 3), probably indicating that they can contribute equally to the formation of this minor metabolite, depending on their relative content. The Km values for the formation of 4′-HOS were ∼2- to 5-fold higher than those obtained with 5-HOS. The CLint values for 5-HOS and 4′-HOS formation indicate that, whereas CYP2C19 is capable of forming both the metabolites at approximately similar rates, CYP2C8 (3-fold) and CYP2C9-arg (7.5-fold) form 5-HOS at a faster rate than 4′-HOS.
Kinetics of 5-HOS formation by human liver microsomes and microsomes derived from CYP c-DNA transfected B-lymphoblastoid cells
Kinetics of 4′-HOS formation by microsomes derived from c-DNA transfected B-lymphoblastoid cells
Correlation with CYP Marker Activities.
The formation of 5-HOS by 10 human liver microsomes was studied with the intention of correlating these data with isoform-specific CYP activities. The formation of 5-HOS correlated with multiple isoform-specific CYP activities (table 4), namely erythromycin N-demethylase (CYP3A, r = 0.885, p < 0.01), S-mephenytoin 4′-hydroxylase (CYP2C19, r = 0.954, p< 0.001), coumarin 7-hydroxylase (CYP2A6, r = 0.670,p < 0.05), and tolbutamide methylhydroxylase (CYP2C9/10, r = 0.696, p < 0.05). This indicated the possibility that more than one enzyme is involved in the formation of 5-HOS. Incidentally, the best correlation was obtained with S-mephenytoin 4′-hydroxylase (CYP2C19), which also correlated with coumarin 7-hydroxylase (CYP2A6; r = 0.667; p < 0.05) and erythromycinN-demethylase (CYP3A; r = 0.811;p < 0.01) in this panel of microsomes (table 4). Thus, the high correlation between 5-HOS formation and CYP2C19 activity could partially be due to this coincidence.
Correlation of the formation of 5-HOS with isoform-specific CYP activities
Inhibition of 5-HOS Formation by Isoform-Selective CYP Inhibitors/Substrates.
The effect of isoform-selective CYP inhibitors/substrates was examined at 20 μM seratrodast (table 5). Quinidine (CYP2D6, 2 μM) (14, 15) and 4-methylpyrazole (CYP2E1, 20 μM) (14, 15) did not affect 5-HOS formation, whereas marginal inhibition was obtained with α-naphthoflavone (CYP1A2, 5 μM, 20.7% inhibition (15), 8-methoxypsoralen (CYP2A6, 20 μM, 22.5% inhibition) (16), andS-mephenytoin (CYP2C19, 500 μM, 18.2% inhibition) (17,18). The CYP2C8 substrate retinol (100 μM) (19) inhibited 5-HOS formation by 40.2% CYP2C9/10 inhibitors/substrates produced significant inhibition: sulfaphenazole (5 μM, 38.5% inhibition) (14), tienilic acid (100 μM, 48.4% inhibition) (20), and tolbutamide (1000 μM, 42.0% inhibition) (17). Ketoconazole (CYP3A, 5 μM, 47.5% inhibition) (21) was a better inhibitor of 5-HOS formation than troleandomycin (CYP3A, 200 μM, 25% inhibition) (14). Nifedipine (25 μM, 59.4% inhibition), a substrate/inhibitor of CYP3A (21) and possibly an inhibitor of CYP2C8/9/10 (22, 23, 24), was found to be an effective inhibitor of 5-HOS formation. The inhibition obtained with CYP3A and CYP2C9/10 inhibitors (i.e. ketoconazole and sulfaphenazole or tienilic acid) was additive, indicating that these two isoforms are likely to be the major contributors to the formation of 5-HOS from seratrodast. The IC50 values for the inhibition of 5-HOS formation (performed with FGL852 microsomes) by ketoconazole, sulfaphenazole, and nifedipine were 2.9, 1.9, and 9.7 μM, respectively (data not shown), further confirming the major role of CYP3A and CYP2C9/10 in 5-HOS formation.
Effect of isoform-selective CYP inhibitors/substrates on the formation of 5-HOS by human liver microsomes
Immunoinhibition.
The effect of rabbit anti-CYP3A4 and anti-CYP2C9 immunoglobulin on the formation of 5-HOS was investigated with FGL852 microsomes at varying ratios of anti-CYP IgG and CYP. The antihuman-CYP2C9 antibodies used in this study cross-react with CYP2C8 and CYP2C19, and thus cannot distinguish between these isoforms. The anti-CYP2C9 and anti-CYP3A4 antibodies used in this study have been shown to produce >80% inhibition of tolbutamide methylhydroxylation and nifedipine oxidation, respectively, at a IgG/CYP ratio of 2.5. At an anti-CYP IgG/CYP ratio of 5, anti-CYP2C9 and anti-CYP3A4 antibodies inhibited 70.9% and 59.9% of 5-HOS formation, respectively (fig. 3). This confirms that CYP3A and members of the CYP2C subfamily are the predominant contributors to the formation of the major oxidative metabolite (5-HOS) of seratrodast in human liver microsomes. The antihuman-CYP2A6 monoclonal antibodies, which have been shown to inhibit >95% of coumarin 7-hydroxylase activity at a IgG/CYP ratio of 0.025 by the manufacturer, did not affect 5-HOS formation even at a ratio of 1.0 (data not shown), thus indicating that CYP2A6 is probably not involved in 5-HOS formation.

Inhibition of 5-HOS formation by rabbit antihuman-CYP3A4 and antihuman-CYP2C9 antibodies.
Effect of Seratrodast on Isoform-Specific CYP Activities.
The effect of seratrodast on isoform-specific CYP activities was examined using HGD057 microsomes. Seratrodast inhibited several isoform-specific CYP activities in human liver microsomes (fig.4). The IC50 values for the inhibition of CYP2C9 (IC50 = 60 μM), CYP2C19 (IC50 = 50 μM), and CYP2A6 (IC50 = 95 μM) were lower than the IC50 values obtained for other activities. This indicates the potential for interaction between seratrodast and CYP2C9/10, CYP2C19, and CYP2A6 substrates. Interestingly, seratrodast was found to be a very weak inhibitor of CYP3A-mediated terfenadine oxidation (IC50 > 200 μM), which is in agreement with low affinity of this enzyme for seratrodast, as demonstrated by aKm > 300 μM for formation of 5-HOS by CYP3A4 lymphoblastoid cell microsomes.

Effect of seratrodast on isoform-specific CYP activities.
The CYP isoform for which the activity is specific for, the concentration of the substrate used, and the IC50 value are presented in parentheses: □, ethoxyresorufin O-deethylase (CYP1A2; 0.2 μM; IC50 = 180 μM); ◊, coumarin 7-hydroxylase (CYP2A6; 0.5 μM; IC50 = 95 μM); ○, tolbutamide methylhydroxylase (CYP2C9/10; 100 μM; IC50 = 60 μM); ▵, S-mephenytoin 4′-hydroxylase (CYP2C19; 80 μM; IC50 = 50 μM); ✳, dextromethorphanO-demethylase (CYP2D6; 10 μM; IC50 = 200 μM); ⧫, p-nitrophenol hydroxylase (CYP2E1; 30 μM; IC50 = >200 μM); ⊕, terfenadine oxidase (CYP3A; 10 μM; IC50 = >200 μM).
Discussion
In this study, using several complementary techniques, the CYP isoforms involved in the formation of the major human liver microsomal metabolite of seratrodast have been identified. The formation of 5-HOS is predominantly catalyzed by CYP2C9/10 and CYP3A, with minor contributions from CYP2C8 and CYP2C19.
The initial studies with CYP-expressed B-lymphoblastoid microsomes indicated the intrinsic capability of CYP3A4 and CYP2C8/9/19 enzymes to form 5-HOS. The Km for 5-HOS formation by human liver microsomes (15.5 μM) was approximately the same as that determined with CYP2C9-arg microsomes (16.5 μM), but was ∼20-fold lower than the Km obtained with CYP3A4 microsomes (311.7 μM), with intermediate values for CYP2C8 and CYP2C19 microsomes. The formation of 5-HOS in a panel of human liver microsomes correlated with CYP2A6-, CYP2C9-, CYP2C19-, and CYP3A-specific activities, indicating the possibility of involvement of multiple enzymes. The inability of CYP2A6 microsomes to form 5-HOS and lack of inhibition by highly specific anti-CYP2A6 monoclonal antibodies indicated the noninvolvement of CYP2A6 in 5-HOS formation, although 8-methoxypsoralen, a CYP2A6 inhibitor (16), produced a marginal inhibition and a weak correlation was obtained with coumarin 7-hydroxylase activity. CYP2B6, which formed 5-HOS at a lower rate than the CYP2C/CYP3A4 microsomes, is not likely to be a significant contributor to the formation of 5-HOS in human liver microsomes, because it accounts for only ∼0.2% of the total CYP in human liver microsomes (25). The CYP2C9/10 and CYP3A inhibitors/substrates were found to be effective inhibitors of 5-HOS formation with fairly low IC50 values, further indicating the major role of these enzymes in this pathway. The IC50 values for inhibition of 5-HOS formation by ketoconazole (15) and sulfaphenazole (15) are several-fold higher than the values reported for the inhibition of the metabolism of isoform-specific substrates. The additive nature of inhibition by ketoconazole and sulfaphenazole or tienilic acid further confirmed the predominant role of CYP3A and CYP2C9/10 in 5-HOS formation. The CYP2C8 substrate retinol was found to be a fairly potent inhibitor of 5-HOS formation, whereas the CYP2C19 substrate (S)-mephenytoin was only marginally inhibitory. The ability of CYP2C8 and CYP2C19 B-lymphoblastoid microsomes to form 5-HOS indicate that these two enzymes are capable of forming 5-HOS. But, the potent inhibition of 5-HOS formation obtained with sulfaphenazole (IC50 = 1.9 μM), a selective inhibitor of CYP2C9/10 (13,15, 23), and marginal inhibition by (S)-mephenytoin (500 μM), which competitively inhibits a CYP2C19-mediated biotransformation with a Ki of ∼20 μM (18), indicate that, in human liver microsomes, CYP2C19 is likely to be only a minor contributor to the formation of 5-HOS. Even though retinol, a CYP2C8 substrate, was inhibitory and the comparable kinetic parameters obtained with CYP2C8 microsomes to those obtained with CYP2C9-arg microsomes, the potent inhibition obtained with sulfaphenazole, which does not affect CYP2C8-mediated biotransformations (23), indicates that CYP2C8 is also likely to be a minor contributor to the formation of 5-HOS in human liver microsomes. It is likely that, like tolbutamide, seratrodast is a substrate of CYP2C8/9/19, albeit with different affinities (26). It has been shown that the members of the CYP2C subfamily constitute ∼20% of the total CYP in adult human liver (25). Because all three CYP2C enzymes are capable of forming the metabolites of seratrodast, the actual contribution of each of these enzymes will depend on their relative levels. Because CYP2C9/10 is the predominant enzyme of the CYP2C subfamily,2it is likely that it is the major contributor to the oxidative biotransformation of seratrodast among CYP2C subfamily members. Even though CYP3A4 B-lymphoblastoid microsomes exhibited very weak ability to form 5-HOS, data obtained with chemical and immunoinhibition experiments strongly suggest that CYP3A contributes substantially to the formation of 5-HOS in human liver microsomes. This could be due to the relative abundance of CYP3A (30–50% of total CYP) in human liver, compared with relatively lower levels of CYP2C enzymes (25).
The multidirectional approach used in this study indicates that CYP3A and CYP2C9/10 are major contributors to the formation of 5-HOS, with a likely minor contribution from CYP2C8 and CYP2C19. The most convincing evidence for the involvement of these enzymes is the potent inhibition obtained with nifedipine, an inhibitor of CYP3A (21), CYP2C9 (22), and CYP2C8 (23, 24). Even though extensive data could not be obtained with 4′-HOS, due to the lower rate of formation, the studies conducted with B-lymphoblastoid microsomes indicate that all three isoforms of CYP2C can form 4′-HOS at comparable rates. The contribution of each of these isoforms to 4′-HOS formation will depend on the relative levels in an individual.
The peak and steady-state plasma concentrations of seratrodast after a 240 mg/day dose are 85 μM and 44 μM, respectively.3 These concentrations are several-fold higher than the Km value obtained for the formation of 5-HOS and is in the same range as the IC50 values for the inhibition of CYP2C9/10-, CYP2C19-, and CYP2A6-mediated biotransformations, indicating the potential for drug interactions. In vivo studies in animal models have indicated that conjugative metabolism (glucuronidation and sulfation) is more important in the overall metabolic clearance of seratrodast than the oxidative metabolism. If animal data are predictive of human disposition, it is likely that the oxidative pathways are minor contributors to the overall disposition of seratrodast. Data obtained in this study indicate that the inhibitors of CYP3A (e.g.ketoconazole) and CYP2C9/10 (e.g. sulfaphenazole and tienilic acid) are likely to inhibit the formation of 5-HOS in vivo in humans. Pharmacokinetic changes due to this inhibition are likely to be minor because of the low fraction of seratrodast metabolized by this pathway. Seratrodast can potentially inhibit CYP2C9/10-mediated (e.g. warfarin and tolbutamide), CYP2C19-mediated (e.g. S-mephenytoin and omeprazole), and CYP2A6-mediated (e.g. coumarin) biotransformations, thus resulting in adverse clinical interactions. More importantly, inducers of CYP3A and/or CYP2C (e.g. rifampicin, phenytoin, and glucocorticoids) (27-29) could potentially induce the formation of 5-HOS in humans, leading to significant pharmacokinetic changes.
Footnotes
-
Send reprint requests to: Dr. Gondi N. Kumar, Biotransformation Department, D-46V, AP9, Abbott Laboratories, 100 Abbott Park Road, Abbott Park, IL 60064-3500.
-
This study was supported by TAP Holdings, Inc. E.D. was the recipient of an Abbott Summer (1995) Research Internship.
-
↵2 Dr. J. M. Lasker, personal communication, 1996.
-
↵3 Dr. E. Samara, personal communication, 1996.
- Abbreviations used are::
- 5-HOS
- 5-methylhydroxy seratrodast
- 4′-HOS
- 4′-hydroxy seratrodast
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- CYP
- cytochrome P450
- IgG
- immunoglobulin G
- CLint
- intrinsic clearance
- Vmax
- maximum initial velocity
- Km
- Michaelis constant
- IC50
- concentration that inhibited 50% of the activity
- Received June 28, 1996.
- Accepted October 10, 1996.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}