


Abstract
Sodium periodate reacts with UDP-glucuronic acid (UDP-GlcUA) to generate a reactive derivative [periodate-oxidized UDP-GlcUA (o-UDP-GlcUA)]. The ability of this analog of UDP-GlcUA to inactivate and label the human recombinant UDP-glucuronosyltransferase (UGT) UGT1A6 via the UDP-GlcUA binding site was investigated. At an o-UDP-GlcUA concentration of 20 mM, the enzymatic activity of UGT1A6 was totally inactivated after 30 min of incubation at pH 7.4. Inhibition was irreversible, time-dependent, and concentration-dependent and exhibited pseudo-first order kinetics (kinact = 4.0 M−1·min−1). Cosubstrate protection with UDP-GlcUA was biphasic, with no protection in the first phase and almost total protection in the second phase, suggesting that at least 65% of the cross-linking occurs at the cosubstrate binding site. Partial inactivation byo-UDP-GlcUA led to a decrease inVmax, suggesting thato-UDP-GlcUA can act as an active site-directed inhibitor. Furthermore, proteins, including the UGTs, from membrane fractions of a recombinant V79 cell line expressing the UGT1A6 enzyme and from rat liver microsomes were cross-linked by in situ periodate oxidation of [β-32P]UDP-GlcUA. The present results suggest that periodate-oxidized UDP-GlcUA, which inactivates UGT1A6 by the possible formation of a Schiff base adduct with active site lysyl residues, can be used as a new affinity label for the UDP-GlcUA binding site.
Glucuronidation is a major biotransformation pathway for thousands of endogenous and xenobiotic compounds. This biotransformation is catalyzed by a family of enzymes, the UGTs,2 which are anchored in the membrane of the ER. The proposed transmembrane topology of UGTs describes proteins oriented predominately in the lumen of the ER, with a single α-helical membrane-spanning segment at the carboxyl terminus and a short sequence (positively charged) projecting into the cytoplasm (Jansen et al., 1992). It has been proposed that the UDP-GlcUA binding site is located in the conserved carboxyl-terminal region of UGTs, whereas the variable amino-terminal region directs aglycone specificity (Mackenzie, 1990) and dimerization (Meech and Mackenzie, 1997).
For structural investigations, experimental tools that can be used for rapid characterization of UGTs in cellular extracts and membrane preparations are indispensable. Photoaffinity labeling of UGTs with [β-32P]5N3-UDP-GlcUA has been developed for probing the UDP-GlcUA binding site. We have investigated the UDP-GlcUA binding domain of human UGT2B4 by expression in Escherichia coli of two peptides (amino acids 14–150 and 299–446) (Pillot et al., 1993), as Staphylococcus aureus protein A fusion proteins. Photoaffinity labeling experiments suggest that the uridine binding site of UDP-GlcUA is located between amino acids 299 and 446, whereas the glucuronic acid binding site is in the amino-terminal sequence of amino acids 14–150.
Periodate-oxidized nucleotides, such as oxidized ATP and others, have been used extensively to label the active sites of various proteins (Colman, 1983). These compounds are known to modify lysyl residues more specifically than do other residues, by forming Schiff bases or dihydroxymorpholino adducts (Lowe and Beechey, 1982), although the guanidino group of arginyl residues can also be cross-linked (Kanaani et al., 1995). Periodate-oxidized nucleotides are effective affinity labels for nucleotide-binding proteins, for the following reasons: the synthesis is usually easily accomplished, the structural analogy with the nucleotide is most often close enough for specific binding to the nucleotide site of the protein, and, finally, lysyl (or arginyl) residues present in the nucleotide binding site allow covalent binding of the oxidized ribose moiety.
We report here the covalent modification of UGT1A6, in membrane fractions from a recombinant V79 cell line expressing human liver UGT1A6, by a periodate-oxidized derivative of UDP-GlcUA. Our data indicate that inactivation of the enzyme results from covalent binding of o-UDP-GlcUA to the protein and that this binding occurs, at least partially, at the UDP-GlcUA binding site of recombinant human UGT1A6. We also provide evidence that o-UDP-GlcUA serves as an affinity label for UGT1A6.
Materials and Methods
Materials.
UDP-GlcUA (sodium salt), n-butyric acid (sodium salt), and 4-methylumbelliferone (free acid) were from Sigma Chemical Co. (St. Louis, MO). All other reagents were of the highest grade commercially available.
Cell Cultures and Membrane Fraction Preparation.
The V79 recombinant cell line expressing the human liver UGT1A6 was cultured as described previously (Battaglia et al., 1994). Membrane fractions of ER were obtained according to the protocol described by Battaglia et al. (1994) and were stored at −80°C in 5 mM HEPES, 0.25 M sucrose, 20 mM MgCl2, pH 7.4. No decrease in the enzymatic activity of the recombinant protein was observed for up to 6 months under these conditions.
o-UDP-GlcUA Synthesis.
o-UDP-GlcUA was synthesized as described by Prehm (1985), with minor modifications; 154 μmol of UDP-GlcUA (sodium salt; Sigma) was dissolved in 0.5 ml of 200 mM sodium phosphate buffer, pH 6.8. Oxidation was initiated by the addition (dropwise) of a 1.2-fold molar excess of a solution of sodium periodate (Sigma) dissolved in 0.5 ml of 200 mM sodium phosphate buffer, pH 6.8 (concentration of sodium periodate, 80 mg/ml). The reaction was carried out on ice in the dark, with continuous stirring. Oxidation of UDP-GlcUA was complete in <5 min under these conditions (as verified by HPLC; see below). Glycerol (50 μl of a 50%, v/v, solution) was added to terminate the oxidation, and the mixture was maintained under the same conditions for an additional 30 min, to scavenge unreacted periodate.
o-UDP-GlcUA was purified by ion-exchange chromatography on DE52 resin (Whatman, Maidstone, England). A 2- × 15-cm gel column was equilibrated using 10 bed-volumes of water. The purification was performed at 4°C, with protection from light. The oxidized derivative of UDP-GlcUA was eluted from the column with a 1-hr linear gradient of 0.5 M NaCl, at a flow rate of ∼3 ml/min; 4-ml fractions were collected. Iodate and unreacted periodate in the fractions were detected as described by Hinrichs and Eyzaguirre (1982). The purity of o-UDP-GlcUA was checked by HPLC (Rainin Instruments, Woburn, MA) with a Lichrosphere 5 RP18e column (250 × 4.0 mm; Phenomenex, Torrance, CA). The mobile phase consisted of 50 mM ammonium phosphate/phosphoric acid, pH 3.0, with 2.5% (v/v) methanol, and the product was eluted at a flow rate of 0.3 ml/min. UV absorbance was monitored at 260 nm. In this system, which was also used to check completion of the oxidation reaction before purification on the DE52 column, UDP-GlcUA and o-UDP-GlcUA exhibited retention times of 9.1 and 6.8 min, respectively. Fractions eluted from the DE52 column (free of iodine and presenting one HPLC peak, with a retention time of 6.8 min) were pooled and concentrated to dryness under vacuum. The residue was extracted with methanol, the extract was dried, and the residue was resuspended in water and stored at −80°C. The concentration of o-UDP-GlcUA was determined using an extinction coefficient of 10,000 cm−1·M−1. The stability of the oxidized derivative after storage was monitored by HPLC as described above.
Inactivation of Recombinant Human Liver UGT1A6 byo-UDP-GlcUA.
Inactivation of UGT1A6 was performed at 20°C under reduced lighting and was initiated by mixing membrane fractions of the recombinant cell line (4.9 mg of protein/ml) with o-UDP-GlcUA (2–25 mM) in 50 mM HEPES, 0.25 M sucrose, 20 mM MgCl2, pH 7.4. After incubation for various times (2–30 min), the inactivation was stopped by the addition of a 100-fold volume excess of 100 mM Tris-HCl, 20 mM MgCl2, pH 7.4, containing a 20-fold molar excess of sodium borohydride (over o-UDP-GlcUA). In preliminary experiments, NaBH4 alone did not inhibit the glucuronidation activity of UGT1A6. Enzymatic glucuronidation activity (3 μg of protein) toward 4-methylumbelliferone was assayed as previously described (Battagliaet al., 1994), using a Perkin-Elmer fluorometer (excitation/emission, 320/380 nm). A control experiment in whicho-UDP-GlcUA was omitted was performed simultaneously, to determine the fractional activity for a given time of inactivation. The inactivation kinetics were expressed as Equation 1where A/Ao is the fractional activity after t min of inactivation andkobs is the pseudo-first order inactivation constant for a given concentration of inactivator. For an inactivation process with an affinity label, kobs is expressed according to
Equation 1where A/Ao is the fractional activity after t min of inactivation andkobs is the pseudo-first order inactivation constant for a given concentration of inactivator. For an inactivation process with an affinity label, kobs is expressed according to
The apparent KM for UDP-GlcUA was determined for native and partially o-UDP-GlcUA-inactivated UGT1A6 with a constant saturating concentration of 4-methylumbelliferone (1 mM), over a UDP-GlcUA concentration range of 0.05–5.0 mM. Data fitting Michaelis-Menten kinetics were analyzed using EnzymeKinetics software (Trinity Software, Campton, NH).
Labeling with In Situ Periodate-Oxidized [β-32P]UDP-GlcUA.
[β-32P]UDP-GlcUA was synthesized as previously described (Battaglia et al., 1996), resulting in a radiolabeled cosubstrate with a specific activity of approximately 2.5 mCi/μmol. Membrane fractions (50 μg of protein) from nontransfected V79 cells, recombinant UGT1A6, or rat (Sprague-Dawley) liver microsomes were incubated with 5 μM [β-32P]UDP-GlcUA in 100 mM HEPES, 15 mM MgCl2, pH 7.4, for 1 min at room temperature.In situ oxidation was initiated with 40 mM sodium periodate. After 1 min, NaBH4 (final concentration, 200 mM) was added and the tubes were kept on ice for 1 hr; NaBH4 was added again and incubation on ice was continued for an additional 1 hr. Proteins were separated by 10% SDS-PAGE, and rat liver UGTs were identified by Western blotting with a rat anti-p-nitrophenol-UGT antibody, a generous gift from M. Green and Dr. T. Tephly (University of Iowa, Iowa City, IA). UGT1A6 was identified by Western blotting, as previously described (Ouzzineet al., 1994).
Preparative Gel Electrophoresis.
The Bio-Rad Prep Cell model 491 was used essentially as described previously (Battaglia et al., 1997); briefly, a 12% SDS-polyacrylamide running gel (pH 8.8, approximately 6 × 3 cm) was polymerized overnight. The stacking gel (pH 6.8, approximately 2 × 3 cm) was polymerized just before loading of the samples. Membrane fractions (5 mg of protein) from the recombinant cell line expressing the UGT1A6 enzyme were incubated with [β-32P]UDP-GlcUA (5 μM), oxidized with sodium periodate, and then reduced with NaBH4, as described above. Proteins were diluted 40-fold in water, concentrated by ultrafiltration through Centricon-30 membranes (Amicon, Berverly, MA), precipitated by addition of a 5-fold excess volume of 10% trichloroacetic acid, and mixed with prestained molecular mass markers (Sigma) [triose phosphate isomerase from rabbit muscle (35.2 kDa) and pyruvate kinase from chicken muscle (75.2 kDa)] in a denaturing buffer (3.6 M urea, 20 mM Tris, 0.14 M dithiothreitol, 5%, w/v, SDS, bromophenol blue, pH 8.0). Electrophoretic separation was performed as previously described (Radominska and Drake, 1994). Selected fractions containing radiolabeled protein were subjected to analytical electrophoresis followed by Western blotting, as previously described (Ouzzine et al., 1994).
Results
Preparation and Purification of o-UDP-GlcUA.
o-UDP-GlcUA was prepared according to a published procedure (Prehm, 1985). It is known that cis-glycols are oxidized more quickly than are trans-glycols (Glick, 1969); therefore, this procedure cleaves and oxidizes the ribose ring of UDP-GlcUA between the 2′- and 3′-carbon atoms and leaves the glucuronic acid moiety intact (Prehm, 1985). After 5 min of periodate oxidation, UDP-GlcUA could not be detected by HPLC. The reaction was then quenched with an excess of glycerol. An improved purification procedure was developed using anion-exchange chromatography on a DE52 column and elution with a gradient of sodium chloride. The previously published method (size-exclusion chromatography) failed to separate iodate and unreacted periodate from o-UDP-GlcUA, which is critical because of the possible inhibitory effect of periodate on UGT activities. The postulated reaction mechanism of o-UDP-GlcUA binding is presented in fig. 1. Periodate-oxidized nucleotides react with amino groups of proteins, resulting in the formation of Schiff bases, which can be further stabilized by reduction with sodium borohydride (Löwet al., 1992).

Structure of o-UDP-GlcUA and postulated reaction mechanism of o-UDP-GlcUA binding.
Time and Concentration Dependence of the Inactivation of UGT1A6 byo-UDP-GlcUA.
Inactivation was performed at pH 7.4, to prevent β-elimination from periodate-oxidized nucleotides (Löw et al., 1992). Preliminary experiments using a concentration range of 5–20 mMo-UDP-GlcUA and 30-min inactivation demonstrated concentration-dependent inhibition of UGT1A6 activity (data not shown). Total inactivation was observed at a concentration of 20 mMo-UDP-GlcUA. Binding of the UDP-GlcUA analog to the UGT1A6 enzyme was irreversible, because extensive dilution of theo-UDP-GlcUA-treated membrane fractions did not suppress inhibition. To further characterize the effect of this UDP-GlcUA analog on enzyme activity, the time and concentration dependence of inactivation was studied (fig. 2). The linearity of the curves presented on a semilogarithmic scale is an indication of pseudo-first order inactivation (eq. 1). We also observed that the inactivation rate was enhanced in the presence of NaBH4. Therefore, it appears that at least a fraction of the adduct exists as a Schiff base, which can be reduced to a stable secondary amine by incubation with NaBH4. Inactivation also occurred without NaBH4 but was less effective (approximately two thirds of the inactivation rate; data not shown), possibly because of the slow reversibility of the enzyme-inhibitor complex in the absence of a reducing agent under the experimental conditions used for enzymatic assays. The slopes of the curves represent the pseudo-first order inactivation constants for given concentrations of inactivator, and a replot of kobs as a function of the concentration of o-UDP-GlcUA yielded a value of 4.0 min−1·M−1 for the second order inactivation rate constant. Thekobs values were proportional too-UDP-GlcUA concentrations (2–25 mM) (fig. 2,inset), and higher concentrations of inhibitor could not be used because of limited solubility.

Time-dependent inactivation of UGT1A6 by o-UDP-GlcUA.
Membrane fractions of UGT1A6 (4.9 mg of protein/ml) were incubated with 20 mM o-UDP-GlcUA for 2–15 min. Inactivation was determined as a function of time after 100-fold dilution, as described in Materials and Methods. The fractional activity was calculated after adjustment for the control assay, which did not contain o-UDP-GlcUA. Inset, plot of the inactivation constant (kobs) as a function of the concentration of the inactivator (2–25 mM).
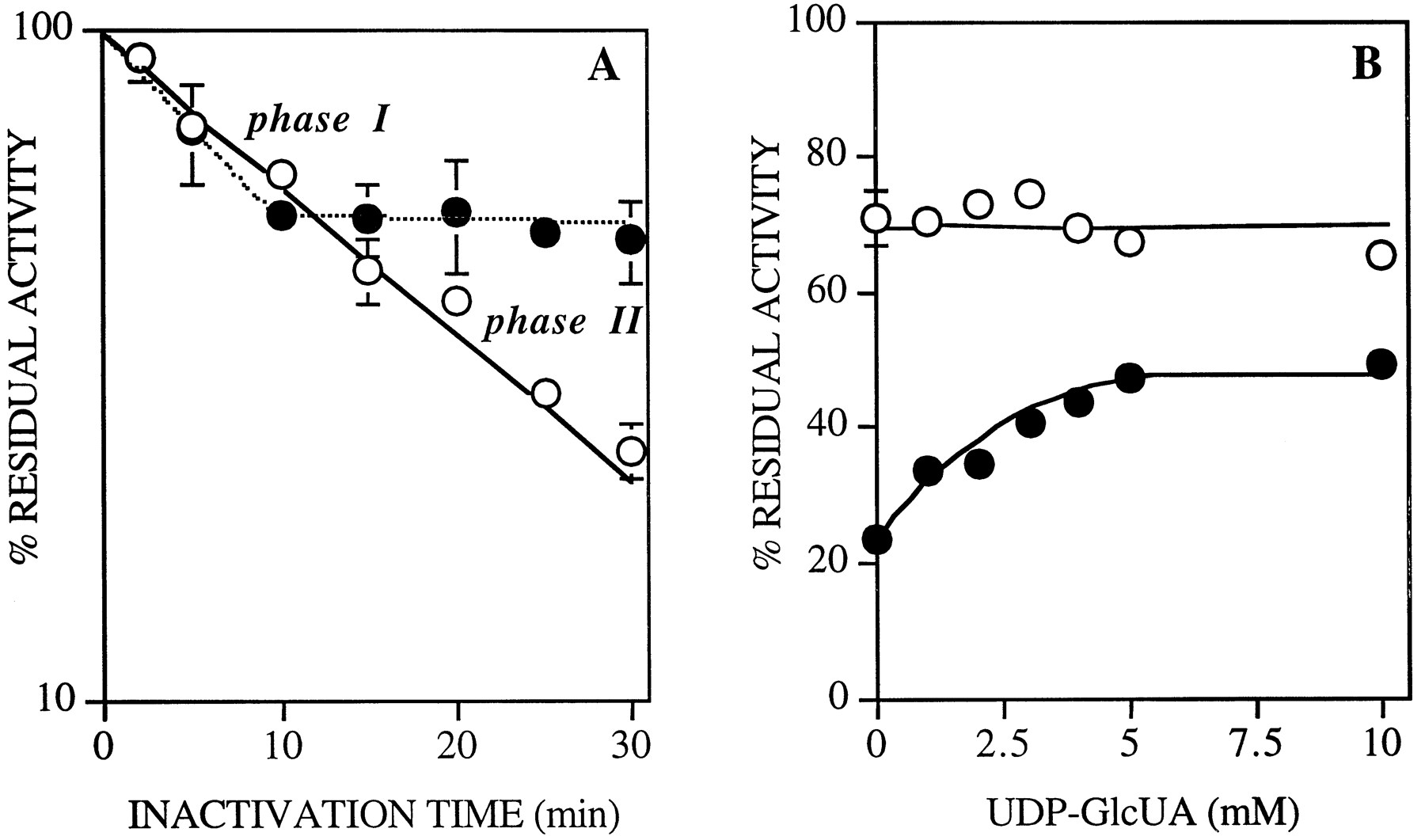
Partial UDP-GlcUA Protection of UGT1A6 from Inactivation byo-UDP-GlcUA.
The influence of preincubation of the UGT1A6 enzyme with UDP-GlcUA on the inactivation by o-UDP-GlcUA was studied. Fig.3A shows a biphasic effect of preincubation with the cosubstrate on the rate of inactivation byo-UDP-GlcUA. The first inactivation phase (phase I, ∼0–10 min with 5 mM o-UDP-GlcUA), which accounts for approximately 35% of the inhibition, was not affected by UDP-GlcUA, whereas in the second phase (phase II, >10 min) UDP-GlcUA provided almost total protection from inactivation. This protective effect was further analyzed by evaluating the influence of increasing UDP-GlcUA concentrations on the residual activity observed after 5 min (phase I) and 30 min (phase II) of inactivation with 5 mM o-UDP-GlcUA (fig. 3B). Fig. 3B shows that UDP-GlcUA decreased the o-UDP-GlcUA inactivation of UGT1A6 in phase II (in a concentration-dependent and saturable manner), whereas identical UDP-GlcUA concentrations did not affect the inactivation in phase I. Therefore, after an initial nonspecific inactivation phase, almost total protection was observed with saturating concentrations of UDPGlcUA.

Effect of preincubation of UGT1A6 with UDP-GlcUA on inactivation kinetics.
A, Membrane fractions (4.9 mg of protein/ml) were treated with 5 mM o-UDP-GlcUA, with (•) or without (○) preincubation with 5 mM UDP-GlcUA. At the times indicated, aliquots were obtained and diluted 100-fold (as described inMaterials and Methods), and enzyme activity was measured. A control reaction with no inactivator was carried out under the same conditions. B, Residual activity was evaluated after 5 min (○) or 30 min (•) of inactivation by 5 mMo-UDP-GlcUA, in the presence of increasing concentrations of UDP-GlcUA (1–10 mM).
Effects of Partial Inactivation with o-UDP-GlcUA on Some Kinetic Parameters of UGT1A6.
Membrane fractions were inactivated with 15 mM o-UDP-GlcUA for 5 min, and the kinetic parameters for UDP-GlcUA were evaluated and compared with those of the native enzyme. ApparentKM (UDP-GlcUA) andVmax values were 197 ± 30 μM and 45 ± 3 nmol/min/mg (mean ± SD, N = 3) for the partially inactivated UGT1A6, compared with values of 133 ± 58 μM and 107 ± 10 nmol/min/mg (mean ± SD,N = 3), respectively, for the native enzyme. Therefore, partial inactivation of UGT1A6 by o-UDP-GlcUA appears to decrease the catalytic rate.
Labeling of UGTs with In Situ Periodate-Oxidized [β-32P]UDP-GlcUA.
We previously synthesized [β-32P]UDP-GlcUA (Battaglia et al., 1996). Here we have developed a new method for the affinity labeling of UGTs, by in situperiodate oxidation in the presence of this radiolabeled cosubstrate. The procedure for in situ labeling involved the incubation of membrane fractions from recombinant V79 cells expressing human liver UGT1A6 or rat liver microsomes with [β-32P]UDP-GlcUA, oxidation with sodium periodate, and reduction of the derivatized protein with an excess of NaBH4. Substrate-protection experiments (fig. 3) showed that some of the modified residues were not in the active site of the enzyme, raising the potential problem of (some) nonspecific radiolabeling of proteins. In situ labeling of nucleotide-binding proteins has been shown to improve the binding specificity, compared with preoxidized nucleotides (Peter et al., 1993). A low concentration of [β-32P]UDP-GlcUA (5 μM) was also used to reduce nonspecific binding (Löw et al., 1992). Covalent incorporation of the radiolabel into UGTs, as well as several other ER membrane proteins, was observed (fig.4). Rat liver microsomal proteins, in the range (50–54 kDa) known to include the UGTs (Drake et al., 1991), were labeled, as documented by autoradiography of gels after SDS-PAGE (fig. 4A). Detergent treatment is known to release UGT activity latency in rat liver microsomes. Detergent treatment before in situ labeling increased the overall background levels, providing a less clear pattern of radiolabeled proteins (data not shown). Furthermore, detergent treatment did not increase the labeling of UGTs, compared with intact microsomes, possibly because thein situ labeling method unsealed the vesicles or because of rapid transport of [β-32P]UDP-GlcUA into the lumen of the microsomes (Drake et al., 1992). Fig.4B shows the Coomassie staining (fig. 4B,lane 1), Western blot analysis (fig. 4B,lane 2), and corresponding autoradiographic analysis (fig.4B, lane 3) of purified32P-labeled o-UDP-GlcUA-UGT1A6 complex obtained by preparative electrophoresis. From these results, in situ periodate oxidation of [β-32P]UDP-GlcUA appears to be an efficient method to radiolabel UGT1A6. The radiolabeling was not enhanced when gentler protein precipitation conditions (using organic solvents with no boiling and no acid or base) (Wessell and Flugge, 1984) were used before electrophoresis. The stability of the cross-linked products (particularly under acidic conditions), combined with the enhanced inactivation by o-UDP-GlcUA in the presence of NaBH4, suggested that the covalent binding was most likely achieved by means of a reduced Schiff base. Because we used [β-32P]UDP-GlcUA for in situperiodate oxidation and proteins were still labeled, it appears that the adduct does not undergo β-elimination (Lowe and Beechey, 1982).o-UDP-GlcUA is a homobifunctional cross-linking reagent, and this raised the possibility of inhibition by multiple intermolecular cross-links. The UGT1A6 enzyme was detected at its expected molecular mass by Western blotting after SDS-PAGE of membrane fractions treated with o-UDP-GlcUA under reducing conditions, excluding the possibility of multiple intermolecular cross-links (fig. 4Band results not shown).

Affinity labeling of rat liver microsomal proteins and recombinant human liver UGT1A6 with in situperiodate-oxidized [β-32P]UDP-GlcUA.
A, Rat liver microsomal proteins (50 μg) were incubated with [β-32P]UDP-GlcUA (5 μM) beforein situ oxidation with sodium periodate, followed by reduction with NaBH4 (as described in Materials and Methods). The radiolabeled proteins were separated by SDS-PAGE and analyzed by autoradiography (lane 1). The radiolabeled UGTs identified by Western blotting with anti-p-nitrophenol-UGT antibody (lane 2) are indicated (arrowhead). The densitometric scan of the autoradiograph is shown on the left. B, Membrane fractions (5 mg of protein) of recombinant V79 cells expressing the UGT1A6 enzyme were incubated with [β-32P]UDP-GlcUA (5 μM), oxidized, and then reduced as described for A. The radiolabeled UGT1A6 polypeptides were purified by preparative electrophoresis (as described inMaterials and Methods). Fractions containing the UGT1A6 polypeptides were identified by Western blot analysis, concentrated by ultrafiltration, pooled, and analyzed on SDS-PAGE (lane 1, Coomassie staining), by Western blotting with a polyclonal, monospecific, anti-UGT1A6 antibody (lane 2), and by autoradiography (lane 3). The same amount of protein (10 μg) was loaded in each lane.
Discussion
We have used UDP-GlcUA analogs (reversible inhibitors and photoaffinity probes) to characterize the cosubtrate binding site of UGTs (Drake et al., 1992; Battaglia et al., 1995). Although o-UDP-GlcUA has been used to label hyaluronate synthase (Prehm, 1985; Prehm and Mausolf, 1986), to the best of our knowledge this UDP-GlcUA analog has not been tested on UGTs. We demonstrated here that o-UDP-GlcUA cross-links with amino acid residues located in the UDP-GlcUA binding site.
The inactivation process obeyed pseudo-first order kinetics in the range of o-UDP-GlcUA concentrations used. However, no saturation kinetics could be observed under these conditions (fig. 2), suggesting that there was no reversible binding before inactivation. Therefore, no dissociable complexes between the UGT1A6 enzyme and the inactivator would be detectable. This was unexpected, considering the structural analogy of o-UDP-GlcUA with the enzyme cosubstrate (Prehm, 1985). Similar results have been recently observed with other compounds designed to be affinity labels (Nakamura et al., 1995) or mechanism-based inhibitors (Braun et al., 1995). In both cases, the most likely explanation for the observed nonsaturable inactivation was the relatively highKi of the inhibitor. Therefore, considering eq. 2 described in Materials and Methods, forKi ≫ [I],kobs tends tokinact·[I]/Ki and no saturation is apparent in the range of inactivator concentrations used, even in the presence of a dissociable intermediate complex. Higher concentrations of o-UDP-GlcUA could not be used because of limited solubility of the inactivator. However, partial inactivation of the enzyme affected Vmax, suggesting that the binding of the probe impaired catalysis, as would be expected for a bulky ligand covalently bound within the UDP-GlcUA binding site.
Important evidence that o-UDP-GlcUA binds at the catalytic site can be obtained by cosubstrate-protection experiments. In the present studies, it was documented that the loss of glucuronidation activity produced by o-UDP-GlcUA could be prevented, in large part, by preincubation of the enzyme with unmodified UDP-GlcUA. Fig. 3 shows that the nonspecific binding (approximately 35%) of the inactivator was followed by specific binding of o-UDP-GlcUA within the active site. This dual effect of UDP-GlcUA on the inactivation of UGT1A6 by o-UDP-GlcUA suggests that the inhibitor modifies two classes of lysyl residues concomitant with the loss of activity. One class of reactive residues is not located within the UDP-GlcUA binding site, as evidenced by the lack of substrate protection depicted as phase I in fig. 3. The second class of residues reacts more slowly with the inactivator (phase II in fig. 3) and is protected against modification by UDP-GlcUA preincubation. This strongly suggests that this second class of modified residues is embedded in the active site of the enzyme. Substrate-protection experiments showed that the residues that are cross-linked most quickly are not protected by UDP-GlcUA. Because these residues are the first to react with the relatively hydrophilic inhibitor, they could be located on, or closer to, the protein surface, possibly in an area surrounding the active site. A similar observation has been made for the chemical modification by butanedione of arginyl residues of the UDP-GlcUA binding site of rat liver UGTs (Zakim et al., 1983). This phenomenon has also been observed with periodate-oxidized nucleotides (Lowe and Beechey, 1982; Prehm, 1985; Rao et al., 1991;Hilden et al., 1995).
An additional application of the in situ oxidation of nucleotides involves their potential use as radiolabeled affinity probes to identify active site residues of the UGTs. Therefore, we studied the covalent incorporation of 32P-labeledo-UDP-GlcUA into UGTs in rat liver microsomes and recombinant UGT1A6 in membrane fractions. The specificity of the labeling toward UDP-GlcUA-utilizing proteins was evaluated by comparison of the affinity labeling using o-UDP-GlcUA with the photoaffinity labeling using [β-32P]5N3-UDP-GlcUA. Photoaffinity labeling with [β-32P]5N3-UDP-GlcUA was used previously for the characterization of UDP-GlcUA-binding proteins (Drake et al., 1991, 1992; Drake and Elbein, 1992;Radominska et al., 1994). Comparison of the labeling of rat liver microsomes using [β-32P]5N3-UDP-GlcUA (Drake et al., 1992) with the autoradiograph in fig.4A shows that in situ periodate-oxidized [β-32P]UDP-GlcUA cross-linked the same proteins in rat liver microsomes as did the photoaffinity label; among these, the UGTs were predominant. The UGT1A6 enzyme in membranes from the recombinant cell line was also specifically labeled by in situ periodate-oxidized [β-32P]UDP-GlcUA (fig. 4B). In spite of the lower level of expression of the single UGT1A6 in this system (compared with total rat liver UGTs) and relatively high background labeling, significant radiolabeling of the recombinant enzyme was observed after purification by preparative electrophoresis (fig. 4B). In situ periodate oxidation of radiolabeled UDP-GlcUA avoids derivatization of the native sugar nucleotide to generate an affinity label, the extended side chain of which can sometimes preclude binding within the active site. Use of this ligand can be considered an alternative approach to the identification and characterization of UDP-GlcUA-utilizing proteins.
In the present study, we have shown that o-UDP-GlcUA, in addition to 5N3-UDP-GlcUA (Drake et al., 1992), is a useful affinity label for characterization of the active site of UGTs. Different amino acids within the active site can be identified with each of these active site-directed probes, based on their structural analogies with UDP-GlcUA and their reactivities toward amino acid residues of the active site. o-UDP-GlcUA probes an area of the active site surrounding the ribofuranose moiety of the cosubstrate, with high specificity for lysyl residues. 5N3-UDP-GlcUA, which carries the photoreactive azido group at the 5′-position of the uridine moiety of the cosubstrate, covalently reacts with amino acid residues of the active site surrounding the uracil base of UDP-GlcUA. Reliable data can be generated by probing the UDP-GlcUA binding site with both of these complementary, 32P-labeled, affinity probes.
Our preliminary peptide-mapping studies of UGTs photolabeled with [β-32P]5N3-UDP-GlcUA support a site of cross-linking of the photoprobe with UGT1A6 between Val350 and Glu403. Alignment of this sequence with the amino acid sequences of known UDP-glycosyltransferases shows that this region is highly conserved (Hundle et al., 1992). Several lysyl and arginyl residues are present in this region of UGT1A6. One of the residues, Lys351, is especially interesting, because it is highly conserved in the UGTs. Strong conservation of Lys351 among UDP-glycosyltransferases, in combination with our results indicating that this residue might be located in the active site, suggests that this residue might have an important function in the protein. Additional studies will be necessary to identify the amino acid(s) involved in the cross-linking in the active site of this enzyme.
Acknowledgments
We thank Prof. B. Burchell for providing the cDNA used to express UGT1A6 and J. Little for critically reviewing the manuscript.
Footnotes
-
Send reprint requests to: Anna Radominska-Pandya, Division of Gastroenterology, University of Arkansas for Medical Sciences, 4301 W. Markham, Slot 567–1, Little Rock, AR 72205.
-
↵1 This work was completed while N.T. was a student at Laboratoire de Biochimie Métabolique et Cellulaire, UFR SciFA, Université de Metz (Metz, France).
-
This work was supported in part by National Institutes of Health Grants DK45123 and DK49715 (to A.R.-P.).
- Abbreviations used are::
- UGT
- UDP-glucuronosyltransferase
- ER
- endoplasmic reticulum
- SDS
- sodium dodecyl sulfate
- PAGE
- polyacrylamide gel electrophoresis
- UDP-GlcUA
- UDP-glucuronic acid
- o-UDP-GlcUA
- periodate-oxidized UDP-glucuronic acid
- 5N3-UDP-GlcUA
- 5-azido-UDP-glucuronic acid
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- Received December 22, 1997.
- Accepted April 17, 1998.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}